

Abstract
AMP-activated-protein-kinase (AMPK) is a key sensor and regulator of cellular and whole-body energy metabolism and plays a key role in regulation of lipid metabolism. Since lipid metabolism has been implicated in neuronal amyloid-β (Aβ) homeostasis and onset of Alzheimer’s disease, we investigated the involvement of AMPK in neuronal lipid metabolism and Aβ production. We observed in cultured rat cortical neurons that Aβ production was significantly reduced when the neurons were stimulated with AMPK activator, 5-aminoimidazole-4-carboxamide-1-D-ribofuranoside (AICAR), but increased when AMPKα2 was knocked out, thus indicating the role of AMPK in amyloidogenesis. Although the detailed mechanisms by which AMPK regulates Aβ generation is not well understood, AMPK-mediated alterations in cholesterol and sphingomyelin homeostasis and in turn the altered distribution of Aβ precursor-protein (APP) in cholesterol and sphingomyelin rich membrane lipid rafts participate in Aβ generation. Taken together, this is the first report on the role of AMPK in regulation of neuronal amyloidogenesis.
Keywords: AMPK, Amyloid-β, Amyloid precursor protein, Cholesterol, Lipid rafts, Sphingomyelin
1. Introduction
Amyloid-β (Aβ), a peptide of 39–43 amino acids, is the main constituent of amyloid plaques in the brains of Alzheimer’s disease patients. The Aβ peptide is generated from its precursor protein (APP) by sequential proteolysis mediated by β- and γ-secretases [1,2]. Recent studies have described that β- and γ-secretases exhibit their optimum activities in bilateral structure of enriched cholesterol and sphingolipids within the membrane, called “detergent insoluble/resistant membrane micro-domains (DIM/DRM)” or “lipid rafts” [1,2]. Therefore, the cellular metabolism of cholesterol and sphingolipids might influence the metabolism of APP for Aβ generation. Indeed, experimental sequestration of cholesterol or blockade of sphingolipid synthesis was reported to inhibit APP processing for Aβ generation [3,4]. In addition, we recently demonstrated that inhibition of cholesterol biosynthesis pathway by 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitor lovastatin reduced APP distribution in lipid rafts and inhibited Aβ generation [5].
Studies from our laboratory and others have demonstrated that AMP-activated-protein-kinase (AMPK; EC 2.7.1.109) plays a critical role in cellular and whole-body energy and lipid metabolism, and its dysfunction directly contributes to metabolic imbalance associated with obesity [6,7]. Obesity has been implicated in increased incidence of Alzheimer’s disease [8]. However, the role for AMPK in Alzheimer’s disease is not well understood.
As named, AMPK senses intracellular and systemic energy states by sensing cellular AMP/ATP ratio and/or metabolic cytokines (adipokines; i.e. leptin, ghrelin and adipostatin) and endo-cannabinoids (See review [9]). Once activated, AMPK induces/promotes ATP producing catabolic processes but inhibits ATP consuming synthetic processes by regulating gene expressions and activities of enzymes that catalyze key metabolic branch points [9]. Studies have shown that AMPK regulates activities of HMG-CoA reductase, ceramide synthase 5 and serine palmitoyl transferase, and thus modulates cholesterol and sphingolipid synthesis [9–12]. Neurons express high levels of AMPK due to their high energy demand [13]. Therefore, changes in neuronal AMPK activity could influence integrity and function of lipid rafts by altering neuronal cholesterol and sphingolipid homeostasis, and thus affect APP metabolism for generation of Aβ. However, no study has yet focused on a possible role of AMPK in neuronal lipid metabolism and amyloidogenesis.
In this study, we report that activation of neuronal AMPK activity inhibits Aβ generation by reducing sphingomyelin levels as well as APP distribution in lipid raft fractions. On the other hand, deletion of AMPKα2 enhances Aβ generation by altering the cholesterol and sphingomyelin levels and thus increasing APP distribution in the lipid raft fractions. This is the first report for the role of AMPK in neuronal lipid metabolism associated with APP processing leading to generation of Aβ.
2. Materials and methods
2.1. Primary rat cortical neuron culture and drug treatments
For neuron cell culture, brain cortices derived from embryonic day 17 rats (Sprague–Dawley) or AMPKα2 knockout mice [14] or their wild type control mice (C57BL/6) were treated with 0.125% trypsin, and the dissociated cells were cultured on poly-D-lysine coated culture plates or dishes in Neurobasal medium (Invitrogen, San Diego, CA, USA) containing 2% B27 supplement (Invitrogen), L-glutamine (0.5 mM; Sigma–Aldrich Co., St. Louis, MO, USA), L-glutamate (25 μM; Sigma–Aldrich Co.) and penicillin/streptomycin mixture (Invitrogen). The cultures were maintained in 5% CO2 at 37 °C for 7 days and exchanged with B27 free Neurobasal medium for drug treatment. AICAR (BioMol, Plymouth Meeting, PA, USA) was prepared in distilled dimethylsulfoxide (DMSO; Sigma–Aldrich Co.).
2.2. Analysis of α- and β-secretase activity and Aβ40/Aβ42 release
The activities of α- and β-secretases in post-nuclear cell extracts or lipid raft fractions were measured using fluorogenic assay kits purchased from R&D Systems Inc. (Minneapolis, MN, USA). The activities were measured by SPECTRAmax® Gemini XS® fluorimeter with SOFTmax PRO® software (Molecular Devices, Chicago, IL, USA) with excitation at 345 nm and emission detection at 500 nm. For quantification of Aβ in media, culture media was centrifuged (1000g for 10 min at 4 °C) and 100 μl supernatant was used for colorimetric ELISA by using human Aβ (1–40) and (1–42) assay kits purchased from IBL Co., Ltd. (Japan) or Wako chemicals (Japan) which are fully compatible with rat Aβ40 or Aβ42.
2.3. Western blot analysis and antibodies
Western blot analysis was performed using antibodies against N-terminal APP695 (22C11, Chemicon, Temecula, CA, USA), C-terminal APP (Ab18813, Abcam, Cambridge, MA), BACE1 (Chemicon), ADAM10 (SantaCruz Biotech.), flotillin-1 (SantaCruz Biotech.), clathrin (SantaCruz Biotech.), PrP (SantaCruz Biotech.), CD71 (SantaCruz Biotech.), pan- and phospho-AMPK (Thr172) (Cell Signaling Tech. Inc., Panvers, MA, USA), pan- and phospho-ACC (Ser79) (Cell Signaling Tech. Inc.).
2.4. AMPKα activity assay
AMPKα activity was assayed as described previously [6] in homogenized neuron cell lysates in lysis buffer (50 mM Tris–HCl, pH 7.4, containing 50 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 10% glycerol, 1% Triton X-100 and protease inhibitor mixture). Approximately 200 μg of cell lysate was incubated with anti-AMPKα antibody for 2 h, then 30 μl of protein A/G plus agarose was added and incubated for an additional 1 h at 4 °C. The immune complexes were washed twice in lysis buffer and twice in kinase buffer (62.5 mM HEPES, pH 7.0, 62.5 mM NaCl, 62.5 mM NaF, 6.25 mM sodium pyrophosphate, 1.25 mM EDTA, 1.25 mM EGTA and 1 mM dithiothreitol), incubated at 30 °C in 30 μl of kinase assay buffer containing 200 μM AMP/ATP mixture and recombinant ACC protein (Millipore, Billerica, MA) for 20 min. The reaction was terminated by addition of SDS–PAGE sample loading buffer and boiling. The resultant phosphorylated ACC levels were analyzed by Western blot analysis.
2.5. Extraction of membrane micro-domains
The cultured cells were washed in ice cold PBS twice and lysed in 0.4 ml MBS buffer [25 mM MES, pH 6.5, 150 mM NaCl, 1 mM Na3VO4 and protease inhibitor cocktail (Roche, Indianapolis, IN)] containing 0.5% Lubrol WX (ICN Biochemicals, Cleveland, OH, USA) for 30 min on ice and homogenized by 10 strokes up and down in a tightly fitted Dounce homogenizer. The homogenates were centrifuged at 1000g for 10 min at 4 °C and the resultant supernatants were analyzed for protein quantity. The same protein quantities of post-nuclear lysates were mixed with the same volume of 80% Nycodenz (Nycomed, Roskilde, Denmark) in MBS buffer with 0.5% Lubrol WX. The resulting 40% Nycodenz containing lysate mixtures were overlaid with two volumes of 30% and one volume of 5% Nycodenz in MBS with 0.5% Lubrol WX as described previously [15]. Following centrifugation for 2 h at 80,000g in a TLV-100 rotor (Beckman, Fullerton, CA, USA) 10 equal volumes of fractions were collected.
2.6. Quantification of cholesterol and sphingomyelin
Total lipids were extracted from cultured neurons by the Folch method in a mixture of 2:1 chloroform/methanol (vol/vol). The extract was washed with 0.2 volumes of saline (NaCl 0.9%) and centrifuged at 2000 rpm for 10 min. The organic phase was used for analysis of neutral lipids (i.e. cholesterol) and acidic lipids (sphingomyelin and other phospholipids) were analyzed using HPTLC as described earlier [16]. All lipid levels were quantified by densitometric scanning using an Imaging Densitometer (Model GS-670, Bio-Rad), and software provided with the instrument by the manufacturer.
3. Results
3.1. AMPK activator decreases Aβ generation in cultured cortical neurons
Recent studies have shown that neurons express high levels of AMPK due to their high energy demand [13]. Because neurons express the amyloidogenic form of APP (APP695) and produce Aβ peptide, we designed experiments to examine whether AMPK is implicated in the regulation of neuronal Aβ generation. For it, primary cultured embryonic cortical neurons were treated with AI-CAR. Fig. 1A-i shows that AICAR dose dependently reduced neuronal Aβ40/42 production without altering cell viability significantly (Fig. 1A-ii). The Western analysis using antibodies against phospho-Thr172-AMPKα, an active form of AMPK, or phospho-Ser79-ACC, a substrate of AMPK (Fig. 1B-i), and in vitro kinase assay for AMPK (Fig. 1B-ii) indicates that AICAR treatment of neurons in culture induces AMPK activation. These data indicate that AMPK activation may negatively regulate Aβ generation in neurons.
Fig. 1.

AMPK activators decrease Aβ generation in cultured neuron cells. To examine the effect of AMPK activators on Aβ secretion, the primary cultured rat cortical neurons were incubated with indicated concentration of AICAR (5-aminoimidazole-4-carboxamide-1-D-ribofuranoside) for 36 h, and then Aβ40/42 levels in culture media were measured by ELISA (A-i). Under the same experimental conditions, neuronal viability was assayed by MTT assay (A-ii). The effect of AICAR (0.5 mM/3 h) on protein levels of p-AMPK, p-ACC and β-actin (B-i) and AMPK enzyme activity (B-ii) were measured by Western blot and by in vitro kinase assay using recombinant (rec.) ACC as a substrate. The vertical bar on each group indicates the standard error of mean (*P < 0.05, ***P < 0.001 compared to control group).
3.2. AICAR modulates APP β-cleavage by altering APP distribution in the specific lipid raft domains
APP processing by β-secretases (i.e. BACE1) is a first step in Aβ generation [1]. The resultant C99 fragment (C-terminal fragment of APP generated by β-secretase) from this step is further processed by γ-secretase to toxic Aβ peptides [1]. In this study, we observed that AICAR treatment down-regulates neuronal C99 levels but without altering APP or BACE1 levels (Fig. 2A-i) and α- and β-secretase activities (Fig. 2A-ii), suggesting that AICAR-induced reduction in APP β-cleavage is independent of cellular levels of APP and BACE1 and the activities of α- and β-secretases. Since APP β-cleavage is predominantly mediated in cholesterol and sphingomyelin rich specific membrane domains, called “lipid rafts”, we then examined the effect of AMPK activation on the distribution of APP and BACE1 in lipid rafts (Fig. 2B-i and ii). The lipid rafts were purified from neurons as described previously [5]. The lipid raft gradient fractions were analyzed for the distribution of APP as well as lipid raft markers. As shown in Fig. 2B-i and ii, AICAR treatment of neurons reduced APP levels in lipid raft fractions that overlap with PrP (a marker for lipid rafts containing glycosylphosphatidylinositol-anchored proteins) and flotillin-1 (a marker for caveolae) double positive fractions (# 1–3), but not with PrP negative and flotillin-1 positive fractions (# 4–7), and also not with CD71 and clathrin double positive fractions (non-lipid raft fractions; # 8–10). AICAR treatment did not alter PrP and flotillin-1 levels (Fig. 2B-i) and total protein levels (Fig. 2B-iii) in fractions # 1–3 indicating that the observed reduction in APP levels in fractions # 1–3 may not be due to differences in protein loading on the gel. In contrast to decreased APP, no alteration in β-secretase activities or BACE1 levels in lipid raft fractions was observed in AICAR treated cells (Fig. 2C). Taken together, these data indicate that AI-CAR treatment reduces Aβ generation possibly by reducing APP levels in the lipid rafts and thus reduced production of Aβ (Fig. 1), rather than modulating α- or β-secretase activity (Fig. 2C).
Fig. 2.
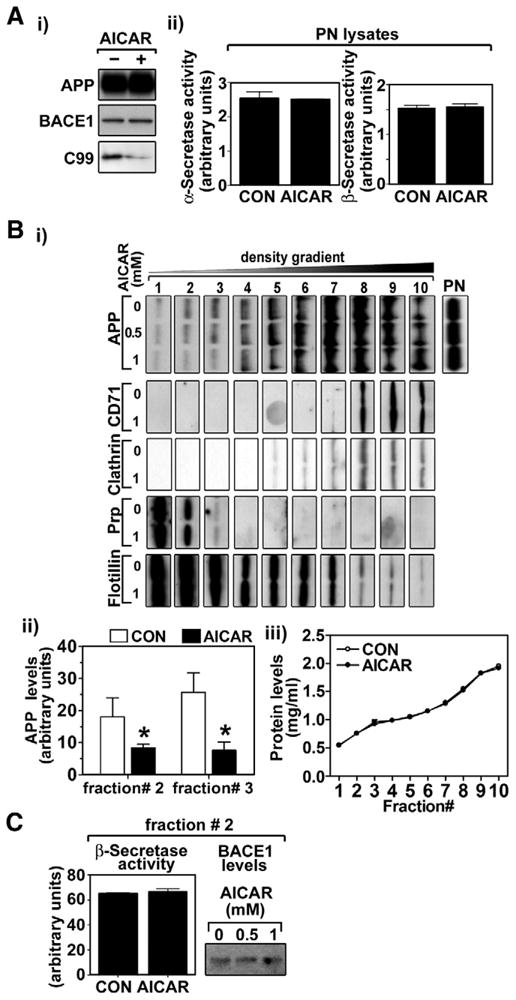
AICAR modulates APP β-cleavage through reducing APP lipid raft distribution. The effect of AICAR (0.5 and/or 1 mM/36 h) on cellular levels of APP, BACE1 and C99 (C-terminal fragment of APP generated by β-secretase; β-CTF) (A-i), activities of α- and β-secretases in post-nuclear fractions (A-ii), APP distribution in membrane micro-domains (B-i) were analyzed in primary cultured rat cortical neurons. The distribution of APP in the lipid raft fractions was determined by comparing protein levels of APP and non-lipid raft markers (CD71 and clathrin), marker for glycosylphosphatidylinositol-anchored protein containing lipid rafts (PrP) or caveolae marker (flotillin-1) (B-i). The protein levels of APP in fractions # 2 and 3 (control and 0.5 mM AICAR) were quantified and represented on bar graph (B-ii). The lack of differences in protein quantities in each fraction between control (CON) and AICAR treated groups (B-iii) indicate that the observed alterations in APP levels are not due to the differences of protein amounts. The effects of AICAR on α-and β-secretase activities and BACE1 protein levels in lipid raft fractions (fraction # 2) were also measured (C). The vertical bar on each group indicates the standard error of mean (*P < 0.05 compared to control group in each fraction).
3.3. Aβ generation is increased in AMPKα2 knockout neurons
The above studies indicate that pharmacological activation of AMPK negatively regulates generation of Aβ. To further support the role of AMPK in the regulation of neuronal Aβ generation, we next examined the effect of AMPK deficiency on neuronal Aβ production and APP distribution in the lipid raft fractions from neurons of AMPKα knockout (KO) mice. Since neurons primarily express the α2 catalytic subunit of AMPK as shown in Fig. 3A-i, neurons cultured from AMPKα2 KO mice and their wild type (WT) control mice were used in these experiments. In both neuronal cultures, there were no detectable differences in cell morphology and viability as shown in Fig. 3A-ii. However, neurons cultured from AMPKα2 KO mice produced significantly higher levels Aβ40/42 peptides (Fig. 3B). Further, to understand the relationship between Aβ generation and APP distribution, we prepared lipid raft fractions from neurons cultured from AMPKα2 KO and WT mice. As expected, AICAR treatment of WT mouse neurons reduced the distribution of APP in lipid raft fractions (Fig. 3C). On the other hand, the increased production of Aβ40/42 peptides in AMPKα2 KO neurons correlated with higher APP levels in lipid raft fractions (fractions # 1–3) from AMPKα2 KO neurons (Fig. 3C). These data document that AMPKα2 negatively regulates APP processing to Aβ peptides by reducing the distribution of APP in lipid rafts.
Fig. 3.
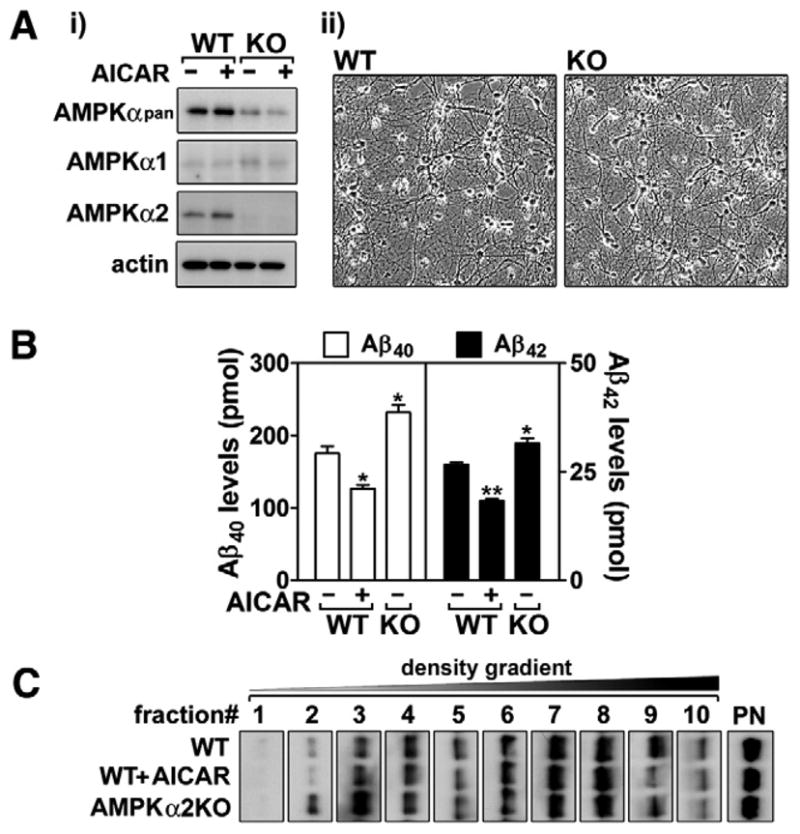
Knockout of AMPKα2 increases neuronal Aβ production. Following the culture of cortical neurons from wild type (WT) and AMPKα2 knockout (KO) mice, protein levels of AMPKα1 and α2 were analyzed to examine a major AMPKα isoform (α1 vs. α2) (A-i). In addition, the difference in neuronal morphology between WT and KO was also examined (A-ii). Under these experimental conditions, the role of AMPKα2 in the production of neuronal Aβ40/42 (B) and APP distribution in lipid raft fractions (C) were analyzed. The vertical bar on each group indicates the standard error of mean; **P < 0.01; ***P < 0.001 for comparison to vehicle (VHC, dimethylsulfoxide) treated WT or KO neurons; +P < 0.01 for comparison to VHC treated WT neurons.
3.4. AMPK regulates neuronal cholesterol and sphingomyelin levels
Cholesterol and sphingomyelin are the major lipid components in lipid rafts which play pivotal role in APP metabolism leading to Aβ generation. Since AMPK has been implicated in control of biosynthesis of cholesterol and sphingomyelin [9,11], we examined whether AMPK down-regulates Aβ generation by affecting the neuronal levels of cholesterol and sphingomyelin. As expected, we observed that the neurons from AMPKα2 KO mice had higher levels of sphingomyelin as compared to neurons from WT mice (Fig. 4A-i). In addition, sphingomyelin levels were decreased in neuron cultures treated with AICAR (Fig. 4A-i). To further understand the effect of AICAR on sphingomyelin distribution in different membrane micro-domains, we examined the levels of sphingomyelin in gradient of control and AICAR treated neurons (Fig. 4A-ii). We observed that AICAR treatment of neurons reduced sphingomyelin levels in lipid raft fractions as similar to APP distribution in lipid raft fractions (Fig. 2B).
Fig. 4.
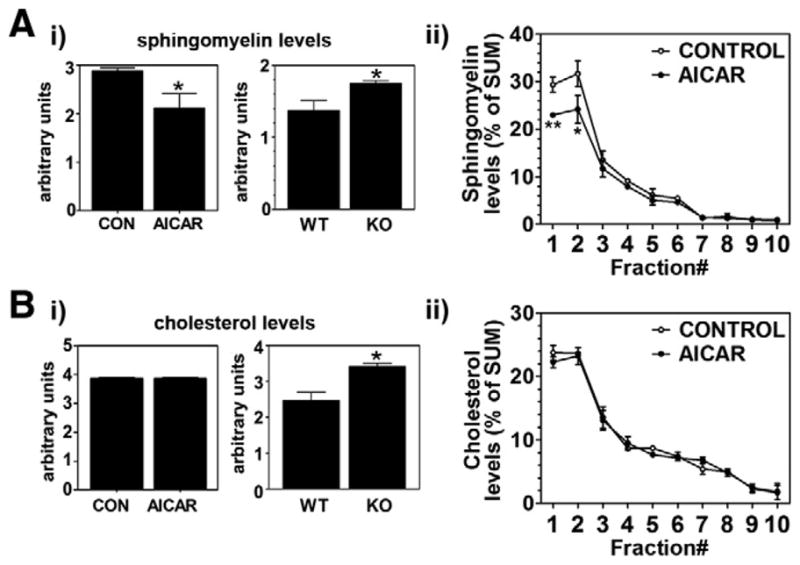
AICAR regulates neuronal levels of sphingomyelin and cholesterol. Following the culture of wild type and AMPKα2 knockout cortical neurons, the neurons were incubated with/without AICAR (0.5 mM for 36 h), and then levels of sphingomyelin (A) and cholesterol (B) in post-nuclear extracts (i) and in lipid raft fractions (ii) were analyzed as described under materials and methods. The vertical bar on each group indicates the standard error of mean (*P < 0.05 and **P < 0.01 compared to control group).
Activation of AMPK has been implicated in inhibition of cholesterol synthesis by down-regulating enzyme activity of HMG-CoA reductase [9]. Accordingly, we observed increased cholesterol levels in the neurons cultured from AMPKα2 KO mice (Fig. 4B-i). This data suggests that AMPK inactivation could cause overload of both sphingomyelin and cholesterol in neurons. Despite these data, however, stimulation of wild type neurons with AICAR for activation of AMPK did not affect cholesterol levels in whole cell lysate (Fig. 4B-i) as well as in lipid raft fractions (Fig. 4B-ii). Taken together, these data suggest that although the reduction in the basal levels of cholesterol is very tightly regulated, AMPK is involved in the regulation of neuronal cholesterol and sphingomyelin levels, which, in turn, regulates APP distribution in the lipid rafts and thus its metabolism leading to generation of Aβ.
4. Discussion
AMPK is a key sensor and regulator of intracellular and whole-body energy metabolism and plays a key role in regulation of lipid metabolism [9]. AMPK regulates activities and gene expressions of HMG-CoA reductase, serine palmitoyl transferase and ceramide synthase 5, and thus controls cholesterol and sphingolipid metabolism [7,9,11,12]. Cholesterol and sphingolipids are the major constituents of lipid rafts which provide optimum environment for APP metabolism leading to generation of Aβ [1,2]. Therefore, AMPK is likely to be involved in Aβ generation and thus Alzheimer’s pathology by regulating lipid raft homeostasis. However, no study to date has tested this hypothesis. Here we demonstrate that AMPK controls Aβ generation through modulating neuronal cholesterol and sphingomyelin levels, and thus distribution of APP in lipid rafts.
Membrane proteins are assigned to three categories: those that are mainly found in the rafts, those that are present in the liquid-disordered phase (non-lipid raft fractions), and those that represent an intermediate state, moving in and out of rafts [17]. Studies documented that β-secretase (BACE) and γ-secretase complexes are localized in lipid rafts, and that is critical for their actions on APP metabolism leading to Aβ [1,2]. On the other hand, APP is observed to be localized in both lipid raft and non-lipid raft fractions [15], and alteration in distribution of APP between non-raft and raft fractions has been implicated in altered Aβ generation [1,2]. In the present study, we observed that AMPK activation selectively inhibited APP distribution (but not BACE1) in low density lipid raft fractions (fractions # 1–3 in Fig. 2B and C). Although mechanism underlying differential effect of AMPK on the distributions of APP and BACE1 in lipid rafts is not understood at present, the reported strong tendency of S-palmitoylated BACE1 to localize in lipid rafts [18] may play a role in unaltered BACE1 distribution in lipid rafts under AMPK activated conditions.
AMPK has been implicated in regulation of cholesterol and sphingolipid biosynthesis by regulating gene expressions and activities of associated enzymes [9,11,12]. Consistent with these studies, we observed that AMPK activation reduced the sphingomyelin levels in lipid rafts as well as whole neuronal lysates (Fig. 4A-i and ii). However, cholesterol levels in neuronal lysates and purified lipid rafts were not altered by AICAR treatment under the same experimental conditions (Fig. 4B-i and ii). We recently reported similar observations in hippocampal neurons treated with lovastatin (an inhibitor of cholesterol synthetic pathway), where cholesterol levels were not altered by lovastatin treatment up to 36 h [5]. The observed unaltered cholesterol levels under lovastatin or AICAR treated conditions may be due to the long half life of pre-existing cholesterol and/or the observed reduction in synthetic rates are still able to maintain cellular cholesterol homeostasis. Interestingly, the observed increased levels of cholesterol and sphingomyelin with loss of AMPK activity in cultured AMPKα2 KO neurons (Fig. 4A and B) suggest that AMPK inactivation could cause an overload of both cholesterol and sphingomyelin in neurons. In turn, these data also suggest that activation of AMPK may efficiently reduce cholesterol overload but may not reduce below the basal levels because of alternate mechanisms for maintaining cholesterol homeostasis. On the other hand, both the overloaded and the basal sphingomyelin levels were efficiently reduced by AMPK activation (Fig. 4A and B), thus suggesting a role for neuronal sphingomyelin homeostasis in lipid raft function for APP metabolism and generation of Aβ.
In summary, these studies describe for the first time that AMPK controls neuronal Aβ generation by modulating distribution of APP in lipid raft membrane micro-domains, and thus raising the possibility that AMPK could be a potential therapeutic target for Alzheimer’s disease. Although detailed mechanism is not known, the reported role of AMPK in regulation of cholesterol and sphingolipid biosynthesis [9,11,12] and our observations demonstrating the increased sphingomyelin and cholesterol levels under AMPK deficient conditions and the decreased sphingomyelin levels under AMPK activated conditions suggest a role for AMPK in lipid metabolism associated with lipid raft function and integrity, and APP distribution in lipid rafts and thus Aβ generation.
Supplementary Material
Acknowledgments
We thank Ms. Joyce Bryan for laboratory assistance and Ms. Chara Williams for secretarial assistance. This study was supported in part by grants from National Institute of Health (NS-22576, NS-34741, NS-37766, AG-25307, RR018823 and RR015455). We would like to thank Dr. Benoit Viollet in INSERM U567 for providing AMP-Kα2 knockout mice.
References
- 1.Kaether C, Haass C. A lipid boundary separates APP and secretases and limits amyloid beta-peptide generation. J Cell Biol. 2004;167:809–812. doi: 10.1083/jcb.200410090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheng H, Vetrivel KS, Gong P, Meckler X, Parent A, Thinakaran G. Mechanisms of disease: new therapeutic strategies for Alzheimer’s disease-targeting APP processing in lipid rafts. Nat Clin Pract Neurol. 2007;3:374–382. doi: 10.1038/ncpneuro0549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Puglielli L, Ellis BC, Saunders AJ, Kovacs DM. Ceramide stabilizes beta-site amyloid precursor protein-cleaving enzyme 1 and promotes amyloid beta-peptide biogenesis. J Biol Chem. 2003;278:19777–19783. doi: 10.1074/jbc.M300466200. [DOI] [PubMed] [Google Scholar]
- 4.Simons M, Keller P, De Strooper B, Beyreuther K, Dotti CG, Simons K. Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc Natl Acad Sci USA. 1998;95:6460–6464. doi: 10.1073/pnas.95.11.6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee JK, Won JS, Singh AK, Singh I. Statin inhibits kainic acid-induced seizure and associated inflammation and hippocampal cell death. Neurosci Lett. 2008;440:260–264. doi: 10.1016/j.neulet.2008.05.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Giri S, Rattan R, Haq E, Khan M, Yasmin R, Won JS, Key L, Singh AK, Singh I. AICAR inhibits adipocyte differentiation in 3T3L1 and restores metabolic alterations in diet-induced obesity mice model. Nutr Metab (Lond) 2006;3:31. doi: 10.1186/1743-7075-3-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hardie DG. AMP-activated protein kinase as a drug target. Annu Rev Pharmacol Toxicol. 2007;47:185–210. doi: 10.1146/annurev.pharmtox.47.120505.105304. [DOI] [PubMed] [Google Scholar]
- 8.Ogden CL, Carroll MD, Curtin LR, McDowell MA, Tabak CJ, Flegal KM. Prevalence of overweight and obesity in the United States, 1999–2004. JAMA. 2006;295:1549–1555. doi: 10.1001/jama.295.13.1549. [DOI] [PubMed] [Google Scholar]
- 9.Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- 10.Sullivan JE, Carey F, Carling D, Beri RK. Characterisation of 5′-AMP-activated protein kinase in human liver using specific peptide substrates and the effects of 5′-AMP analogues on enzyme activity. Biochem Biophys Res Commun. 1994;200:1551–1556. doi: 10.1006/bbrc.1994.1627. [DOI] [PubMed] [Google Scholar]
- 11.Blazquez C, Geelen MJ, Velasco G, Guzman M. The AMP-activated protein kinase prevents ceramide synthesis de novo and apoptosis in astrocytes. FEBS Lett. 2001;489:149–153. doi: 10.1016/s0014-5793(01)02089-0. [DOI] [PubMed] [Google Scholar]
- 12.Jin J, Mullen TD, Hou Q, Bielawski J, Bielawska A, Zhang X, Obeid LM, Hannun YA, Hsu YT. AMPK inhibitor Compound C stimulates ceramide production and promotes Bax redistribution and apoptosis in MCF7 breast carcinoma cells. J Lipid Res. 2009;50:2389–2397. doi: 10.1194/jlr.M900119-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Culmsee C, Monnig J, Kemp BE, Mattson MP. AMP-activated protein kinase is highly expressed in neurons in the developing rat brain and promotes neuronal survival following glucose deprivation. J Mol Neurosci. 2001;17:45–58. doi: 10.1385/JMN:17:1:45. [DOI] [PubMed] [Google Scholar]
- 14.Viollet B, Andreelli F, Jørgensen SB, Perrin C, Geloen A, Flamez D, Mu J, Lenzner C, Baud O, Bennoun M, Gomas E, Nicolas G, Wojtaszewski JF, Kahn A, Carling D, Schuit FC, Birnbaum MJ, Richter EA, Burcelin R, Vaulont S. The AMP-activated protein kinase α2 catalytic subunit controls whole-body insulin sensitivity. J Clin Invest. 2003;111:91–98. doi: 10.1172/JCI16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Won JS, Im YB, Khan M, Contreras M, Singh AK, Singh I. Lovastatin inhibits amyloid precursor protein (APP) beta-cleavage through reduction of APP distribution in Lubrol WX extractable low density lipid rafts. J Neurochem. 2008;105:1536–1549. doi: 10.1111/j.1471-4159.2008.05283.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khan M, Contreras M, Singh I. Endotoxin-induced alterations of lipid and fatty acid compositions in rat liver peroxisomes. J Endotoxin Res. 2000;6:41–50. doi: 10.1177/09680519000060010601. [DOI] [PubMed] [Google Scholar]
- 17.Simons K, Ehehalt R. Cholesterol, lipid rafts, and disease. J Clin Invest. 2002;110:597–603. doi: 10.1172/JCI16390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vetrivel KS, Meckler X, Chen Y, Nguyen PD, Seidah NG, Vassar R, Wong PC, Fukata M, Kounnas MZ, Thinakaran G. Alzheimer disease Abeta production in the absence of S-palmitoylation-dependent targeting of BACE1 to lipid rafts. J Biol Chem. 2009;284:3793–3803. doi: 10.1074/jbc.M808920200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.