

Abstract
The global dissemination of drug-resistant Plasmodium falciparum is spurring intense efforts to implement artemisinin (ART)–based combination therapies for malaria, including mefloquine (MFQ)–artesunate and lumefantrine (LUM)–artemether. Clinical studies have identified an association between an increased risk of MFQ, MFQ-artesunate, and LUM-artemether treatment failures and pfmdr1 gene amplification. To directly address the contribution that pfmdr1 copy number makes to drug resistance, we genetically disrupted 1 of the 2 pfmdr1 copies in the drug-resistant FCB line, which resulted in reduced pfmdr1 mRNA and protein expression. These knockdown clones manifested a 3-fold decrease in MFQ IC50 values, compared with that for the FCB line, verifying the role played by pfmdr1 expression levels in mediating resistance to MFQ. These clones also showed increased susceptibility to LUM, halofantrine, quinine, and ART. No change was observed for chloroquine. These results highlight the importance of pfmdr1 copy number in determining P. falciparum susceptibility to multiple agents currently being used to combat malaria caused by multidrug-resistant parasites.
Plasmodium falciparum drug resistance is seriously hindering public health efforts to control infection and is contributing to a global increase in the burden of malaria. In addition to resistance to chloroquine (CQ) and sulfadoxine-pyrimethamine (SP), the former linchpins of malaria treatment, studies have revealed parasite resistance to alternatives, such as mefloquine (MFQ), in Southeast Asia [1, 2]. In areas where MFQ resistance is prevalent, reduced efficacy can extend to other antimalarial drugs, including lumefantrine (LUM), halofantrine (HF), and quinine (QN), which share variable degrees of cross-resistance [3]. Reliable molecular markers of resistance play a vital, sentinel role in the surveillance of drug efficacy [4]. For example, screening for the pfcrt K76T mutation, which is strongly associated with CQ resistance in vitro and with CQ treatment failure in clinical settings, has documented the rapid worldwide dissemination of CQ resistance and high-lighted the need for alternative first-line drugs in Africa [5–7]. Surveys of single-nucleotide polymorphisms in the P. falciparum dihydropteroate synthase and dihydrofolate reductase genes are similarly vital to monitoring resistance to SP and, together with clinical investigations, have revealed a rapid decrease in SP efficacy [2]. The progression of CQ and SP resistance leaves few alternative treatment strategies that are affordable. Current antimalarial strategies are based on ART-based combination therapies (ACTs), which usually include an ART (such as artesunate, artemether, or dihydroartemisinin) as a fast-acting component, and MFQ, LUM, a quinoline, or an antifolate as the partner drug [8].
The P. falciparum multidrug (MDR) resistance gene (pfmdr1) has been implicated in altering parasite susceptibility to a variety of currently available antimalarial drugs. This gene, located on chromosome 5, encodes a predicted 12-transmembrane-domain protein, PfMDR1 (also known as “Pgh-1”) [9, 10]. PfMDR1 localizes to the parasite digestive vacuole, which is the site of action of CQ and possibly of other quinoline-based antimalarial drugs, including QN [11–13]. A member of the ABC transporter family, PfMDR1 is a homologue of mammalian P glycoprotein, which is a determinant of MDR resistance in mammalian tumor cells [14]. Point mutations in pfmdr1 have been associated with changes in parasite susceptibility to CQ, QN, MFQ, and ART derivatives in both laboratory lines and clinical isolates, but these mutations have limited use as molecular markers [1, 14, 15].
Amplification of pfmdr1 has been implicated in MDR resistance in both in vitro and clinical studies. Early studies on the in vitro selection of MFQ-resistant culture-adapted lines identified increases in pfmdr1 copy number, which correlated with elevated pfmdr1 transcript and protein levels [9–11, 16–20]. In vitro selection studies also observed an inverse relationship between MFQ and CQ susceptibility that was associated with changes in pfmdr1 copy number [19–21]. Analyses of field isolates confirmed the association between pfmdr1 copy number and parasite susceptibility to MFQ in most studies, although not in all of them [17, 22–26]. Recently, a comprehensive prospective study in Thailand provided compelling evidence that increased pfmdr1 copy number is a determinant of MFQ treatment failure and also increases the risk of failure of MFQ-artesunate combination therapy [27]—indeed, in multivariate analysis, pfmdr1 copy number was the most important predictor of failure, and this was not altered by the addition of point-mutation data.
In the present study, we sought to define the role played by pfmdr1 copy number in P. falciparum resistance to MFQ and to extend this analysis to other drugs currently being used to treat malaria caused by CQ- and SP-resistant parasites. To do this, we genetically disrupted 1 of the 2 copies of pfmdr1 present in the drug-resistant P. falciparum FCB line and assessed the subsequent alterations in drug susceptibility. The data from our experiments—and their implications for ACT—are presented below.
MATERIALS AND METHODS
Parasites and transfection
The P. falciparum FCB line was cultured and transfected as described elsewhere [28]. Episomally transfected parasites were selected with 2.5 μg/mL blasticidin HCl (Invitrogen). Plasmid integration into the endogenous pfmdr1 locus was detected by polymerase chain reaction (PCR) and was confirmed by Southern blot analysis (see below). Recombinant parasites were cloned by limiting dilution and identified by their expression of parasite lactate dehydrogenase, as described elsewhere [7].
DNA constructs
A 1.6-kb pfmdr1 coding-sequence fragment (nt 1372–2983 of the 4.3-kb pfmdr1 gene; PlasmoDB identification no. PFE1150w [available at: http://www.plasmoDB.org/]) was amplified by PCR from FCB genomic DNA with the primers 5′-AAGGATCCGGAGTTGTTAGTCAAGATCCAT-3′ (BamHI site underscored) and 5′-AAGCGGCCGCATGCATATTATAAAATGCTTCCTGT-3′ (NotI site underscored). This fragment was subcloned into BamHI/NotI-digested pcamBSD [28]. This transfection plasmid expresses the blasticidin-S-deaminase (bsd) selectable marker, which is under the control of a 0.6-kb P. falciparum calmodulin 5′ untranslated region (UTR) and a 0.6-kb hrp2 3′ UTR. The resulting 6.1-kb plasmid was designated “pcamBSDKD/mdr.”
DNA analysis
P. falciparum DNA was purified from saponin-released trophozoite pellets by use of DNeasy tissue kits (QIAGEN). PCR-based detection of integration (figure 1) used the pfmdr1-specific primers P1 (5′-TTAGAACAAGTGAGTTCAGGAAT-3′) and P4 (5′-AATTTTCCAGCATAACTACCAGT-3′) and the pBluescript-specific primers P2 (5′-CAATTAACCCTCA-CTAAAGGG-3′) and P3 (5′-GCGTAATACGACTCACTATAGGGC-3′). For Southern blot analysis, 1–2 μg of DNA was digested by use of BamHI, electrophoresed, and transferred onto nylon membranes. Hybridizations were performed with a hexamer-primed [32P]-labeled probe that was prepared from full-length bsd. Plasmid rescue was performed by electroporating Escherichia coli (DH5α strain) with 100 ng of parasite DNA. The rescue efficiency, calculated as the number of colony-forming units (cfu) per microgram of genomic DNA, is a measure of the number of episomally replicating plasmids in individual lines [29].
Figure 1.

pfmdr1 knockdown strategy and molecular characterization of clones. The transfection plasmid pcamBSDKD/mdr contains a 1.6-kb pfmdr1 fragment (Δmdr ) centrally located in the coding sequence and a blasticidin-S-deaminase (bsd) selectable marker cassette (A). Single-site crossover between the plasmid and 1 of the 2 endogenous pfmdr1 copies results in inactivation of the copy, leaving 1 functional pfmdr1 copy remaining (B and C ). The disrupted locus contains an upstream pfmdr1 fragment lacking the 3′ end of the gene and the 3′ untranslated region (UTR) as well as a downstream pfmdr1 fragment lacking the 5′ UTR and the 5′ start of the gene, with these 2 fragments separated by the bsd selectable marker. Polymerase chain reaction (PCR) primers (P1–P4) and BamHI (B) fragment sizes are indicated. Square brackets delineate the plasmid sequence that can integrate as tandem linear copies (n ≥ 1). PCR analyses of the parental FCB line and the knockdown clones (KD1mdr1, KD2mdr1, and KD3mdr1) with primers specific for either the upstream truncated locus (P1+P2) or the downstream remnant (P3+P4) confirmed the disruption of 1 copy of pfmdr1 (D and E ). Southern blot hybridization of BamHI-digested genomic DNA samples with a bsd probe revealed 12.5-kb and 6.1-kb bands in the recombinants only, which is consistent with integration of tandem plasmid copies into the pfmdr1 locus (F). The no. of plasmid copies that integrated in tandem was estimated by densitometry to be 3 for KD1mdr1 and KD2mdr1 and 2 for KD3mdr1, indicating that the integration event that occurred in KD3mdr1 was distinct from that which occurred in the 2 other clones. pBS, pBluescript.
RNA preparation and quantitative real-time reverse-transcription (RT)–PCR assays
Parasites were tightly synchronized using consecutive rounds of sorbitol lysis performed on ring-stage parasites. Samples were collected from 5 time points over the following generation, corresponding to early rings, mid rings, late rings/early trophozoites, mid-late trophozoites, and schizonts (harvested at ~10, 18, 26, 34, and 42 h after invasion). RNA was prepared after Trizol treatment (Invitrogen) of saponinlysed parasite pellets and was treated with 2 rounds of DNaseI (Ambion), to remove contaminating genomic DNA (confirmed with intron-spanning pfcrt and β-tubulin gene primers; data not shown). cDNA was prepared from purified RNA by reverse transcription with oligo-dT primers. Quantitative reactions to determine relative transcript levels between lines were performed using real-time PCR [27]. Primers and probes for pfmdr1 and the β-tubulin gene (PlasmoDB identification no. PF10_0084) have been reported elsewhere [27]. Note that the pfmdr1 real-time RT-PCR is specific for the 3′ end of this gene and detects full-length transcript but not the truncated sequence present in the upstream, nonfunctional locus in the knockdown clones (figure 1). For the 13-exon pfcrt gene (PlasmoDB identification no. MAL7P1.27), the primers used were 5′-AATATAAAAAATGGTTTCGCATGTTTA-3′ (exon 7) and 5′-AGAAGGAAAACAATGCGAAGGTT-3′ (exon 9), resulting in amplification of nt 844–973 of the pfcrt coding sequence, and the FAM-labeled internal probe was 5′-TCCATGCTCCGTCACAATCATCACAT-3′ (exon 8). pfmdr1 and pfcrt transcript levels were normalized against that of the single-copy β-tubulin gene. All probes had a TAMRA (6-carboxytetramethylrhodamine) label at their 3′ end. Values were calibrated by performing parallel assays with DNA from the reference 3D7 line, which has a single copy of pfmdr1. Data were quantified using the 2−ΔΔCt method [27].
Protein analysis
Protein extracts were prepared from sodium deoxycholate–treated, sorbitol-synchronized trophozoite-stage parasites, as described elsewhere [28]. For each sample, protein from ~1 × 106 parasites was loaded per well, electrophoresed on 10% SDS-PAGE gel, and transferred onto poly-vinylidene difluoride membranes (Bio-Rad). Membranes were probed with rabbit anti-PfMDR1 antibodies (diluted 1:500), which were raised against a C-terminal fragment containing the last 168 aa [11], followed by incubation with horseradish peroxidase–conjugated donkey anti–rabbit IgG (diluted 1:5000; Santa Cruz Biotech). Rabbit anti–PfERD2 antibodies (MR4, ATCC; http://www.malaria.MR4.org/) were used at a 1:500 dilution [30]. Bands were visualized using the ECL Western blot analysis system (Amersham Biosciences). Protein levels were quantified by densitometric analysis of autoradiograph data, using NIH Image (version 1.6.2; available at: http://rsb.info.nih.gov/nih-image/). PfMDR1 band intensities were normalized against the PfERD2 bands, to correct for minor differences in protein loading.
In vitro antimalarial drug assays
MFQ, LUM, and HF were provided by W. Ellis and W. Milhous (Walter Reed Army Institute of Research, Silver Spring, MD). ART, QN, and CQ were purchased from Sigma. Parasite susceptibilities to anti-malarial drugs were measured in vitro by [3H]-hypoxanthine assays [31]. Briefly, predominantly ring-stage cultures that had been sorbitol synchronized were seeded in duplicate in 96-well plates at a parasitemia and hematocrit of 0.4% and 1.6%, respectively. [3H]-hypoxanthine (0.5 μCi/well) was added after 48 h, and cells were harvested after another 24 h. IC50 values were determined as described elsewhere [31]. For statistical comparisons between lines, 1-way analysis of variance tests with Bonferroni posttests were performed for each drug.
RESULTS
Generation of knockdown clones containing 1 less copy of pfmdr1
To study the contribution that pfmdr1 amplification makes to P. falciparum resistance to antimalarial drugs, we applied a gene-disruption strategy to the P. falciparum FCB line. This line has two 100-kb amplicons that each contain 1 copy of pfmdr1 (expressing the haplotype 86Y/184Y/1034S/1042N/1246D, which is frequently found in Southeast Asia) and has low and moderately high levels of resistance to MFQ and CQ, respectively (mean ± SE IC50 values of 22.1 ± 0.6 and 216.8 ± 34.6 nmol/L, respectively) (table 1) [32, 33]. The genetic strategy is outlined in figure 1. Briefly, we constructed the 6.1-kb pcamBSDKD/mdr plasmid to contain an internal 1.6-kb pfmdr1 fragment from the central region of this gene and a bsd selectable marker cassette (figure 1A). Single-site crossover between this plasmid insert and the homologous pfmdr1 sequence in one of the tandem endogenous copies was predicted to disrupt a single copy, leaving a single functional locus (figure 1B and 1C).
Table 1.
Antimalarial IC50 values for pfmdr1 knockdown lines
| Drug | Parasite lines |
|||
|---|---|---|---|---|
| FCB | KD1mdr1 | KD2mdr1 | KD3mdr1 | |
| Mefloquine | 22.1±0.6 | 6.6±0.8 | 6.0±0.8 | 6.5±1.7 |
| p value | <0.0001 | <0.0001 | 0.0006 | |
| # of assays | 3 | 4 | 4 | 4 |
| Lumefantrine | 89.6±16.9 | 18.1±2.7 | 22.8±5.5 | 23.7±4.4 |
| p value | 0.0059 | 0.0095 | 0.0093 | |
| # of assays | 4 | 4 | 4 | 4 |
| Artemisinin | 34.3±4.0 | 18.8±2.5 | 18.5±3.3 | 21.7±3.5 |
| p value | 0.0107 | 0.0159 | 0.0435 | |
| # of assays | 5 | 5 | 5 | 5 |
| Quinine | 418.6±47.2 | 238.5±18.5 | 216.4±18.7 | 216.9±21.5 |
| p value | 0.0121 | 0.0073 | 0.0081 | |
| # of assays | 4 | 4 | 4 | 4 |
| Halofantrine | 1.17±0.10 | 0.54±0.01 | 0.52±0.03 | 0.61±0.06 |
| p value | 0.0007 | 0.0007 | 0.0031 | |
| # of assays | 4 | 4 | 4 | 4 |
| Chloroquine | 216.8±34.6 | 242.2±43.9 | 204.1±17.7 | 231.1±17.3 |
| p value | 0.6659 | 0.7553 | 0.7239 | |
| # of assays | 4 | 4 | 4 | 4 |
IC50 values were derived by curve fitting analysis of drug inhibition data generated from 72 h [3H]-hypoxanthine incorporation assays performed in duplicate. Values indicate mean ± SEM, shown in nM. p values were calculated from unpaired two-tailed t tests.
FCB parasites transfected with the pcamBSDKD/mdr plasmid were screened for integration into the pfmdr1 locus by PCR, and knockdown clones were subsequently obtained by limiting dilution. These clones were named “KD1mdr1,” “KD2mdr1,” and “KD3mdr1.” Plasmid rescue of genomic DNA prepared from these clones confirmed that they no longer harbored episomes (rescue efficiencies of <100 cfu/μg of genomic DNA for these clones vs. efficiencies of 1 × 104–1 × 105 cfu/μg of genomic DNA for episomally transfected control lines; data not shown). The disruption of 1 pfmdr1 copy was demonstrated by PCR with primer pairs specific for the upstream and downstream truncated fragments, which produced the expected 2.6-kb and 3.5-kb bands with the primer pair P1 and P2 and the primer pair P3 and P4, respectively (figure 1D). Disruption of this second endogenous pfmdr1 copy was confirmed by Southern blot analysis. Hybridization of BamHI-digested genomic DNA samples with a bsd probe revealed 12.5-kb and 6.1-kb bands in the recombinant clones (figure 1F), as was expected for single-site crossover into this locus. The presence of the 6.1-kb plasmid band in the knockdown clones provided evidence that the plasmid had integrated into the endogenous locus as multiple tandem copies. The numbers of integrated copies differed between KD3mdr1 and the 2 other clones, indicating independent integration events.
Reduced transcript and protein expression in pfmdr1 knockdown clones
The effect that the genetic disruption of 1 pfmdr1 copy had was determined at both the transcript level and the protein level. To compare transcript levels, cultures of FCB parasites and the knockdown clones were tightly synchronized and used to prepare RNA from 5 time points over a single 48-h generation. cDNA was then prepared and subjected to quantitative PCR analysis. pfmdr1 transcript levels for the FCB parasites and the knockdown clones were normalized against those for the single-copy β-tubulin gene and the reference 3D7 line, which carries a single copy of pfmdr1 [27]. For comparison, quantitative multiplex PCRs were also performed with primers for pfcrt, whose transcription profile is similar to that of pfmdr1 (see http://www.plasmoDB.org/). pfcrt transcript levels were also normalized against β-tubulin and 3D7. For both pfmdr1 and pfcrt, the highest normalized transcript levels were observed in early ring stages and revealed an ~80% reduction in pfmdr1 transcript levels for the KD1mdr1 and KD2mdr1 clones, compared with that for the parental FCB line. In contrast, pfcrt transcript levels remained unchanged for the pfmdr1 knockdown clones, compared with those for the FCB line (figure 2).
Figure 2.

Quantitative real-time reverse-transcription polymerase chain reaction (RT-PCR) analysis of the parental FCB line and the pfmdr1 knockdown clones. The effect that disruption of 1 copy of pfmdr1 had on transcript levels was determined by quantitative real-time PCR using cDNA prepared from tightly synchronized parasites. pfmdr1 transcript levels for each are presented as mean ± SE normalized genome equivalents [27]. Panel A shows an ~80% reduction in pfmdr1 transcript levels for the clones KD1mdr1 and KD2mdr1, compared with that for the FCB line, as determined from early ring-stage preparations (when transcription is maximal). Results for KD3mdr1 were excluded, because of low real-time PCR yields. Panel B shows the results for parallel quantitative real-time RT-PCRs, which revealed no significant change in pfcrt transcript levels between the parental line and the knockdown clones.
To compare PfMDR1 expression levels between the recombinant clones and the parental line, Western blot analysis was performed using polyclonal anti-PfMDR1 antibodies. This detected an ~160-kDa protein, as has been previously reported [11]. These same protein samples were also probed with control polyclonal antibodies to PfERD2, a P. falciparum ER-Golgi transport protein [30]. Comparison of PfMDR1 signal intensities (normalized against these PfERD2 results) revealed a reduction in PfMDR1 expression levels in the order of 40%–60% for the knockdown clones, which is consistent with expression of 1 of the 2 original gene copies (figure 3).
Figure 3.
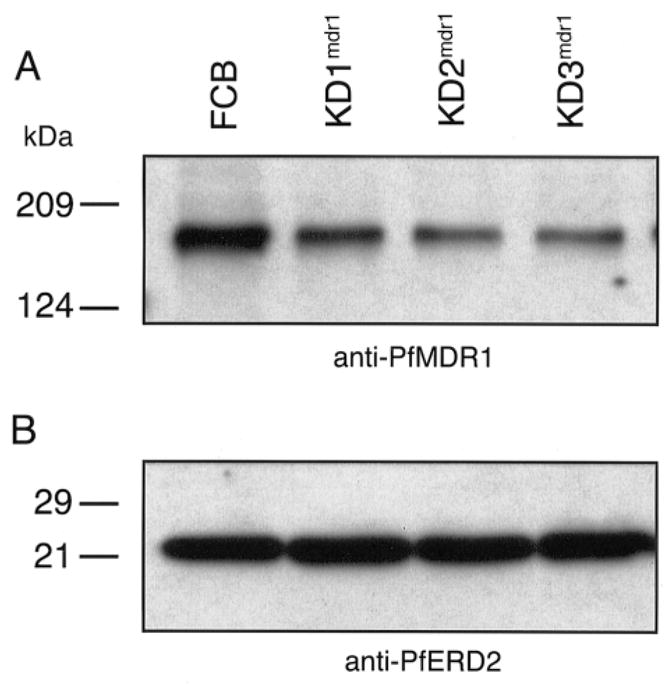
Western blot analysis of the parental FCB line and the pfmdr1 knockdown clones. Probing of sorbitol-synchronized trophozoite-stage proteins with anti-PfMDR1 antibodies revealed an ~160-kDa band in both the parental line and the clones (A). Densitometric analysis revealed that PfMDR1 expression levels (normalized to PfERD2 expression levels) were reduced by 38%, 61%, and 52% for the KD1mdr1, KD2mdr1, and KD3mdr1 clones, respectively. Densitometric analysis of these PfERD2 bands was indicative of essentially equivalent protein loadings across the parental FCB line and the knockdown clones, with the clones displaying signals that were 85%–106% of those of the FCB line (B).
Drug response of pfmdr1 knockdown clones
IC50 values for these parasite clones were obtained for MFQ, LUM, HF, QN, ART, and CQ and are shown in figure 4 (numerical values are provided in table 1). Of note, the disruption of 1 copy of pfmdr1 resulted in a 3-fold decrease in MFQ IC50 values, which decreased from a mean of 22 nnol/L for the parental FCB line to 6–7 nmol/L for the knockdown clones (P < .0001). The response to LUM was also pronounced, with the knockdown parasites displaying a 4–5-fold decrease in IC50 values, which decreased from 90 nmol/L for the FCB line to 18–24 nmol/L for the knockdown clones (P < .001). A decrease was also observed for HF, with IC50 values reduced from 1.2 nmol/L for the FCB line to 0.5–0.6 nmol/L for the knockdown clones (P < .0001).
Figure 4.

In vitro antimalarial drug response of the pfmdr1 knockdown clones. In vitro [3H]-hypoxanthine incorporation assays (72 h) were performed with the knockdown clones and the parental FCB line, which were tested in duplicate against each antimalarial drug on 3–5 separate occasions. IC50 values (shown as means ± SEs) were derived by regression analysis. Numerical values are listed in table 1. For statistical comparisons, 1-way analysis of variance tests with Bonferroni posttests were performed. *P < .05, **P < .01, and ***P < .001, compared with the FCB parental line.
Significant decreases in IC50 values were also observed for ART (decreasing from 34 nmol/L for the FCB line to 19–22 nmol/L for the knockdown clones; P < .05) and for QN (decreasing from 419 nmol/L for the FCB line to 216–239 nmol/L for the knockdown clones; P < .001). These results indicate that amplification of pfmdr1 directly contributes to reduced in vitro susceptibility to these antimalarial drugs. There were no significant changes in CQ IC50 values for the knockdown clones, compared with those for the FCB line (all values ranged from 204 to 242 nmol/L).
DISCUSSION
pfmdr1 copy number has a pronounced effect on the response of P. falciparum to a number of drugs currently being used to treat malaria caused by CQ- and SP-resistant parasites. In the present study, ablation of a second functional copy of pfmdr1 in the MDR-resistant FCB line and phenotypic comparison of the resulting knockdown clones with the parental line has demonstrated that pfmdr1 gene amplification significantly reduces parasite susceptibility to MFQ, LUM, HF, ART, and QN. Our data on MFQ complement those of a recent clinical study that found an association between increased pfmdr1 copy number and increased risk of failure of MFQ monotherapy or MFQ-artesunate combination therapy [27]. That study observed a gene dose effect, whereby incremental increases in copy number translated into increasing risks of treatment failure; for both treatment regimens, however, the transition from 1 to 2 copies of pfmdr1 signaled the highest increase in the risk of treatment failure as well as the largest shift in IC50 values in the sampled Thai parasite populations [27]. The present results for LUM also agree with those from a separate clinical trial in Thailand [34], which found that increased pfmdr1 copy number was associated with parasite recrudescence after a low-dose course of LUM-artemether. That effect, however, was not detected with an increased LUM-artemether dose regimen [34]. In the present study, the 3- and 5-fold decreases in IC50 values observed for MFQ and LUM, respectively, underscore the marked effect that pfmdr1 copy number has on their in vitro potency and highlight the importance of surveying for pfmdr1 copy number to monitor the spread of resistance to these agents.
Both MFQ and LUM are important partners of ACTs. The present study revealed a 2-fold increase in ART potency after genetic ablation of a second copy of pfmdr1, confirming earlier reports of an in vitro association between pfmdr1 copy number and ART susceptibility [24, 26, 27]. This association, however, does not extend to clinical resistance, which to date has not been observed with the ART family of endoperoxide antimalarial drugs [8, 35]. Our data suggest that PfMDR1 expression levels might modulate the disposition of ART in the parasites and thereby alter in vitro susceptibility.
Our phenotypic data on the arylaminoalcohol antimalarial drugs MFQ, LUM, and HF are consistent with previous reports of cross-resistance between these drugs in both field isolates and laboratory lines [17, 19, 20, 27, 36, 37]. These findings confirm that pfmdr1 copy number contributes to their cross-resistance patterns. Other factors that could play a role in resistance to these drugs in vitro include the presence of other genetic polymorphisms in pfmdr1, as evidenced, for example, by allelic-exchange studies in culture-adapted lines that demonstrated that the C-terminal S1034C, N1042D, and D1246Y mutations had an effect on in vitro parasite responses to MFQ, HF, QN, and ART [28, 38]. Field studies supporting this include one that was recently conducted in Zanzibar and that indicated that late treatment failures for LUM-artemether were associated with selection for parasites harboring the pfmdr1 86N mutation [39, 40]. pfcrt mutations can also alter in vitro susceptibilities to MFQ and HF, as has been demonstrated in allelic-exchange and drug-selection experiments [7, 41, 42]. Additional determinants may exist, as suggested by a study in Thailand that identified in vitro MFQ resistance in 16 of the 85 isolates harboring a single copy of pfmdr1 [27]. High-level MFQ resistance observed in vitro may also involve separate genes, as seen in the laboratory-adapted MFQ-resistant FAC8 and W2mef lines (each of which contained 3 copies of pfmdr1), which, on selection for high-level resistance, showed no further pfmdr1 amplification [19, 43].
Earlier studies of MFQ selection produced resistant lines that were found to have undergone sizeable amplifications of chromosome 5 segments harboring the pfmdr1 locus [19, 20]. Mapping of amplicon breakpoints from these lines and other culture-adapted field isolates has identified multiple origins of amplification, with amplicons ranging in size from 20 to 200 kb [16, 23, 32]. The present data imply that the alteration in pfmdr1 copy number was the causal factor accounting for changes in response to MFQ. Interestingly, several selection studies have also found that amplification or deamplification of this chromosome 5 region affected CQ response, such that increased pfmdr1 copy number was associated with increased CQ susceptibility [19–21]. This association has been observed in some, although not all, studies conducted with field isolates [24, 27]. Our knockdown clones, however, showed no significant difference in response to CQ. We suggest that this may reflect either a strain-specific contribution of pfmdr1 copy number to CQ resistance or an attenuation of its effect by the combined contribution of point mutations in pfcrt and pfmdr1. Of note, the FCB line used in our experiments carries the Asian/African pfcrt allele that confers CQ resistance as well as the pfmdr1 N86Y mutation that has been associated with resistance to CQ and with reduced MFQ IC50 values in some, but not all, studies [5, 7, 14, 27, 33]. This N86Y mutation has been found very rarely in field isolates harboring multiple pfmdr1 copies in studies conducted to date [17, 24, 26, 27, 34], suggesting that parasites harboring multicopy 86Y alleles fare poorly in natural infection. Attempts to transfect the MFQ-resistant Tm91C235 Thai isolate (which harbors 2 copies of wild-type pfmdr1 [44]) with the knockdown transfection plasmid used here for the FCB line have, to date, proven to be unsuccessful (data not shown).
Our present findings also imply a role for pfmdr1 copy number in in vitro QN resistance (defined as an IC50 >450 nmol/L [38]), which is in agreement with the findings of some, although not all, studies that have tested for this association in vitro [17, 19, 23]. QN resistance, which is relatively rare and is mostly restricted to pockets of Southeast Asia [2], is nevertheless clearly multifactorial. Point mutations in pfmdr1 and pfcrt [7, 42, 45] also contribute, as does a locus on chromosome 13 that was identified via analysis of a genetic cross and that may correspond to the parasite sodium-proton exchanger pfnhe (although this needs to be confirmed experimentally [46]). Interestingly, an earlier study in West Africa identified MFQ resistance in areas where this drug had not been used and suggested that this may have resulted from QN use and cross-resistance [47]. The present findings suggest that pfmdr1 copy number may play a role and highlight the potential utility of assessing pfmdr1 copy number in areas where ACTs involving MFQ or LUM are being considered for wide scale use against malaria caused by MDR-resistant parasites. This will be particularly important in areas such as Southeast Asia, where increased pfmdr1 copy number is frequently observed. Even though increased pfmdr1 copy number is rare in Africa at present, it will be useful to monitor its emergence there prospectively, especially given that increased copy number has been detected after MFQ selection pressure in Gabon [48].
The World Health Organization has endorsed ACTs, which pair highly potent and fast-acting ART derivatives with a partner drug, as the preferred policy for treatment of malaria caused by CQ-resistant P. falciparum [49]. Current ACT regimens include MFQ-artesunate, LUM-artemether, dihydroartemisinin-piperaquine (Artekin, which combines the active metabolite of ART with a bisquinoline that is active against CQ-resistant malarial parasites), SP-artesunate, and amodiaquine-artesunate. These regimens, although costly, benefit from the very rapid reductions in parasite biomass afforded by ART derivatives. Nevertheless, ART derivatives have relatively short plasma elimination half-lives in vivo, and their success also depends on the efficacy of the longer-acting partner [8]. Our genetic evidence indicating that pfmdr1 copy number reduces in vitro susceptibility to MFQ, ART, and LUM emphasizes the importance of monitoring this marker of reduced drug susceptibility as efforts rapidly proceed to implement ACTs globally for the treatment of malaria.
Acknowledgments
Financial support: New York Community Trust (Blood Diseases Research Program grant to D.A.F.); Wellcome Trust (grant 066201 to S.K.).
Footnotes
Presented in part: Keystone Symposium on Malaria: Functional Genomics to Biology to Medicine, Taos, New Mexico, 28 February–5 March 2006 (abstract 325).
Potential conflicts of interest: none reported.
References
- 1.Uhlemann AC, Krishna S. Antimalarial multi-drug resistance in Asia: mechanisms and assessment. Curr Top Microbiol Immunol. 2005;295:39–53. doi: 10.1007/3-540-29088-5_2. [DOI] [PubMed] [Google Scholar]
- 2.Baird JK. Effectiveness of antimalarial drugs. N Engl J Med. 2005;352:1565–77. doi: 10.1056/NEJMra043207. [DOI] [PubMed] [Google Scholar]
- 3.Wongsrichanalai C, Pickard AL, Wernsdorfer WH, Meshnick SR. Epidemiology of drug-resistant malaria. Lancet Infect Dis. 2002;2:209–18. doi: 10.1016/s1473-3099(02)00239-6. [DOI] [PubMed] [Google Scholar]
- 4.Uhlemann A-C, Yuthavong Y, Fidock DA. Mechanisms of antimalarial drug action and resistance. In: Sherman I, editor. Molecular approaches to malaria. 2. Washington, DC: ASM Press; 2005. pp. 229–61. [Google Scholar]
- 5.Fidock DA, Nomura T, Talley AK, et al. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol Cell. 2000;6:861–71. doi: 10.1016/s1097-2765(05)00077-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Djimde A, Doumbo OK, Steketee RW, Plowe CV. Application of a molecular marker for surveillance of chloroquine-resistant falciparum malaria. Lancet. 2001;358:890–1. doi: 10.1016/S0140-6736(01)06040-8. [DOI] [PubMed] [Google Scholar]
- 7.Sidhu AB, Verdier-Pinard D, Fidock DA. Chloroquine resistance in Plasmodium falciparum malaria parasites conferred by pfcrt mutations. Science. 2002;298:210–3. doi: 10.1126/science.1074045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ashley EA, White NJ. Artemisinin-based combinations. Curr Opin Infect Dis. 2005;18:531–6. doi: 10.1097/01.qco.0000186848.46417.6c. [DOI] [PubMed] [Google Scholar]
- 9.Foote SJ, Thompson JK, Cowman AF, Kemp DJ. Amplification of the multidrug resistance gene in some chloroquine-resistant isolates of P. falciparum. Cell. 1989;57:921–30. doi: 10.1016/0092-8674(89)90330-9. [DOI] [PubMed] [Google Scholar]
- 10.Wilson CM, Serrano AE, Wasley A, Bogenschutz MP, Shankar AH, Wirth DF. Amplification of a gene related to mammalian mdr genes in drug-resistant Plasmodium falciparum. Science. 1989;244:1184–6. doi: 10.1126/science.2658061. [DOI] [PubMed] [Google Scholar]
- 11.Cowman AF, Karcz S, Galatis D, Culvenor JG. A P-glycoprotein homologue of Plasmodium falciparum is localized on the digestive vacuole. J Cell Biol. 1991;113:1033–42. doi: 10.1083/jcb.113.5.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Foley M, Tilley L. Quinoline antimalarials: mechanisms of action and resistance and prospects for new agents. Pharmacol Ther. 1998;79:55–87. doi: 10.1016/s0163-7258(98)00012-6. [DOI] [PubMed] [Google Scholar]
- 13.O’Neill PM, Bray PG, Hawley SR, Ward SA, Park BK. 4-Aminoquinolines—past, present, and future: a chemical perspective. Pharmacol Ther. 1998;77:29–58. doi: 10.1016/s0163-7258(97)00084-3. [DOI] [PubMed] [Google Scholar]
- 14.Duraisingh MT, Cowman AF. Contribution of the pfmdr1 gene to antimalarial drug-resistance. Acta Trop. 2005;94:181–90. doi: 10.1016/j.actatropica.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 15.Woodrow CJ, Krishna S. Antimalarial drugs: recent advances in molecular determinants of resistance and their clinical significance. Cell Mol Life Sci. 2006 May 15; doi: 10.1007/s00018-006-6071-1. electronically published ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Triglia T, Foote SJ, Kemp DJ, Cowman AF. Amplification of the multidrug resistance gene pfmdr1 in Plasmodium falciparum has arisen as multiple independent events. Mol Cell Biol. 1991;11:5244–50. doi: 10.1128/mcb.11.10.5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilson CM, Volkman SK, Thaithong S, et al. Amplification of pfmdr 1 associated with mefloquine and halofantrine resistance in Plasmodium falciparum from Thailand. Mol Biochem Parasitol. 1993;57:151–60. doi: 10.1016/0166-6851(93)90252-s. [DOI] [PubMed] [Google Scholar]
- 18.Volkman SK, Wilson CM, Wirth DF. Stage-specific transcripts of the Plasmodium falciparum pfmdr1 gene. Mol Biochem Parasitol. 1993;57:203–11. doi: 10.1016/0166-6851(93)90196-5. [DOI] [PubMed] [Google Scholar]
- 19.Peel SA, Bright P, Yount B, Handy J, Baric RS. A strong association between mefloquine and halofantrine resistance and amplification, overexpression, and mutation in the P-glycoprotein gene homolog (pfmdr) of Plasmodium falciparum in vitro. Am J Trop Med Hyg. 1994;51:648–58. doi: 10.4269/ajtmh.1994.51.648. [DOI] [PubMed] [Google Scholar]
- 20.Cowman AF, Galatis D, Thompson JK. Selection for mefloquine resistance in Plasmodium falciparum is linked to amplification of the pfmdr1 gene and cross-resistance to halofantrine and quinine. Proc Natl Acad Sci USA. 1994;91:1143–7. doi: 10.1073/pnas.91.3.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barnes DA, Foote SJ, Galatis D, Kemp DJ, Cowman AF. Selection for high-level chloroquine resistance results in deamplification of the pfmdr1 gene and increased sensitivity to mefloquine in Plasmodium falciparum. EMBO J. 1992;11:3067–75. doi: 10.1002/j.1460-2075.1992.tb05378.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Basco LK, Le Bras J, Rhoades Z, Wilson CM. Analysis of pfmdr1 and drug susceptibility in fresh isolates of Plasmodium falciparum from subsaharan Africa. Mol Biochem Parasitol. 1995;74:157–66. doi: 10.1016/0166-6851(95)02492-1. [DOI] [PubMed] [Google Scholar]
- 23.Chaiyaroj SC, Buranakiti A, Angkasekwinai P, Looressuwan S, Cowman AF. Analysis of mefloquine resistance and amplification of pfmdr1 in multidrug-resistant Plasmodium falciparum isolates from Thailand. Am J Trop Med Hyg. 1999;61:780–3. doi: 10.4269/ajtmh.1999.61.780. [DOI] [PubMed] [Google Scholar]
- 24.Pickard AL, Wongsrichanalai C, Purfield A, et al. Resistance to anti-malarials in Southeast Asia and genetic polymorphisms in pfmdr1. Antimicrob Agents Chemother. 2003;47:2418–23. doi: 10.1128/AAC.47.8.2418-2423.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nelson AL, Purfield A, McDaniel P, et al. pfmdr1 genotyping and in vivo mefloquine resistance on the Thai-Myanmar border. Am J Trop Med Hyg. 2005;72:586–92. [PubMed] [Google Scholar]
- 26.Price RN, Cassar C, Brockman A, et al. The pfmdr1 gene is associated with a multidrug-resistant phenotype in Plasmodium falciparum from the western border of Thailand. Antimicrob Agents Chemother. 1999;43:2943–9. doi: 10.1128/aac.43.12.2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Price RN, Uhlemann AC, Brockman A, et al. Mefloquine resistance in Plasmodium falciparum and increased pfmdr1 gene copy number. Lancet. 2004;364:438–47. doi: 10.1016/S0140-6736(04)16767-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sidhu AB, Valderramos SG, Fidock DA. pfmdr1 mutations contribute to quinine resistance and enhance mefloquine and artemisinin sensitivity in Plasmodium falciparum. Mol Microbiol. 2005;57:913–26. doi: 10.1111/j.1365-2958.2005.04729.x. [DOI] [PubMed] [Google Scholar]
- 29.Fidock DA, Wellems TE. Transformation with human dihydrofolate reductase renders malaria parasites insensitive to WR99210 but does not affect the intrinsic activity of proguanil. Proc Natl Acad Sci USA. 1997;94:10931–6. doi: 10.1073/pnas.94.20.10931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Elmendorf HG, Haldar K. Identification and localization of ERD2 in the malaria parasite Plasmodium falciparum: separation from sites of sphingomyelin synthesis and implications for organization of the Golgi. EMBO J. 1993;12:4763–73. doi: 10.1002/j.1460-2075.1993.tb06165.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fidock DA, Nomura T, Wellems TE. Cycloguanil and its parent compound proguanil demonstrate distinct activities against Plasmodium falciparum malaria parasites transformed with human dihydrofolate reductase. Mol Pharmacol. 1998;54:1140–7. doi: 10.1124/mol.54.6.1140. [DOI] [PubMed] [Google Scholar]
- 32.Kidgell C, Volkman SK, Daily J, et al. A systematic map of genetic variation in Plasmodium falciparum. PLoS Pathog. 2006;2:e57. doi: 10.1371/journal.ppat.0020057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Su X-Z, Kirkman LS, Wellems TE. Complex polymorphisms in a ~330 kDa protein are linked to chloroquine-resistant P. falciparum in Southeast Asia and Africa. Cell. 1997;91:593–603. doi: 10.1016/s0092-8674(00)80447-x. [DOI] [PubMed] [Google Scholar]
- 34.Price RN, Uhlemann AC, van Vugt M, et al. Molecular and pharmacological determinants of the therapeutic response to artemether-lumefantrine in multidrug-resistant Plasmodium falciparum malaria. Clin Infect Dis. 2006;42:1570–7. doi: 10.1086/503423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krishna S, Woodrow CJ, Staines HM, Haynes RK, Mercereau-Puijalon O. Re-evaluation of how artemisinins work in light of emerging evidence of in vitro resistance. Trends Mol Med. 2006;12:200–5. doi: 10.1016/j.molmed.2006.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Basco LK, Le Bras J. In vitro activity of halofantrine and its relationship to other standard antimalarial drugs against African isolates and clones of Plasmodium falciparum. Am J Trop Med Hyg. 1992;47:521–7. doi: 10.4269/ajtmh.1992.47.521. [DOI] [PubMed] [Google Scholar]
- 37.Basco LK, Bickii J, Ringwald P. In vitro activity of lumefantrine (benflumetol) against clinical isolates of Plasmodium falciparum in Yaounde, Cameroon. Antimicrob Agents Chemother. 1998;42:2347–51. doi: 10.1128/aac.42.9.2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reed MB, Saliba KJ, Caruana SR, Kirk K, Cowman AF. Pgh1 modulates sensitivity and resistance to multiple antimalarials in Plasmodium falciparum. Nature. 2000;403:906–9. doi: 10.1038/35002615. [DOI] [PubMed] [Google Scholar]
- 39.Sisowath C, Stromberg J, Martensson A, et al. In vivo selection of Plasmodium falciparum pfmdr1 86N coding alleles by artemether-lumefantrine (Coartem) J Infect Dis. 2005;191:1014–7. doi: 10.1086/427997. [DOI] [PubMed] [Google Scholar]
- 40.Martensson A, Stromberg J, Sisowath C, et al. Efficacy of artesunate plus amodiaquine versus that of artemether-lumefantrine for the treatment of uncomplicated childhood Plasmodium falciparum malaria in Zanzibar, Tanzania. Clin Infect Dis. 2005;41:1079–86. doi: 10.1086/444460. [DOI] [PubMed] [Google Scholar]
- 41.Cooper RA, Ferdig MT, Su XZ, et al. Alternative mutations at position 76 of the vacuolar transmembrane protein PfCRT are associated with chloroquine resistance and unique stereospecific quinine and quinidine responses in Plasmodium falciparum. Mol Pharmacol. 2002;61:35–42. doi: 10.1124/mol.61.1.35. [DOI] [PubMed] [Google Scholar]
- 42.Johnson DJ, Fidock DA, Mungthin M, et al. Evidence for a central role for PfCRT in conferring Plasmodium falciparum resistance to diverse antimalarial agents. Mol Cell. 2004;15:867–77. doi: 10.1016/j.molcel.2004.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lim AS, Galatis D, Cowman AF. Plasmodium falciparum: amplification and overexpression of pfmdr1 is not necessary for increased mefloquine resistance. Exp Parasitol. 1996;83:295–303. doi: 10.1006/expr.1996.0077. [DOI] [PubMed] [Google Scholar]
- 44.Looareesuwan S, Kyle DE, Viravan C, et al. Treatment of patients with recrudescent falciparum malaria with a sequential combination of artesunate and mefloquine. Am J Trop Med Hyg. 1992;47:794–9. doi: 10.4269/ajtmh.1992.47.794. [DOI] [PubMed] [Google Scholar]
- 45.Lakshmanan V, Bray PG, Verdier-Pinard D, et al. A critical role for PfCRT K76T in Plasmodium falciparum verapamil-reversible chloroquine resistance. EMBO J. 2005;24:2294–305. doi: 10.1038/sj.emboj.7600681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ferdig MT, Cooper RA, Mu J, et al. Dissecting the loci of low-level quinine resistance in malaria parasites. Mol Microbiol. 2004;52:985–97. doi: 10.1111/j.1365-2958.2004.04035.x. [DOI] [PubMed] [Google Scholar]
- 47.Brasseur P, Kouamouo J, Moyou-Somo R, Druilhe P. Multi-drug resistant falciparum malaria in Cameroon in 1987–1988. II. Mefloquine resistance confirmed in vivo and in vitro and its correlation with quinine resistance. Am J Trop Med Hyg. 1992;46:8–14. doi: 10.4269/ajtmh.1992.46.8. [DOI] [PubMed] [Google Scholar]
- 48.Uhlemann AC, Ramharter M, Lell B, Kremsner PG, Krishna S. Amplification of Plasmodium falciparum multidrug resistance gene 1 in isolates from Gabon. J Infect Dis. 2005;192:1830–5. doi: 10.1086/497337. [DOI] [PubMed] [Google Scholar]
- 49.World Health Organization. Position of WHO’s Roll Back Malaria Department on malaria treatment policy. [Accessed 7 July 2006]; Available at: http://www.who.int/malaria/docs/who_apt_position.htm.