

Abstract
Neutrophils undergo spontaneous apoptosis, but their survival can be extended during inflammatory responses. αMβ2 is reported either to delay or accelerate neutrophil apoptosis, but the mechanisms by which this integrin can support such diametrically opposed responses are poorly understood. The abilities of closely related αMβ2 ligands, plasminogen and angiostatin, derived from plasminogen, as well as fibrinogen and its two derivative αMβ2 recognition peptides, P1 and P2-C, differed markedly in their effects on neutrophil apoptosis. Plasminogen, fibrinogen, and P2-C suppressed apoptosis via activation of Akt and ERK1/2 kinases, while angiostatin and P1 failed to activate these prosurvival pathways and did not prevent neutrophil apoptosis. Using cells transfected with αMβ2 or its individual αM or β2 subunits, and purified receptors and its constituent chains, we show that engagement of both subunits with prosurvival ligands is essential for induction of the prosurvival response. Hence, engagement of a single integrin by closely related ligands can induce distinct signaling pathways, which can elicit distinct cellular responses.
Neutrophils (polymorphonuclear leukocytes, or PMNs)3 are terminally differentiated cells with a very short half-life, being committed to programmed cell death (apoptosis) (1, 2). PMN apoptosis is critical in the clearance of such cells from inflamed tissue, a process that supports the resolution of protective inflammatory responses. However, in some inflammatory diseases, such as arthritis, meningitis, peritonitis, hypersensitivity reactions, and acute coronary syndromes, PMNs not only accumulate but also survive for extended times, leading to injury in the affected tissues (3–5). These observations support the hypothesis that constitutive apoptosis, also referred to as spontaneous or passive apoptosis, is a default fate of PMNs, which can be delayed or accelerated depending on the balance of proapoptotic and anti-apoptotic signals. Agents that promote PMN responsiveness, such as GM-CSF, IL-8, LPS, and fibrinogen (Fg) delay PMN apoptosis (6 –9). These agents promote PMN survival by activating intracellular signaling pathways, such as MAPK, particularly ERK1/2, and PI3K/Akt pathways (10, 11). In contrast, Fas ligand or TNF-α promote PMN death via interaction with their receptors on the cell surface (12, 13).
Growing evidence demonstrates a role of integrin αMβ2 (CD11b/CD18, Mac-1) in regulation of PMN apoptosis. This member of the β2 integrin family is a heterodimer composed of a unique αM (CD11b) subunit noncovalently linked to the common β2 subunit (CD18), which is shared by other leukocyte integrins. αMβ2 is indispensable for diverse PMN functions crucial for innate immunity, including adhesion, transmigration across the endothelium, phagocytosis, and the oxidative burst (reviewed in Ref. 14). The role of αMβ2 as a key regulator of the PMN lifespan depends on its capacity to either delay or accelerate apoptosis. For example, PMN exposure to immobilized or soluble αMβ2 ligands, Fg and ICAM-1, Ab crosslinking of αMβ2 on these cells, or αMβ2-dependent PMN transmigration through endothelium induces survival signals (9, 15, 16). On the other hand, engagement of αMβ2 with activating Abs in the presence of proapoptotic agents, TNF-α or anti-Fas Ab, accelerates PMN apoptosis (15). Phagocytosis of complement-opsonized targets including Escherichia coli also induces PMN death in an αMβ2-dependent manner (17, 18). Studies of β2-deficient mice demonstrate that they exhibit a neutrophilia. This increase in PMN in the β2-deficient mice has been ascribed to up-regulation of antiapoptotic Bcl-XL, which delays apoptosis (19), but others have suggested that neutrophilia is the result of increased granulocytosis induced by elevated levels of IL-17 and G-CSF without effects on apoptosis (20, 21). Thus, the mechanisms of αMβ2-dependent regulation of PMN apoptosis are complex and unresolved.
Several recent observations point to a potential role of plasminogen (Plg), a major fibrinolytic plasma protein, in PMN apoptosis. First, regulation of the inflammatory response by Plg is observed in vivo. PMN recruitment induced by biopolymer implants is attenuated in Plg−/− mice as compared with wild-type mice (22). Additionally, Plg gene expression is up-regulated by inflammatory cytokines, resulting in an increase in circulating levels of Plg (23, 24). Second, plasmin (Plm), the active enzymatic form of Plg, degrades extracellular matrix proteins, leading to detachment and apoptosis of smooth muscle and neuronal cells as well as fibroblasts (25–27). On the other hand, with nonadherent cells such as monocytes, Plg binding capacity is significantly increased on late apoptotic cells (28) and Plm inhibits TNF-α-induced apoptosis in monocytes via a PAR-1-dependent manner (29).
It has been previously demonstrated by us (30) and others (31, 32) that Plg and its short derivative Ang(1– 4), angiostatin composed of four kringle domains of Plg, are ligands of αMβ2 integrin. As both Plg and αMβ2 are important modulators of leukocyte survival, in the present study we examined how interactions of Plg and Ang(1– 4) with αMβ2 influence PMN apoptosis. Our results led us to compare the effects of other αMβ2 ligands on PMN apoptosis. Ultimately, our studies have identified a molecular and cellular basis to explain how occupancy of the same receptor, αMβ2, by closely related ligands can elicit prosurvival responses in PMN.
Materials and Methods
Antibodies, proteins, and synthetic peptides
Glutamic Plg was isolated from normal human plasma by affinity chromatography on lysine-Sepharose followed by gel filtration (33). Ang(1– 4), Ang(1–3), and Ang(1–5) and BSA were from Calbiochem. mAbs to the αM subunit (44a and 904), to the β2 (TS1/18 and IB4) and to MHC class I (W6/32) were from American Type Culture Collection (ATCC). mAb 24 was kindly provided by N. Hogg (34). Fas receptor-activating mAb was from Upstate Biotechnology. Human Fg was from Enzyme Research Laboratories, and neutrophil inhibitory factor (NIF) was a gift from Corvas International. Peptides corresponding to sequences in the fibrinogen γ-chain, P1, Fgγ(190)GWTVFQKRLDGSV(202) and P2-C, Fgγ(385)-KIIPFNRLTIG(395), were synthesized on an Applied Biosystems model 430A peptide synthesizer and purified on HPLC as described (35). Methyl-β-cyclodextrin (MβCD) (36), cytochalasin D, 2,3-butanedione 2-monoxime (BDM), 1-(5-isoquinolinylsulfonyl)-2-methylpiperazine dihydrochloride (H7) were from Sigma-Aldrich, and myosin light chain kinase inhibitor peptide 18 and calpain inhibitor III were from Calbiochem.
Neutrophil preparation
PMNs were isolated from human peripheral blood of healthy volunteers drawn into sterile acid citrate dextrose (1/7 (v/v) 145 mM sodium citrate (pH 4.6) and 2% dextrose). Isolation was performed by density gradient centrifugation onto Ficoll-Hypaque (Pharmacia), followed by dextran sedimentation of erythrocytes and hypotonic lysis of residual erythrocytes.
Development of K562 cells expressing wild-type and mutant forms of αMβ2
Two constitutively active mutants of αMβ2 were generated: αM(Ile316→Gly)β2 (37) and αM(D294C/Q311C)β2 (38) using the QuikChange Multi Site-Directed Mutagenesis kit (Stratagene) according to the manufacturer’s instructions. Cytoplasmic tails were removed from αM and β2 cDNAs by replacing the codons for Lys1120 in αM and Leu729 in β2 with stop codons by PCR to generate αM(1119Δ) and β2(728Δ), which were subsequently cloned to pcDNA3.1 vector using a TOPO cloning kit (In-vitrogen). The αM/αLβ2 chimera cDNA encoding the extracellular/transmembrane regions of αM and cytoplasmic tail of αL (39) was recloned to pcDNA3.1 vector using a TOPO kit.
Human erythroleukemic K562 cells were transiently transfected with 10 μg of pcDNA 3.1 (Invitrogen) containing respective cDNAs for wild-type or mutant αM and/or β2 or vector alone (mock-transfected) using Cell Line Nucleofector Kit V (Amaxa) and the O-17 program. Forty-eight hours after transfection, receptor expression and activation status were analyzed routinely by flow cytometry (FACS) using a FACSCalibur instrument (BD Biosciences) and mAbs 44a, 904, IB4, TS1/18, and 24 recognizing the activation-dependent epitope of the integrin. The data were analyzed with the CellQuest program (BD Biosciences).
Apoptosis assays
PMNs or K562 cells expressing various variants of αMβ2 or mock cells (48 h after transfection) were resuspended in HBSS buffer containing 1 mM CaCl2, 1 mM MgCl2, and 0.1% BSA (pH 7.4) at density of 1 × 106 PMNs/ml or 1.5 × 106 K562 cells/ml and incubated in the absence or presence of increasing concentrations of human Plg, Ang(1– 4), Ang(1–5), NIF, Fg (0 –10 μM), and P1, P2-C, P2-Cscr peptides (0 –500 μM) for 4 –24 h at 37°C in suspension with rotation. In inhibition experiments, PMN were pretreated for 20 min at 37°C with 10 μM Akt inhibitor from Calbiochem, 50 μM ERK1/2 inhibitor, PD 98059 (Calbiochem), or 50 nM NIF before addition of αMβ2 ligands, and these inhibitors were present for the 16 h of incubation. The inhibitors of integrin clustering MβCD (5 mM), myosin L chain kinase (MLCK) inhibitor peptide 18 (0.5 μM), H7 (300 μM), BDM (20 mM), cytochalasin D (10 μM), and calpain inhibitor III (10 μM) were added simultaneously with αMβ2 ligands and cell mixtures were incubated for 16 h. Cell apoptosis was estimated using annexin V FITC apoptosis detection kit (Calbiochem), according to the manufacturer’s instructions, and analyzed by FACS. As defined, cells that were annexin V+propidium iodide (PI)− were in early apoptosis, and cells that were annexin V+PI+ were in late apoptosis. Alternatively, cell pellets, obtained by centrifugation, were lysed in Chaps Cell Extract Buffer (Cell Signaling Technology), and 40 μg of total protein from each sample was analyzed by Western blot using Abs recognizing cleaved products of caspase-3 (BD Biosciences), caspase-8 (Cell Signaling Technology), or to bax (BD Biosciences). Anti-GAPDH (Chemicon International) was used as a loading control. Blots were developed using the secondary HRP-conjugated goat anti-rabbit or anti-mouse IgG (Calbiochem) and SuperSignal West Pico chemiluminescent substrate (Pierce).
Cell signaling assays
K562 cells or PMNs stimulated with 1 nM PMA were incubated in the absence or presence of the αMβ2 ligands in HBSS buffer containing 1 mM CaCl2, 1 mM MgCl2, and 0.1% BSA (pH 7.4) for 30 min at 37°C and then lysed in ice-cold lysis buffer (10 mm Tris (pH 7.5), 5 mm EDTA, 50 mm sodium pyrophosphate, 50 mm NaF, 50 mm NaCl, 0.5% Triton X-100, 0.1% SDS, 1% Nonidet P-40, 0.1 mm Na3VO4, and 1 mm PMSF. In the inhibition experiments, PMNs were pretreated for 20 min at 37°C with 10 μM Akt inhibitor or 50 μM ERK1/2 inhibitor (PD 98059, Calbiochem), 50 nM NIF, F(ab′)2 fragments of anti-αM mAb (clone 44a) or anti-MHC-1 (cloneW6/32) (20 μg/ml) before addition of Plg. Cell lysates were clarified by centrifugation, and protein concentration was measured in supernatants using the Bio-Rad DC protein assay (Bio-Rad Laboratories). Equal amounts of total protein from cell lysates were analyzed by Western blots using PathScan Multiplex Western Cocktail I, anti-phospho-Akt, anti-Akt, anti-phospho-ERK1/2, anti-ERK1/2, or anti-actin Abs (Cell Signaling Technology) and were developed as described above. Since such phosphorylation responses occur rapidly after integrin engagement and often diminish with time, we used a low dose of PMA (1 nM) as a stimulus of integrin activation to enhance binding of αMβ2 ligands, whose interactions with PMN (except NIF) are dependent on receptor activation. Other experiments such as apoptosis and integrin clustering assays did not require presence of PMA, since there was enough time (4 –16 h) for the integrin-ligand interactions to occur, more likely due to increasing αMβ2 activation.
αMβ2 clustering analysis
PMNs were incubated in the absence or presence of the αMβ2 ligands as described under “Apoptosis assay” for 4 h at 37°C, fixed with 4% paraformaldehyde for 15 min at 22°C, and stained with anti-αM mAb (10 μg/ml) (clone OKMI) and Alexa 488-conjugated goat anti-mouse IgG (1/500). The inhibitors of integrin clustering (final concentrations as specified above) were added simultaneously with the ligands. Following washing with HBSS, the cells were cytospun onto Superfrost Plus microscope slides and mounted using Vectashield mounting medium (Vector Laboratories). The samples were observed using a Leica DMR microscope equipped with ×5/0.12 numeric aperture (NA), ×10/0.4 NA, ×20/0.5 NA, or ×40/0.7 NA objective lenses (Leica Microsystems). Images were photographed with a Qimaging Retiga ExiFas camera using Image Pro 5.1 software (Media Cybernetics).
Cell attachment/adhesion and soluble ligand binding assays
Non-tissue culture-treated 96-well Falcon plates (BD Biosciences) were coated with 100 μl of Plg, Ang(1– 4) at 0.2 μM, or with Fg (1 μg/ml) in PBS overnight at 4°C and then postcoated with 0.5% polyvinylpyrrolidone (PVP) for 1 h at room temperature (40). Control wells were coated with PVP only. Before use, the plates were rinsed three times with PBS. Transfected K562 cells were harvested, washed with HBSS, and resuspended in DMEM F-12. The cells were seeded at 1.5–2 × 105 cells/well onto the assay plates and incubated at 37°C for 30 min. The plates were washed three times with HBSS and adherent cells were quantified using the Cy-Quant cell proliferation assay kit (Molecular Probes) according to the manufacturer’s instructions. Data are presented as relative fluorescence units (RFU) ± SEM of three independent experiments. PMNs, which were stimulated with 1 nM PMA, or transfected K562 cells were incubated with soluble human Alexa 488-conjugated Plg or Ang(1– 4) in HBSS buffer containing 1 mM CaCl2 and 1 mM MgCl2 for 40 min at 37°C followed by two washings with the same buffer. In the inhibition experiments, the cells were pretreated with function-blocking mAbs to αM or β2 subunit (20 μg/ml) or NIF (20 nM) for 20 min at 37°C.
Solid-phase binding assays
αMβ2, αM, and β2 recombinant integrin subunits were purified as previously described (35). These were coated onto 96-well Immulon 4HBX microtiter plates (Dynatech Laboratories) in TBS containing 10 mM n-octyl-β-D-glucopyranoside overnight at 4°C and postcoated with 0.5% PVP for 1 h at 37°C. The αMβ2 ligands were biotinylated using EZLink sulfo-NHS-LC-biotin (Pierce) according to the manufacturer’s instructions. Next, increasing concentrations (0 –10 μM) of biotinylated ligands were added to the respective wells and incubated for 2 h at 37°C in TBS containing 10 mM n-octyl-β-D-glucopyranoside and 1 mM CaCl2/1 mM MgCl2. The bound ligands were detected using alkaline phosphatase-conjugated ImmunoPure avidin (Pierce Chemicals) and para-nitrophenylphosphate (Pierce Chemicals) as the substrate, and absorbance at 405 nm (A405) was measured. In binding isotherm studies, the Kd values of Plg and Ang(1– 4) binding to αMβ2 and its subunits were estimated using the SigmaPlot software (SPSS) in which a one-site binding equation was used to fit the data.
Data analysis
The data are expressed as means ± SEM. To determine the significance of differences between two groups, a two-tailed Student’s t test was performed using the SigmaPlot software program (SPSS); p < 0.05 was considered significant.
Results
Plasminogen, but not Ang(1–4), delays PMN apoptosis
Integrin αMβ2 is not only involved in PMN adhesion and migration but also controls PMN apoptosis and may accelerate or suppress cell death in specific settings (reviewed in Ref. 41). We previously demonstrated that αMβ2 interacts with Plg by enhancing Plg activation on the PMN surface (30). Fg, also a ligand of αMβ2, has been shown to delay apoptosis via its engagement of the integrin (9), and we first sought to determine whether these two interrelated plasma protein ligands exerted similar effects on PMN survival. In parallel, we determined if a shorter Plg fragment Ang(1– 4), which is composed of the first four kringle domains of Plg and has antiangiogenic and antiinflammatory properties (32, 42), behaved similar to its parent molecule with respect to PMN apoptosis. As an initial monitor of apoptosis, we measured FITC-conjugated annexin V binding and PI staining by FACS. Increasing concentrations of these αMβ2 ligands were added to human PMNs and incubated for 4 or 16 h at 37°C with gentle rotation. After 4 h in the absence of αMβ2 ligands, 20 ± 5% were in early apoptosis (annexin V+PI−) (Fig. 1A). The percentage of early apoptotic cells was reduced by Plg in a dose-dependent manner; at 1 μM Plg, apoptosis was only 3.5 ± 1.5%. The protective effect of Plg was similar to that observed with Fg; that is, 5 ± 2% (Fig. 1A, left panel). In contrast to intact Plg, Ang(1– 4) failed to protect PMNs against apoptosis at any concentration tested, even as high as 20 μM. Additionally, NIF, a high-affinity ligand of αMβ2, known to block αMβ2-dependent PMN functions (43), also failed to reduce apoptosis. Two peptides derived from Fg γ-chain have been shown to interact with αMβ2. These peptides, designated P1γ(190)GWTVFQKRLDGSV(202) and P2-Cγ(385)KIIPFNRLTIG(395), both interact with the I domain within the αM sub-unit of αMβ2 (35, 44). In view of the distinct effects of Plg and Ang(1– 4) on PMN spontaneous apoptosis, we compared the effects of the two Fg peptides on cell survival. As shown in Fig. 1A (right panel), the P2-C peptide reduced PMN apoptosis in a dose-dependent manner while the P1 and scrambled P2-C peptides did not have any prosurvival effect (17–22 ± 5% of early apoptotic cells).
FIGURE 1.

Regulation of PMN spontaneous apoptosis by αMβ2 ligands. A and B, Human PMN were incubated in HBSS buffer supplemented with 1 mM CaCl2, 1 mM MgCl2, and 0.1% BSA in the absence or presence of Plg, Ang(1– 4), NIF, Fg (0 –10 μM), or Fg-derived P1, P2-C, and P2-Cscr peptides (0 –500 μM) for 4 h (A) or 16 h (B) at 37°C with gentle rotation. The percentage of early apoptotic (annexin V+PI−) (A) or early/late (annexin V+PI−/annexin V+PI+) apoptotic cells (B) was assessed by FACS analysis using the annexin V-FITC apoptosis detection kit. The data represent means ± SEM from three independent experiments. C, PMNs were incubated in the absence or presence of various αMβ2 ligands (proteins at 1 μM, peptides at 100 μM) for 16 h and were lysed in Chaps Cell Extract Buffer, and 40 μg of total protein from cell lysates was analyzed by Western blot using Abs to cleaved forms of Bax, caspase-3, and caspase-8. Next, the Western blots were stripped and probed with anti-GAPDH Abs to confirm equal loading. The Bax blot, which was probed with anti-GAPDH, is shown as an example. The data are representative of six independent experiments performed on PMN isolated from blood of six different donors.
At 4 h only 2–3% of PMNs were in late apoptosis (annexin V+PI+). At 16 h, a robust increase of not only early (annexin V+PI−) but also of late apoptotic PMNs was observed. The data in Fig. 1B examine all annexin V+ cells and demonstrate that different αMβ2 ligands still exert distinct effects on PMN survival. Plg, Fg, and P2-C reduced PMN apoptosis in a dose-dependent manner (20 –30 ± 4 –10% apoptotic cells) as compared with PMNs incubated without αMβ2 ligands (90 ± 20% apoptotic cells). On the other hand, Ang(1– 4), NIF, P1, and P2-Cscr did not rescue PMNs from apoptosis (80 –90 ± 5–15% apoptotic cells). In the presence of the prosurvival ligands, the percentage of apoptotic PMNs still increased from 4 to 16 h (3–5% apoptotic PMNs at 4 h vs 20 –30% apoptotic PMNs at 16 h), suggesting that the prosurvival αMβ2 ligands delay rather than prevent apoptosis.
Additionally, the αMβ2 prosurvival ligands reduced not only spontaneous, but also Fas-induced PMN apoptosis. When PMNs were incubated with mAb activating the Fas receptor for 4 h, 50 ± 8% PMNs were in late and early apoptosis (annexin V+), whereas in the presence of Plg, Fg, or P2-C, only 4 –15 ± 2–5% of the treated PMNs were apoptotic. The ligands that did not affect spontaneous PMN apoptosis also failed to reduce the Fas-dependent apoptosis (data not shown). In control experiments, when Plg was added to apoptotic PMNs, it did not compete with annexin V-FITC binding at any concentration tested (0 –10 μM) (data not shown).
Another characteristic phenotype of apoptotic cells is their activation of caspases, a family of cysteine proteases that coordinates the structural dismantling of the cell (45). Activation of caspase-8 and caspase-3, which regulate PMN spontaneous apoptosis, and cleavage of Bax, a proapoptotic member of the Bcl-2 family, were assessed. As shown on Fig. 1C, the 22-kDa Bax and caspase-8 fragments, as well as the 18-kDa caspase-3 fragment, were detected in the lysates of PMNs incubated for 16 h in the buffer alone or in the presence of Ang(1– 4), the P1 peptide, and control P2-Cscr, while these apoptotic markers were absent in cells treated with the prosurvival αMβ2 ligands, Plg, Fg, and P2-C. These results provide independent corroboration of the differential effects of the various αMβ2 ligands on PMN survival.
Prosurvival ligands of αMβ2 activate Akt and ERK1/2 in PMNs
Activation of Akt and/or ERK1/2 has been implicated in antiapoptotic signaling in PMNs and other cells (9, 15, 46). Thus, we sought to determine whether the prosurvival αMβ2 ligands selectively induced phosphorylation of these kinases. As shown in Fig. 2A, Plg, Fg, and P2-C triggered robust Akt phosphorylation as compared with untreated cells, while Akt phosphorylation was negligible in the presence of Ang(1– 4), P1, and P2-Cscr. Similarly, Plg, Fg, and P2-C induced ERK1/2 activation, whereas Ang(1– 4), P1, and control P2-Cscr did not (Fig. 2B). The data shown in Fig. 2 are representative of at least three experiments performed with PMNs from different donors and distinguish the prosurvival ligands from those that do not prolong PMN survival.
FIGURE 2.

Prosurvival ligands of αMβ2 induce Akt and ERK1/2 activation via their integrin receptor. A and B, Human PMNs were stimulated with 1 nM PMA and incubated in the absence or presence of the αMβ2 ligands as indicated (concentrations are the same as for apoptosis assay in Fig. 1C) for 30 min at 37°C and subsequently lysed in ice-cold lysis buffer. Equal amounts of total protein in PMN lysates were analyzed by SDS-PAGE (8% gels) and Western blot using the PathScan Multiplex Western Cocktail I containing the anti-phospho-Akt Ab (A) or anti-phospho-ERK1/2 Ab (B). The Western blots were subsequently stripped and probed with Abs recognizing total Akt (A) or total ERK1/2 (B). C and D, PMNs were pretreated with 10 μM Akt inhibitor, 50 μM ERK1/2 inhibitor (PD 98059), 50 nM NIF, with F(ab′)2 fragments of anti-αM (clone 44a) or anti-MHC-1 (clone W6/32) mAbs (20 μg/ml) for 20 min at 37°C and incubated with Plg (1 μM) for 30 min at 37°C. Equal amounts of total protein in PMN lysates were analyzed by SDS-PAGE and Western blot using Abs to phospho-Akt (C) or to phospho-ERK1/2 (D). The Western blots were subsequently stripped and probed with Abs recognizing total Akt (C) or total ERK1/2 (D) to ensure equal loading. The results are representative of three experiments performed on PMNs from three different donors.
αMβ2 is a critical receptor mediating Plg-dependent PMN survival and activation of Akt and ERK1/2
Activation of Akt and ERK1/2 by the prosurvival ligands of αMβ2 suggests that these kinases may be directly involved in the delay of PMN spontaneous apoptosis. Indeed, when PMNs were incubated with Plg and an Akt inhibitor, a phosphatidylinositol ether analog, the Plg-mediated PMN protection from apoptosis was completely abrogated (32 ± 10% with Plg, 85 ± 22% (n = 6) with Plg + Akt inhibitor), indicating a critical role of Akt in PMN survival (Table I). Additionally, complete inhibition of ERK1/2 activity with PD 98059 (as shown on the Western blot in Fig. 2D) also reduced Plg-dependent PMN survival, but to a lesser extent than the Akt inhibitor (64 ± 14% (n = 6) apoptotic cells). Consistent with these effects on apoptosis, these inhibitors significantly blocked Plg-dependent Akt and ERK1/2 activation in PMNs as assessed by Western blots (Fig. 2, C and D).
Table I.
Plg-dependent PMN survival is mediated via engagement of αMβ2 and Akt/ERK1/2 activationa
| PMN Treatment | PMN Early/Late apoptosis (% Annexin V+ Cells) |
|---|---|
| Media alone | 90 ± 21 |
| Plg (1μM) | 32 ± 105 |
| Plg (1μM) + anti-αM (44a) | 80 ± 20 |
| Plg (1μM) + anti-MHC-1 | 30 ± 10 |
| Plg (1μM) + NIF | 90 ± 15 |
| Plg (1μM) + Akt inhibitor | 85 ± 22 |
| Plg (1μM) + ERK1/2 inhibitor | 64 ± 14 |
PMNs were pretreated with the blocking reagents as indicated for 30 min at room temperature and incubated in the presence or absence of 1 μM Plg for 16 h at 37°C. The data represent means ± SEM (n = 6).
Since αMβ2 was identified as a major Plg receptor on human PMNs, we sought to verify that this integrin participates in Plg-dependent PMN survival. As shown in Table I, F(ab′)2 fragments of an anti-αM mAb, but not of the control anti-MHC-1 mAb, completely inhibited Plg-dependent PMN survival to the level observed in the absence of Plg (Plg + anti-αM: 80 ± 20% vs Plg + anti-MHC-1: 30 ± 10%). Additionally, these reagents substantially reduced Plg-induced activation of Akt and ERK1/2 (Fig. 2, C and D), while the control anti-MHC-1 mAb did not. These results indicate that the engagement of integrin αMβ2 with Plg triggers activation of Akt and ERK1/2, which subsequently delays PMN spontaneous apoptosis. Of note, NIF, which does not have a protective effect on PMNs (Fig. 1, A and B) and inhibits Plg binding to αMβ2 (30), also abrogated the protective effects of Plg (Table I).
The role of αMβ2 clustering in submission of prosurvival signal
In view of previous findings that indicated involvement of αMβ2 clustering in regulation of PMN apoptosis (15), we analyzed αMβ2 clustering in the presence of its various ligands.
As shown in Fig. 3A, the αMβ2 macroclusters, as defined by Kim et al. (47), were detected on the PMN surface in the presence of the antiapoptotic ligands, Fg, Plg and P2-C, while Ang(1– 4) and P1 did not support αMβ2 clustering. With these latter ligands or with PMNs incubated in the absence of ligands, αMβ2 was more uniformly distributed with more intense staining along the rims of the cells. Thus, the ligands that prolong PMN survival trigger αMβ2 clustering, whereas ligands that do not do so also fail to induce integrin macroclustering. Next, we utilized several inhibitors of integrin clustering to assess the role of this response in PMN survival: 1) MβCD, a disruptor of membrane lipid rafts, which are pivotal in integrin clustering (36); 2) inhibitors of MLCK activity, MLCK inhibitor peptide 18 and H7, which inhibit phosphorylation of MLCK (48); 3) modifiers of the cytoskeleton: BDM, an inhibitor of myosin ATPase activity, and actin-myosin interactions (48); 4) cytochalasin D, an disrupter of the actin cytoskeleton (49); and 5) calpain inhibitor III, which releases integrin from its cytoskeleton constraint (49, 50). In preliminary experiments, all of these inhibitors exerted their anticipated effects on αMβ2; that is, they blocked or significantly reduced Plg-dependent αMβ2 clustering in PMNs as assessed by fluorescence microscopy (Fig. 3B). Most of the inhibitors tested did not modify PMN apoptosis in the absence of αMβ2 ligands, and all completely blocked PMN survival dependent on engagement of αMβ2 with Fg, Plg, and P2-C, but they did not affect apoptosis of cells incubated with P1 (Table II). In control experiments, Plg and Fg binding to αMβ2, when they were added to PMNs together with the clustering inhibitors, was not inhibited as assessed by FACS (Table III). Taken together, our results indicate that engagement of αMβ2 with the ligands that protect PMNs from apoptosis, but not with the ligands failing to do so, leads to αMβ2 clustering, which is a crucial step in αMβ2-dependent PMN survival.
FIGURE 3.

The prosurvival ligands of αMβ2 induce clustering of the integrin. A, PMNs were incubated in the absence or presence of the indicated αMβ2 ligands (concentrations are the same as for apoptosis assays in Fig. 1C) for 4 h at 37°C, fixed with 4% paraformaldehyde, and stained with anti-αM mAb (clone OKMI) and Alexa 488-conjugated goat anti-mouse IgG. Cells were analyzed using a fluorescence microscope (magnification, ×1000×, ×40/0.7 numeric aperture objective), bar size 5 μm. The images shown are representative of PMNs obtained from four different donors, although the intensity of the staining varied among the donors. B, PMNs were treated with Plg (1 μM) in the presence or absence of integrin-clustering inhibitors as indicated (concentrations are specified in Materials and Methods) for 4 h at 37°C, fixed, and visualized as described above.
Table II.
αMβ2 clustering is required for submission of prosurvival signala
| Inhibitors | PMN Early/Late Apoptosis (% Annexin V+ cells) |
||||
|---|---|---|---|---|---|
| No Ligand | Fg | Plg | P2-C | P1 | |
| No inhibitor | 65.4 ± 15 | 22.4 ± 5 | 22.3 ± 6 | 20.1 ± 4 | 56.1 ± 13 |
| MβCD | 55.6 ± 17 | 59.3 ± 15 | 75.0 ± 13 | 75.6 ± 17 | 66.3 ± 18 |
| MLCK inhibitor peptide 18 | 70.9 ± 14 | 84 ± 24 | 68.1 ± 16 | 82.0 ± 22 | 65.3 ± 17 |
| H7 | 77.6 ± 18 | 62.3 ± 16 | 79.2 ± 18 | 78.6 ± 17 | 60.1 ± 14 |
| BDM | 98.9 ± 20 | 98.7 ± 24 | 99.2 ± 24 | 97.8 ± 22 | 85.4 ± 20 |
| Cytochalasin D | 58.9 ± 18 | 55.5 ± 18 | 66.6 ± 15 | 77.4 ± 16 | 66.07 ± 15 |
| Calpain inhibitor III | 97.3 ± 24 | 88.9 ± 23 | 97.1 ± 22 | 74.7 ± 19 | 94.5 ± 22 |
PMNs were incubated with inhibitors of integrin clustering pathways and various αMβ2 ligands (proteins at 1 μM, peptides at 100 μM) as indicated for 16 h at 37°C. The data represent means ± SEM (n = 5).
Table III.
Integrin-clustering inhibitors do not inhibit ligand binding to αMβ2 on PMNa
| Inhibitors | Ligand Binding (MFI) |
||
|---|---|---|---|
| No Ligand | Plg-Alexa 488 | Fg-Alexa 488 | |
| No inhibitor | 2.50 ± 0.5 | 240.0 ± 40 | 446.0 ± 60 |
| MβCD | 3.09 ± 0.4 | 303.0 ± 35 | 403.4 ± 55 |
| MLCK inhibitor peptide 18 | 2.70 ± 0.4 | 285.4 ± 35 | 492.4 ± 50 |
| H7 | 3.2 ± 0.6 | 323.4 ± 45 | 423.2 ± 45 |
| BDM | 2.90 ± 0.5 | 232.5 ± 30 | 509.3 ± 72 |
| Cytochalasin D | 2.3 ± 0.3 | 291.4 ± 40 | 476.4 ± 50 |
| Calpain inhibitor III | 3.4 ± 0.4 | 311.5 ± 35 | 415.1 ± 55 |
PMNs were incubated with soluble Alexa 488-labeled Plg or Fg in the presence or absence of integrin-clustering inhibitors as indicated and incubated for 1 h at 37°C in HBSS/Ca2+/Mg2+ buffer. After two washings with the same buffer, the bound ligands were detected by FACS. The data represent means ± SEM (n = 3).
Distinct modes of engagement of integrin αMβ2 by prosurvival ligands
A mechanism was sought to account for the differential effects of specific ligands on PMN apoptosis, focusing on Plg and Ang(1– 4) as being representative of ligands that do or do not alter PMN survival. Human erythroleukemic K562 cells, which do not express αMβ2, were transiently transfected with the cDNAs encoding for the separate αM or β2 subunits or cotransfected with both cDNAs. In previous studies conducted in HEK cells (35), we have shown that this transfection strategy can lead to cell lines expressing the separate subunits or the αMβ2 heterodimer. The generation of such cells in the K562 background was successful and provided cells that undergo spontaneous apoptosis. As assessed by FACS, cells expressing αMβ2 or the original αM or β2 subunits were reactive with appropriate mAbs: αM cells stained with mAbs to the αM subunit (clones 44a, 904) but not with mAbs to the β2 subunit (clones IB4, TS1/18); β2 cells stained with the mAbs to the β2 subunit but not with mAbs to the αM subunit; and αMβ2 cells stained with both sets of mAbs. None of the mAbs reacted with the mock-transfected cells (data not shown). The integrin activation status on these cells was analyzed by FACS using mAb 24, which recognizes an activation-dependent epitope in the αM subunit (34). In the presence of Ca2+/Mg2, the αMβ2 cells (mean fluorescence intensity of 92.6 ± 20) and αM cells (95.7 ± 22) were reactive whereas the mock cells were not (mean fluorescence intensity of 4.28 ± 1.2). Integrin activation was further confirmed in functional studies. The αMβ2, the αM, and the β2 cells bound soluble Fg and adhered to immobilized Fg in the αM- or β2-specific manner; that is, these interactions were inhibited by the appropriate function-blocking mAbs to the αM or the β2 subunit to the level observed with the mock cells (data not shown). Thus, αMβ2 and its subunits are at least partially activated on the K562 cells.
With these characterizations established, the interaction of these cells with Plg and Ang(1– 4) was characterized. Cells expressing the αMβ2 heterodimer or the αM subunit alone adhered to Plg and Ang(1– 4) (Fig. 4A). In contrast, cells expressing the β2 subunit alone adhered to Plg but not to Ang(1– 4). This difference was confirmed by FACS. As shown in Fig. 4B, the K562 cells expressing the β2 subunit bound soluble Plg but failed to bind soluble Ang(1– 4). K562 cells expressing αMβ2 or the αM subunit alone bound both ligands. Finally, we validated this distinction in recognition specificity in direct binding studies using αMβ2, αM, or β2 purified from HEK293 cell lines (35). Using solid phase assays in which the integrin or its subunits were immobilized, various concentrations of biotinylated Plg or Ang(1– 4) were added, and the binding isotherms were analyzed to determine the affinity of each ligand for each receptor component. As summarized in Table IV, both Plg and Ang(1– 4) bound to αMβ2. The affinity of the intact integrin was ~10-fold higher for Plg than for Ang(1– 4). When the binding of these ligands to separate integrin subunits was analyzed, Plg interacted with the individual αM and β2 subunits with similar affinities. In contrast, Ang(1– 4) was recognized only by the αM and not by the β2 subunit. The affinity of Ang(1– 4) for the αM subunit was similar to its affinity for the heterodimeric receptor. Taken together, these data demonstrate a distinct interaction mechanism for Plg and Ang(1– 4) with αMβ2: Plg interacts with both integrin subunits, while the αM subunit predominates in recognition of Ang(1– 4).
FIGURE 4.

Plg and Ang(1–4) exhibit distinct interactions with cells expressing integrin αMβ2 or its individual subunits. A, Ninety-six-well non-tissue culture-treated plates were coated with Plg or Ang(1–4) (0.2 μM) for 16 h at 4°C and postcoated with 0.5% PVP for 1 h at 37°C. K562 cells were seeded at 1.5–2 × 105 cells/well onto the assay plates in DMEM F-12 and incubated at 37°C for 30 min. After three washings, adherent cells were quantified using the CyQuant cell proliferation assay kit according to the manufacturer’s instructions. The background cell adhesion to the PVP-coated wells, which did not exceed 20% of the highest RFU value, has been subtracted. The data are expressed as RFU ± SEM of three independent experiments. B, Transfected K562 cells were incubated in the presence or absence of soluble Alexa 488-labeled Plg or Ang(1–4) for 40 min at 37°C in HBSS/Ca2+/Mg2+ buffer. After two washings with the same buffer, the bound ligands were detected by FACS. The data are representative of three independent experiments.
Table IV.
Comparison of Plg and Ang(1–4) binding to recombinant purified integrin αMβ2 and its separate αM and β2 subunitsa
| Ligand | αMβ2 | αM | β2 |
|---|---|---|---|
| Plg | 6 × 10−8 | 1 × 10−7 | 5 × 10−7 |
| Ang(1– 4) | 5 × 10−7 | 4 × 10−7 | > 10−4 |
| Fg | 1 × 10−8 | 1.2 × 10−7 | 3 × 10−8 |
| Peptide P1 | 5 × 10−8 | 2 × 10−7 | > 10−4 |
| Peptide P2-C | 1.2 × 10−8 | 1 × 10−7 | 5 × 10−8 |
| NIF | 1.3 × 10−9 | 2 × 10−8 | > 10−4 |
The Kd values are expressed in M (mol/L) and were estimated from binding isotherms constructed at 22°C using the SigmaPlot software, in which data points were fit to a single-site binding equation.
We sought to determine whether this difference in recognition specificity extended to Fg and its peptides, which also exert differential effects on PMN survival. The binding affinities of these ligands for αMβ2 and its individual subunits (see Table IV) indicate that Fg and P2-C peptide engage both integrin subunits, while P1 is recognized predominantly by the αM subunit of the αMβ2 heterodimer. Thus, the prosurvival ligands Plg, Fg, and P2-C interacted with the β2 subunit while the ligands that did not affect spontaneous apoptosis, Ang(1– 4) and P1, did not. Falling into the latter category was NIF, which previously has been demonstrated to interact exclusively with the I domain within the αM subunit (51), and consistently NIF bound very poorly to the β2 subunit (Table IV) and failed to prolong PMN survival.
Having observed these recognition differences, we sought to determine whether and how these ligands regulate survival of K562 cell lines. The cells were incubated in the presence or absence of Plg, Ang(1– 4), Fg, and the two Fg peptides in serum-free HBSS buffer for 24 h, and the percentage of early and late apoptotic cells was measured by FACS. As shown on Fig. 5A, all tested cells, αMβ2, αM, and β2, underwent spontaneous apoptosis (56 – 67%, SEM = 10 –16%, n = 3) in the absence of αMβ2 ligands. Apoptosis of the αMβ2 cells was blocked by Plg (5 ± 2% apoptotic cells, n = 3) but not by Ang(1– 4) (55 ± 10% apoptotic cells, n = 3). Fg and P2-C had the same effect as Plg, prolonging the survival of the αMβ2 cells (3 ± 1% and 4 ± 2% apoptotic cells, n = 3), whereas P1 (56 ± 7% apoptotic cells, n = 3) and the P2-Cscr peptide (59 ± 12% apoptotic cells, n = 3), like Ang(1– 4), failed to do so. However, none of tested ligands prevented apoptosis of the αM cells, the β2 cells, and the mock cells (55– 65% apoptotic cells, SEM = 8 –17%, n = 3).
FIGURE 5.

Engagement of both αMβ2 integrin sub-units is crucial for leukocyte survival. A, K562 cells expressing αM, β2, αMβ2, or mock-transfected control cells were suspended at 1.5 × 106 cells/ml in HBSS containing 1 mM CaCl2, 1 mM MgCl2, and 0.1% BSA and incubated in the absence or presence of the αMβ2 ligands (concentrations are the same as in Fig. 1C) for 24 h at 37°C. The percentage of early/late-apoptotic cells was estimated using the annexin V-FITC apoptosis detection kit and analyzed by FACS. B–D, Alternatively, after incubation, the cells were lysed in Chaps Cell Extract buffer and equal amounts of total protein from the cell lysates were analyzed by Western blot using Abs to cleaved forms of Bax (B), caspase-3 (C), and caspase-8 (D). Blots were then stripped and probed with anti-GAPDH Abs to confirm equal loading. The data are expressed as means ± SEM and are representative of three independent experiments.
These results were corroborated when the activation of caspase-3, caspase-8, and Bax were analyzed in cell lysates. As shown on Fig. 5, B–D, the cleavage products of these caspases and Bax were not detected in the αMβ2 transfectants treated with Plg, Fg, and P2-C (Fig. 5, B–D, row 1), while they were present in untreated as well as in Ang(1– 4), P1, and P2-Cscr-treated αMβ2 cells. The mock-transfected cells and cells expressing the single integrin subunits were not protected from apoptosis by any of tested ligands, and cleaved caspase-3, caspase-8, and Bcl-2 protein Bax were found in all these cell lysates.
Interaction of the prosurvival ligands with the αMβ2 heterodimer, but not with single subunits, triggers Akt activation
Since interaction of the prosurvival ligands with αMβ2 induced extensive phosphorylation of Akt and ERK1/2 in PMNs, which was critical to the survival response (see above), we examined activation of these kinases in the K562 cells expressing the various integrin subunits. As in PMN, robust Akt phosphorylation was triggered by Plg, Fg, and P2-C, but only in the K562 cells expressing αMβ2 cells and not in K562 cells expressing the individual αM or β2 subunits (Fig. 6A) or mock-transfected cells. However, Ang(1– 4) and P1, which are recognized predominantly by the αM subunit but not by the β2 subunit, failed to induce Akt activation, not only in the αMβ2 cells but also in the three other cell types tested. In contrast to Akt, ERK1/2 activation was strongly induced by Plg, Fg, and P2-C not only in the αMβ2 cells but also in the αM cells, indicating that engagement of the αM integrin subunit with these ligands is sufficient for induction of ERK1/2 activation. ERK1/2 phosphorylation was poorly induced by these ligands in mock cells and the β2 cells and by Ang(1– 4) and P1 in all transfected K562 cells. Moreover, activation of both kinases was negligible in all untreated cells. Thus, ERK1/2 is necessary for the prosurvival ligands of αMβ2 to be optimally protective, but is not sufficient to induce a survival response.
FIGURE 6.

Interactions of the prosurvival ligands with only the intact αMβ2 heterodimer trigger Akt activation. A, K562 cells expressing αM, β2, αMβ2, or mock-transfected cells were incubated in the absence or presence of the αMβ2 ligands for 30 min at 37°C and subsequently lysed in ice-cold RIPA buffer. Equal amounts of total protein in cell lysates were analyzed by SDS-PAGE and Western blot using the PathScan Multiplex Western Cocktail I. The Western blots were subsequently reprobed with anti-total ERK1/2 Ab. The results are representative of three experiments. B, K562 cells expressing wild-type or mutant αMβ2 or mock-transfected cells were incubated with Plg for 30 min at 37°C and processed as described in A. The Western blots were probed with anti-Akt(PSer473) and subsequently reprobed with anti-total Akt. The results are representative of three experiments.
Both cytoplasmic tails of αMβ2 are critical in transmission of the prosurvival signal
Knowing that only the ligands that engage both integrin subunits of αMβ2 protect K562 cells from apoptosis, we sought to establish the role of αM and β2 cytoplasmic tails in this process. Thus, in addition to wild-type αMβ2, we expressed mutant receptors with cytoplasmic tail truncations either within the αM (αM(1119Δ)β2) or β2 subunit (αMβ2(728Δ)). We also expressed the chimeric receptor, in which the cytoplasmic tail of the αM subunit was replaced with that of the αL subunit (αM/αLβ2), another member of the leukocyte β2 integrin family. As has been assessed by FACS, the expression levels of each of these αMβ2 mutants were similar to those of the wild-type αMβ2 integrin. The activation status of the truncated αMβ2 variants was ~30% higher than of wild-type αMβ2 based on staining with mAb 24 (data not shown). Additionally, as shown in Table V, the αMβ2 tailless mutants and chimeric αM/αLβ2 maintained their abilities to bind ligands; the binding of Alexa 488-labeled Plg or Fg was similar to that of wild-type αMβ2. However, K562 cells expressing αM(1119Δ)β2 or αMβ2(728Δ) lost their ability to survive even in the presence of the prosurvival ligands; for example, the percentage of apoptotic cells in the presence of Plg (~79 ± 10%, n = 5) was similar to that of mock cells (~78 ± 10%, n = 5) or αMβ2 cells without ligand present (72 ± 14, n = 5) (Table VI). K562 cells expressing the αM/αLβ2 chimeric receptor also underwent apoptosis in the presence of prosurvival ligands (76 ± 13%, n = 5), although their interactions with these ligands were not impaired. Additionally, no Akt activation could be detected in cells expressing the tailless variants of αMβ2 or chimeric αM/αLβ2 (Fig. 6B). These data indicate the crucial importance of both αM and β2 cytoplasmic tails in transmission of prosurvival signal and suggest integrin-dependent specificity in regulation of cell survival signals.
Table V.
The αMβ2 cytoplasmic tail modifications do not impair binding of prosurvival ligandsa
| αMβ2 Variants | Ligand Binding (MFI) |
||
|---|---|---|---|
| No Ligand | Plg-Alexa 488 | Fg-Alexa 488 | |
| αMβ2 | 2.08 ± 0.21 | 130.2 ± 20 | 137.3 ± 10 |
| αMβ2(728Δ) | 2.05 ± 0.3 | 152.1 ± 23 | 136.4 ± 15 |
| αM(1119Δ)β2 | 1.95 ± 0.35 | 145.1 ± 25 | 114.2 ± 15 |
| αM/αLβ2 | 2.90 ± 0.2 | 135.4 ± 28 | 129.2 ± 10 |
| Mock | 2.40 ± 0.4 | 16.3 ± 4 | 48.3 ± 5 |
K562 cells 48 h after transfection with the indicated αMβ2 constructs were incubated in the presence or absence of soluble Alexa 488-labeled Plg or Fg for 40 min at 37°C in HBSS/Ca2+/Mg2+ buffer. After two washings with the same buffer, the bound ligands were detected by FACS. The data are means ± SEM (n = 3).
Table VI.
The role of the αMβ2 cytoplasmic tails and integrin activation in K562 survivala
| αMβ2 Variants | K562 Early/Late Apoptosis (% Annexin V+ Cells) |
||||||
|---|---|---|---|---|---|---|---|
| No Ligand | Fg | Plg | P2-C | P1 | P2-Cscr | Ang(1–4) | |
| αMβ2 | 72 ± 14 | 28 ± 5 | 29 ± 4 | 26 ± 4.5 | 82 ± 6 | 71 ± 17 | 71 ± 15 |
| αMβ2(728Δ) | 76 ± 15 | 80 ± 10 | 79 ± 10 | 74 ± 10 | 76 ± 11 | 76 ± 15 | 74 ± 16 |
| αM(1119Δ)β2 | 73 ± 11 | 80 ± 9 | 81 ± 8.5 | 80 ± 16 | 77 ± 9 | 78 ± 12 | 78 ± 14 |
| αM(Ile316→Gly)β2 | 77 ± 12 | 19 ± 2.5 | 16 ± 2.1 | 15 ± 3 | 79 ± 7 | 76 ± 13 | 80 ± 9 |
| αM(D294C/Q311C)β2 | 78 ± 10 | 20 ± 3 | 18 ± 3.2 | 17 ± 3.5 | 80 ± 8 | 79 ± 10 | 76 ± 10 |
| αM/αLβ2 | 77 ± 12 | 80 ± 7.7 | 76 ± 13 | 81 ± 8 | 81 ± 6 | 80 ± 8 | 80 ± 5 |
| Mock | 81 ± 8 | 72 ± 13 | 78 ± 10 | 76 ± 12 | 70 ± 12 | 77 ± 6 | 78 ± 8 |
K562 cells 48 h after transfection with the indicated αMβ2 constructs were incubated in the presence or absence of various αMβ2 ligands (proteins at 1 μM, peptides at 100 μM) for 16 h at 37°C. The data represent means ± SEM (n = 5). Integrin expression levels were monitored by FACS and were similar.
Next, we sought to determine the role of αMβ2 activation in K562 cell survival. Two constitutively active mutants of αMβ2 were αM(Ile316→Gly)β2 and αM(D294C/Q311C)β2. Both mutations stabilize the αM-I domain, the major ligand recognition site, in an open conformation, which results in significant enhancement of αMβ2 activation (37, 38). Indeed, K562 cells expressing these mutants showed enhanced adhesion to Plg as well as soluble Plg binding (3– 4-fold greater than cells bearing wild-type αMβ2), while their expression levels were comparable (data not shown). When apoptosis of these cells was compared in the absence of ligands, the percentage of apoptotic cells was similar in all cells, and in the presence of the prosurvival ligands, the observed subtle differences were not statistically significant ( p = 0.064 – 0.2073) (Table VI). Importantly, the ligands, which did not support survival in αMβ2 cells, also failed to protect the cells bearing the constitutively active variants of αMβ2. Thus, these results indicate that apoptosis is regulated by the integrin engagement with the specific ligands rather than by integrin activation per se.
Discussion
The influence of integrin ligation on the survival of adherent cells is well documented (reviewed in Ref. 52), and growing evidence suggests that integrins may also participate in the survival of circulating cells, such as leukocytes. For example, engagement of the β7 integrins with ligands augments the survival of eosinophils (53), and interaction between α9β1 and vascular cell adhesion molecule-1 (VCAM-1) inhibits PMN apoptosis (54). The involvement of αMβ2 in control of PMN apoptosis also has been examined extensively (reviewed in Ref. 41), but the data are contradictory, showing that αMβ2 can either accelerate or delay PMN apoptosis (9, 15, 16, 19 –21, 55). Thus, molecular mechanisms governing the effects of αMβ2 on PMN apoptosis remain poorly understood. In this study, we demonstrate that different αMβ2 ligands, even very closely related ones, can exert different effects of PMN apoptosis, with a subset of ligands inducing a prosurvival response. Furthermore, we provide a mechanism for these differential effects: ligands that engage both subunits of αMβ2 prolong PMN life by inducing prosurvival signaling pathways, primarily Akt activation, while ligands that engage primarily the αM subunit are unable to induce the prosurvival signaling responses and do not delay PMN apoptosis (Fig. 7).
FIGURE 7.
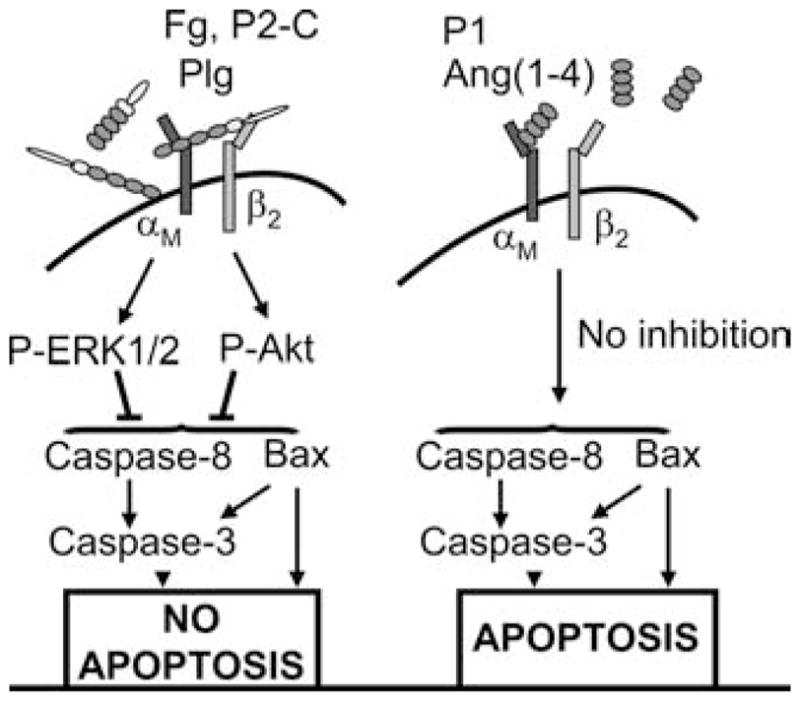
Distinct engagement patterns of αMβ2 by its ligands determine regulation of PMN constitutive apoptosis. Two groups of αMβ2 ligands can be distinguished. The first group, which includes Plg, like Fg and P2-C, is recognized by both αM and β2 subunits, and the second group, which includes Ang(1– 4), P1, and NIF, interacts primarily with the αM subunit but not with the β2 subunit. Interaction with both integrin subunits is necessary to induce αMβ2 clustering and activation of prosurvival ERK1/2 and Akt kinases, which leads to suppression of caspase-3 and caspase-8 activation as well as proapoptotic protein Bax cleavage, resulting in inhibition of apoptosis (left panel). In contrast, interaction of the ligands of the second group is not sufficient to trigger the phosphorylation of ERK1/2 and Akt. Thus, activation of caspases and Bax is not inhibited, and PMNs are not spared from spontaneous apoptosis.
Our previous studies identified Plg, as well as its activator urokinase (uPA), as novel ligands of αMβ2. Their interactions with this integrin enhances uPA-dependent Plg activation on PMN surface, which, in turn, facilitates PMN migration and PMN-mediated fibrinolysis, activities that are directed toward the extracellular environment of the cell (30, 56). The present study demonstrates that interaction between Plg and αMβ2 on PMNs also induces intracellular signaling that results in a prosurvival response. Plg, in contrast to Ang(1– 4), its shorter four-kringle derivative, significantly inhibits spontaneous apoptosis of PMNs. This effect is abrogated when αMβ2 is blocked by F(ab′)2 fragments of function-blocking Abs to either the αM or β2 subunits and by NIF, a high-affinity ligand that blocks many αMβ2-mediated PMN responses (43, 51). Failure of Ang(1– 4) to prolong PMN survival is consistent with the data by Chavakis et al. (32), who showed that Ang(1– 4) has an antiinflammatory effect on leukocytes by inhibiting their migration and adhesion to αMβ2 ligands. These authors also demonstrated that Ang(1– 4) generation in vivo peaks at the last phase of wound healing when PMN apoptosis is needed to terminate the inflammatory response. Thus, it can be envisioned that Ang(1– 4) would bind to αMβ2 and compete with Plg and its other prosurvival ligands. Chavakis et al. (32) identified kringle 4 domain as directly interacting with αMβ2. However, in our experiments, not only Ang(1– 4) but also Ang(1–3) (data not shown), which lacks kringle 4, is recognized by this integrin. Indeed, Ang(1–3) recognition by αMβ2 is physiologically relevant, as biologically active Ang(1–3) is generated from Plg by neutrophil-derived elastase (57). Thus, PMNs have an intrinsic ability to induce their own apoptosis by generating angiostatin derivatives that compete with Plg and by degrading Plg, a prosurvival ligand. αMβ2 is not the only integrin interacting with Ang(1– 4); it is also recognized by α4β1, α9β1, and αVβ3, integrins that have also been implicated in cellular apoptosis.
We hypothesized that one of the mechanisms underlying distinct PMN responses to Plg and Ang(1– 4) may be differences in their mode of αMβ2 engagement. Utilizing purified recombinant integrin and K562 cells expressing αMβ2 or its individual subunits, we found that engagement of both integrin subunits is a prerequisite for induction of prosurvival signals. Ang(1– 4) interacted only with the αM subunit and failed to inhibit apoptosis. This mechanism was generalized by experiments with Fg and its two αMβ2-binding peptides, P1 and P2-C (44, 58). Fg and P2-C exerted cytoprotective effects on PMN and αMβ2 K562 cells, while the P1 peptide failed to inhibit apoptosis in these cells. Fg and P2-C are recognized by the entire heterodimer, whereas P1 is recognized only by the αM subunit (35). Whitlock et al. proposed a model in which αMβ2 clustering inhibits spontaneous and TNF-α-induced apoptosis via activation of Akt and ERK1/2 pathways (15). In view of this model, we assessed αMβ2 clustering in the absence or presence of its ligands and found that cytoprotective ligands interacting with both integrin subunits, such as Plg, Fg, or P2-C, induced αMβ2 clustering while Ang(1– 4) and P1 did not. Furthermore, clustering was pivotal to the effects of the prosurvival ligands of αMβ2, as several clustering inhibitors blocked the prosurvival effects. According to Kim et al. (47), integrins do not cluster spontaneously, but only upon engagement of immobilized ligands. However, in our system ligands are presented in solution and they are still capable of αMβ2 clustering, which is consistent the data by Buensuceso et al. showing that soluble Fg induces αIIbβ3 clustering on platelets (59). Thus, engagement of both αMβ2 integrin subunits with cytoprotective ligands is prerequisite to the receptor clustering and subsequent transmission of prosurvival signals. This may be the case, as integrin interactions with cytoskeleton, which occur primarily via the β subunits, are critical for integrin clustering (60). Indeed, based on our data, both the cytoskeleton and cytoplasmic tails of both subunits of αMβ2 are pivotal in transmission of prosurvival signals. However, we cannot exclude the possibility that distinct patterns of engagement of the integrin subunits by the tested ligands may differentially regulate integrin activation since binding of an activating mAb to αMβ2 has been shown to prevent spontaneous apoptosis (15). The binding of all αMβ2 ligands tested (except NIF) is significantly enhanced by integrin activation. However, integrin activation by itself does not inhibit cell apoptosis in the absence of the ligands, as concluded from our experiments with cells expressing constitutively active αMβ2. Receptor engagement by the appropriate ligands is crucial for induction of the cytoprotective response.
The effect of Plg on PMN apoptosis did not require its conversion to active Plm, as diisopropyl fluorophosphate (DFP)-treated Plg recapitulated the prosurvival effect of untreated Plg. Additionally, we found that active Plm did not exert a cytoprotective function (data not shown). These observations contrast with the recent demonstration that Plm formation and its activation of PAR1 are necessary for induction of monocyte survival by Plg (29) and suggest cell type-specific mechanisms for Plg to influence apoptosis. Plg-dependent PMN survival was reduced by ε-aminocaproic acid, which blocks interaction of the lysine binding sites within the Plg kringles with cell receptors (30, 61).
Activation of Akt and ERK1/2 by engagement of αMβ2 with prosurvival ligands Plg, Fg, and P2-C is consistent with Whitlock et al. (15) and Rubel et al. demonstrating that these kinases are activated downstream of αMβ2 and other integrins (62) and are crucial for PMN survival. ERK1/2 inhibition has been shown to enhance resolution of inflammation by prolonging PMN lifespan in vivo (63). In our study, Plg-mediated PMN survival was completely reversed by Akt inhibition and was only partially reduced by ERK1/2 inhibition, suggesting that Akt is upstream of ERK1/2. This interpretation is supported by our finding that inhibition of Akt reduced Plg-dependent activation of ERK1/2 (data not shown), and by the fact that Akt directly phosphorylates MEK1 and MEK2, which are upstream activators of ERK1/2 (64). The pivotal role of Akt in PMN survival via ERK1/2 activation is further supported by our observation that Plg-dependent ERK1/2 phosphorylation occurs in the absence of Akt activation in αM cells but is not sufficient to rescue these cells from apoptosis. Complete blockade of PMN survival by Akt and only partial blockade by ERK1/2 inhibitors also indicate that there are other ERK1/2-independent downstream targets of Akt. Possible targets of Akt phosphorylation that could inhibit apoptosis include the proapoptotic protein Bad, activation of transcription factors CREB and NF-κB (46), or prevention of cytochrome c release (65).
PMN apoptosis is an important mechanism regulating the extent of an inflammatory response, making it self-limiting and preventing excessive tissue damage. At sites of inflammation, a complex interplay between proapoptotic and survival signals must control the outcome of the response. This study describes a novel mechanistic model by which various αMβ2 ligands may regulate PMN apoptosis and suggests that their subtle balance in the local microenvironment provides an important extrinsic checkpoint in the resolution of inflammation.
Footnotes
Supported by National Institutes of Health Grant R01 HL17964 and P50 HL 081011 (E.F.P.) and by an American Heart Association Scientist Development Grant (E.P.).
Abbreviations used in this paper: PMN, polymorphonuclear leukocyte; Ang(1– 4), angiostatin composed of four kringle domains; BDM, 2,3-butanedione 2-monoxime; Fg, fibrinogen; H7, 1-(5-isoquinolinylsulfonyl)-2-methylpiperazine dihydrochloride; MFI, mean fluorescence intensity; MLCK, myosin L chain kinase; NIF, neutrophil inhibitory factor; MβCD, methyl-β-cyclodextrin; PI, propidium iodide; Plg, plasminogen; Plm, plasmin; PVP, polyvinylpyrrolidone; RFU, relative fluorescence units.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Savill JS, Wyllie AH, Henson JE, Walport MJ, Henson PM, Haslett C. Macrophage phagocytosis of aging neutrophils in inflammation: programmed cell death in the neutrophil leads to its recognition by macrophages. J Clin Invest. 1989;83:865–875. doi: 10.1172/JCI113970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grigg JM, Savill JS, Sarraf C, Haslett C, Silverman M. Neutrophil apoptosis and clearance from neonatal lungs. Lancet. 1991;338:720–722. doi: 10.1016/0140-6736(91)91443-x. [DOI] [PubMed] [Google Scholar]
- 3.Nathan C. Points of control in inflammation. Nature. 2002;420:846–852. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- 4.Ottonello L, Cutolo M, Frumento G, Arduino N, Bertolotto M, Mancini M, Sottofattori E, Dallegri F. Synovial fluid from patients with rheumatoid arthritis inhibits neutrophil apoptosis: role of adenosine and proinflammatory cytokines. Rheumatology. 2002;41:1249–1260. doi: 10.1093/rheumatology/41.11.1249. [DOI] [PubMed] [Google Scholar]
- 5.Garlichs CD, Eskafi S, Cicha I, Schmeisser A, Walzog B, Raaz D, Stumpf C, Yilmaz A, Bremer J, Ludwig J, Daniel WG. Delay of neutrophil apoptosis in acute coronary syndromes. J Leukocyte Biol. 2004;75:828–835. doi: 10.1189/jlb.0703358. [DOI] [PubMed] [Google Scholar]
- 6.Kettritz R, Gaido ML, Haller H, Luft FC, Jennette JC, Falk RJ. Interleukin-8 delays spontaneous and tumor necrosis factor-α -mediated apoptosis in human neutrophils. Kidney Int. 1998;53:84–91. doi: 10.1046/j.1523-1755.1998.00741.x. [DOI] [PubMed] [Google Scholar]
- 7.Lee A, Whyte MK, Haslett C. Inhibition of apoptosis and prolongation of neutrophil functional longevity by inflammatory mediators. J Leukocyte Biol. 1993;54:283–288. [PubMed] [Google Scholar]
- 8.Colotta F, Re F, Polentarutti N, Sozzani S, Mantovani A. Modulation of granulocyte survival and programmed cell death by cytokines and bacterial products. Blood. 1992;80:2012–2020. [PubMed] [Google Scholar]
- 9.Rubel C, Gomez S, Fernandez GC, Isturiz MA, Caamano J, Palermo MS. Fibrinogen-CD11b/CD18 interaction activates the NF-κ B pathway and delays apoptosis in human neutrophils. Eur J Immunol. 2003;33:1429–1438. doi: 10.1002/eji.200323512. [DOI] [PubMed] [Google Scholar]
- 10.Klein JB, Rane MJ, Scherzer JA, Coxon PY, Kettritz R, Mathiesen JM, Buridi A, McLeish KR. Granulocyte-macrophage colony-stimulating factor delays neutrophil constitutive apoptosis through phosphoinositide 3-kinase and extracellular signal-regulated kinase pathways. J Immunol. 2000;164:4286–4291. doi: 10.4049/jimmunol.164.8.4286. [DOI] [PubMed] [Google Scholar]
- 11.Klein JB, Buridi A, Coxon PY, Rane MJ, Manning T, Kettritz R, McLeish KR. Role of extracellular signal-regulated kinase and phosphatidylinositol-3 kinase in chemoattractant and LPS delay of constitutive neutrophil apoptosis. Cell Signal. 2001;13:335–343. doi: 10.1016/s0898-6568(01)00151-6. [DOI] [PubMed] [Google Scholar]
- 12.Liles WC, Kiener PA, Ledbetter JA, Aruffo A, Klebanoff SJ. Differential expression of Fas (CD95) and Fas ligand on normal human phagocytes: implications for the regulation of apoptosis in neutrophils. J Exp Med. 1996;184:429–440. doi: 10.1084/jem.184.2.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Avdi NJ, Nick JA, Whitlock BB, Billstrom MA, Henson PM, Johnson GL, Worthen GS. Tumor necrosis factor-α activation of the c-jun N-terminal kinase pathway in human neutrophils. J Biol Chem. 2001;276:2189–2199. doi: 10.1074/jbc.M007527200. [DOI] [PubMed] [Google Scholar]
- 14.Harris ES, McIntyre TM, Prescott SM, Zimmerman GA. The leukocyte integrins. J Biol Chem. 2000;275:23409–23412. doi: 10.1074/jbc.R000004200. [DOI] [PubMed] [Google Scholar]
- 15.Whitlock BB, Gardai S, Fadok V, Bratton D, Henson PM. Differential roles for αMβ2 integrin clustering or activation in the control of apoptosis via regulation of Akt and ERK survival mechanisms. J Cell Biol. 2000;151:1305–1320. doi: 10.1083/jcb.151.6.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Watson RW, Rotstein OD, Nathens AB, Parodo J, Marshall JC. Neutrophil apoptosis is modulated by endothelial transmigration and adhesion molecule engagement. J Immunol. 1997;158:945–953. [PubMed] [Google Scholar]
- 17.Watson RW, Redmond HP, Wang IH, Condron C, Bouchier-Hayes D. Neutrophils undergo apoptosis following ingestion of Escherichia coli. J Immunol. 1996;156:3986–3992. [PubMed] [Google Scholar]
- 18.Zhang B, Hirahashi J, Cullere X, Mayadas TN. Elucidation of molecular events leading to neutrophil apoptosis following phagocytosis: crosstalk between caspase 8, reactive oxygen species, and MAPK/ERK activation. J Biol Chem. 2003;278:28443–28454. doi: 10.1074/jbc.M210727200. [DOI] [PubMed] [Google Scholar]
- 19.Weinmann P, Scharffetter-Kochanek K, Forlow SB, Peters T, Walzog B. A role for apoptosis in the control of neutrophil homeostasis in the circulation: insights from CD18-deficient mice. Blood. 2003;101:739–746. doi: 10.1182/blood-2002-01-0239. [DOI] [PubMed] [Google Scholar]
- 20.Forlow SB, Schurr JR, Kolls JK, Bagby GJ, Schwarzenberger PO, Ley K. Increased granulopoiesis through interleukin-17 and granulocyte colony-stimulating factor in leukocyte adhesion molecule-deficient mice. Blood. 2001;98:3309–3314. doi: 10.1182/blood.v98.12.3309. [DOI] [PubMed] [Google Scholar]
- 21.Horwitz BH, Mizgerd JP, Scott ML, Doerschuk CM. Mechanisms of granulocytosis in the absence of CD18. Blood. 2001;97:1578–1583. doi: 10.1182/blood.v97.6.1578. [DOI] [PubMed] [Google Scholar]
- 22.Busuttil SJ, Ploplis VA, Castellino FJ, Tang L, Eaton JW, Plow EF. A central role for plasminogen in the inflammatory response to biomaterials. J Thromb Haemost. 2004;2:1798–1805. doi: 10.1111/j.1538-7836.2004.00916.x. [DOI] [PubMed] [Google Scholar]
- 23.Jenkins GR, Seiffert D, Parmer RJ, Miles LA. Regulation of plasminogen gene expression by interleukin-6. Blood. 1997;89:2394–2403. [PubMed] [Google Scholar]
- 24.Bannach FG, Gutierrez A, Fowler BJ, Bugge TH, Degen JL, Parmer RJ, Miles LA. Localization of regulatory elements mediating constitutive and cytokine-stimulated plasminogen gene expression. J Biol Chem. 2002;277:38579–38588. doi: 10.1074/jbc.M202509200. [DOI] [PubMed] [Google Scholar]
- 25.Houard X, Monnot C, Dive V, Corvol P, Pagano M. Vascular smooth muscle cells efficiently activate a new proteinase cascade involving plasminogen and fibronectin. J Cell Biochem. 2003;88:1188–1201. doi: 10.1002/jcb.10460. [DOI] [PubMed] [Google Scholar]
- 26.Chen ZL, Strickland S. Neuronal death in the hippocampus is promoted by plasmin-catalyzed degradation of laminin. Cell. 1997;91:917–925. doi: 10.1016/s0092-8674(00)80483-3. [DOI] [PubMed] [Google Scholar]
- 27.Rossignol P, Ho-Tin-Noe B, Vranckx R, Bouton MC, Meilhac O, Lijnen HR, Guillin MC, Michel JB, Angles-Cano E. Protease nexin-1 inhibits plasminogen activation-induced apoptosis of adherent cells. J Biol Chem. 2004;279:10346–10356. doi: 10.1074/jbc.M310964200. [DOI] [PubMed] [Google Scholar]
- 28.O’Mullane MJ, Baker MS. Loss of cell viability dramatically elevates cell surface plasminogen binding and activation. Exp Cell Res. 1998;242:153–164. doi: 10.1006/excr.1998.4067. [DOI] [PubMed] [Google Scholar]
- 29.Mitchell JW, Baik N, Castellino FJ, Miles LA. Plasminogen inhibits TNFα -induced apoptosis in monocytes. Blood. 2006;107:4383–4390. doi: 10.1182/blood-2005-07-2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pluskota E, Soloviev DA, Bdeir K, Cines DB, Plow EF. Integrin αMβ2 orchestrates and accelerates plasminogen activation and fibrinolysis by neutrophils. J Biol Chem. 2004;279:18063–18072. doi: 10.1074/jbc.M310462200. [DOI] [PubMed] [Google Scholar]
- 31.Lishko VK, V, Novokhatny V, Yakubenko VP, Skomorovska-Prokvolit HV, Ugarova TP. Characterization of plasminogen as an adhesive ligand for integrins αMβ2 (Mac-1) and α5β1 (VLA-5) Blood. 2004;104:719–726. doi: 10.1182/blood-2003-09-3016. [DOI] [PubMed] [Google Scholar]
- 32.Chavakis T, Athanasopoulos A, Rhee JS, Orlova V, Schmidt-Woll T, Bierhaus A, May AE, Celik I, Nawroth PP, Preissner KT. Angiostatin is a novel anti-inflammatory factor by inhibiting leukocyte recruitment. Blood. 2005;105:1036–1043. doi: 10.1182/blood-2004-01-0166. [DOI] [PubMed] [Google Scholar]
- 33.Miles LA, Plow EF. Receptor mediated binding of the fibrinolytic components, plasminogen and urokinase, to peripheral blood cells. Thromb Haemostasis. 1987;58:936–942. [PubMed] [Google Scholar]
- 34.Hogg N, Dransfield I. Regulated expression of Mg2+ binding epitope on leukocyte integrin alpha subunits. EMBO J. 1989;8:3759–3765. doi: 10.1002/j.1460-2075.1989.tb08552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Solovjov DA, Pluskota E, Plow EF. Distinct roles for the α and β subunits in the functions of integrin αMβ2. J Biol Chem. 2005;280:1336–1345. doi: 10.1074/jbc.M406968200. [DOI] [PubMed] [Google Scholar]
- 36.Leitinger B, Hogg N. The involvement of lipid rafts in the regulation of integrin function. J Cell Sci. 2002;115:963–972. doi: 10.1242/jcs.115.5.963. [DOI] [PubMed] [Google Scholar]
- 37.Xiong JP, Li R, Essafi M, Stehle T, Arnaout MA. An isoleucine-based allosteric switch controls affinity and shape shifting in integrin CD11b A-domain. J Biol Chem. 2000;275:38762–38767. doi: 10.1074/jbc.C000563200. [DOI] [PubMed] [Google Scholar]
- 38.Shimaoka M, Lu C, Salas A, Xiao T, Takagi J, Springer TA. Stabilizing the integrin αM inserted domain in alternative conformations with a range of engineered disulfide bonds. Proc Natl Acad Sci USA. 2002;99:16737–16741. doi: 10.1073/pnas.252633099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weber KS, Klickstein LB, Weber C. Specific activation of leukocyte β2 integrins lymphocyte function-associated antigen-1 and Mac-1 by chemokines mediated by distinct pathways via the α subunit cytoplasmic domains. Mol Biol Cell. 1999;10:861–873. doi: 10.1091/mbc.10.4.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang L, Plow EF. Overlapping, but not identical sites, are involved in the recognition of C3bi, NIF, and adhesive ligands by the αMβ2 integrins. J Biol Chem. 1996;271:18211–18216. doi: 10.1074/jbc.271.30.18211. [DOI] [PubMed] [Google Scholar]
- 41.Mayadas TN, Cullere X. Neutrophil β2 integrins: moderators of life or death decisions. Trends Immunol. 2005;26:388–395. doi: 10.1016/j.it.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 42.O’Reilly MS, Holmgren L, Shing Y, Chen C, Rosenthal RA, Moses M, Lane WS, Cao Y, Sage EH, Folkman J. Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell. 1994;79:315–328. doi: 10.1016/0092-8674(94)90200-3. [DOI] [PubMed] [Google Scholar]
- 43.Muchowski PJ, Zhang L, Chang ER, Soule HR, Plow EF, Moyle M. Functional interaction between the integrin antagonist neutrophil inhibitory factor and the I domain of CD11b/CD18. J Biol Chem. 1994;269:26419–26423. [PubMed] [Google Scholar]
- 44.Ugarova TP, Solovjov DA, Zhang L, Loukinov DI, Yee VC, Medved LV, Plow EF. Identification of a novel recognition sequence for integrin αMβ2 within the γ-chain of fibrinogen. J Biol Chem. 1998;273:22519–22527. doi: 10.1074/jbc.273.35.22519. [DOI] [PubMed] [Google Scholar]
- 45.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 46.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 47.Kim M, Carman CV, Yang W, Salas A, Springer TA. The primacy of affinity over clustering in regulation of adhesiveness of the integrin αLβ2. J Cell Biol. 2004;167:1241–1253. doi: 10.1083/jcb.200404160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chrzanowska-Wodnicka M, Burridge K. Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J Cell Biol. 1996;133:1403–1415. doi: 10.1083/jcb.133.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tardif MR, Tremblay MJ. Regulation of LFA-1 activity through cytoskeleton remodeling and signaling components modulates the efficiency of HIV type-1 entry in activated CD4+ T lymphocytes. J Immunol. 2005;175:926–935. doi: 10.4049/jimmunol.175.2.926. [DOI] [PubMed] [Google Scholar]
- 50.Kim SJ, Nair AM, Fernandez S, Mathes L, Laitinen J. Enhancement of LFA-1-mediated T cell adhesion by human T lymphotropic virus type 1 p12I. J Immunol. 2006;176:5463–5470. doi: 10.4049/jimmunol.176.9.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang L, Plow EF. Identification and reconstruction of the binding pocket within αMβ2 for a specific and high affinity ligand, NIF. J Biol Chem. 1997;272:17558–17564. doi: 10.1074/jbc.272.28.17558. [DOI] [PubMed] [Google Scholar]
- 52.Giancotti FG, Ruoslahti E. Integrin signaling. Science. 1999;285:1028–1032. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- 53.Meerschaert J, Vrtis RF, Shikama Y, Sedgwick JB, Busse WW, Mosher DF. Engagement of α4β7 integrins by monoclonal antibodies or ligands enhances survival of human eosinophils in vitro. J Immunol. 1999;163:6217–6227. [PubMed] [Google Scholar]
- 54.Ross EA, Douglas MR, Wong SH, Ross EJ, Curnow SJ, Nash GB, Rainger E, Scheel-Toellner D, Lord JM, Salmon M, Buckley CD. Interaction between integrin α9β1 and vascular cell adhesion molecule-1 (VCAM-1) inhibits neutrophil apoptosis. Blood. 2006;107:1178–1183. doi: 10.1182/blood-2005-07-2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Coxon A, Rieu P, Barkalow FJ, Askari S, Sharpe AH, Von Andrian UH, Arnaout MA, Mayadas TN. A novel role for the β2 integrin CD11b/CD18 in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity. 1996;5:653–666. doi: 10.1016/s1074-7613(00)80278-2. [DOI] [PubMed] [Google Scholar]
- 56.Pluskota E, Solovjov DA, Plow EF. Convergence of the adhesive and fibrinolytic systems: recognition of urokinase by integrin αMβ2 as well as by the urokinase receptor regulates cell adhesion and migration. Blood. 2003;101:1582–1590. doi: 10.1182/blood-2002-06-1842. [DOI] [PubMed] [Google Scholar]
- 57.Scapini P, Nesi L, Morini M, Tanghetti E, Belleri M, Noonan D, Presta M, Albini A, Cassatella MA. Generation of biologically active angiostatin kringle 1–3 by activated human neutrophils. J Immunol. 2002;168:5798–5804. doi: 10.4049/jimmunol.168.11.5798. [DOI] [PubMed] [Google Scholar]
- 58.Altieri DC, Plescia J, Plow EF. The structural motif glycine 190-valine 202 of the fibrinogen gamma chain interacts with CD11b/CD18 integrin alpha M beta 2, (Mac-1) and promotes leukocyte adhesion. J Biol Chem. 1993;268:1847–1853. [PubMed] [Google Scholar]
- 59.Buensuceso C, deVirgilio M, Shattil SJ. Detection of integrin αIIbβ3 clustering in living cells. J Biol Chem. 2003;278:15217–15224. doi: 10.1074/jbc.M213234200. [DOI] [PubMed] [Google Scholar]
- 60.Sánchez-Mateos P, Campanero MR, Balboa MA, Sanchez-Madrid F. Co-clustering of beta 1 integrins, cytoskeletal proteins, and tyrosine-phosphorylated substrates during integrin-mediated leukocyte aggregation. J Immunol. 1993;151:3817–3828. [PubMed] [Google Scholar]
- 61.Miles LA, Dahlberg CM, Plescia J, Felez J, Kato K, Plow EF. Role of cell-surface lysines in plasminogen binding to cells: identification of α -enolase as a candidate plasminogen receptor. Biochemistry. 1991;30:1682–1691. doi: 10.1021/bi00220a034. [DOI] [PubMed] [Google Scholar]
- 62.King WG, Mattaliano MD, Chan TO, Tsichlis PN, Brugge JS. Phosphatidylinositol 3-kinase is required for integrin-stimulated AKT and Raf-1/mitogen-activated protein kinase pathway activation. Mol Cell Biol. 1997;17:4406–4418. doi: 10.1128/mcb.17.8.4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sawatzky DA, Willoughby DA, Colville-Nash PR, Rossi AG. The involvement of the apoptosis-modulating proteins ERK1/2, Bcl-xL and Bax in the resolution of acute inflammation in vivo. Am J Pathol. 2006;168:33–41. doi: 10.2353/ajpath.2006.050058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sato T, Fujita N, Tsuruo T. Involvement of 3-phosphoinositide-dependent protein kinase-1 in the MEK/MAPK signal transduction pathway. J Biol Chem. 2004;279:33759–33767. doi: 10.1074/jbc.M402055200. [DOI] [PubMed] [Google Scholar]
- 65.Kennedy SG, Kandel ES, Cross TK, Hay N. Akt/protein kinase B inhibits cell death by preventing the release of cytochrome c from mitochondria. Mol Cell Biol. 1999;19:5800–5810. doi: 10.1128/mcb.19.8.5800. [DOI] [PMC free article] [PubMed] [Google Scholar]