

Abstract
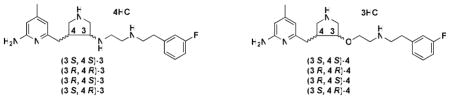
Neuronal nitric oxide synthase (nNOS) represents an important therapeutic target for the prevention of brain injury and the treatment of various neurodegenerative disorders. A series of trans substituted amino pyrrolidinomethyl 2-aminopyridine derivatives (8–34) was designed and synthesized. A structure-activity relationship analysis led to the discovery of low nanomolar nNOS inhibitors [(±)-32 and (±)-34] with more than 1000-fold selectivity for nNOS over eNOS. Four enantiomerically pure isomers of 3′-[2″-(3‴-fluorophenethylamino)ethoxy]pyrrolidin-4′-yl}methyl}-4-methylpyridin-2-amine (4) also were synthesized. It was found that (3′R, 4′R)-4 can induce enzyme elasticity to generate a new “hot spot” for ligand binding. The inhibitor adopts a unique binding mode, the same as that observed for (3′R, 4′R)-3′-[2″-(3‴-fluorophenethylamino)ethylamino]pyrrolidin-4′-yl}methyl}-4-methylpyridin-2-amine ((3′R, 4′R)-3) (J. Am. Chem. Soc. 2010, 132(15), 5437–5442). On the basis of structure-activity relationships of 8–34 and different binding conformations of the cis and trans isomers of 3 and 4, critical structural requirements of the NOS active site for ligand binding are revealed.
Introduction
Nitric oxide (NO),1 an essential signaling molecule, is produced by a family of enzymes called nitric oxide synthase (NOSa, EC 1.14.13.39).2 There are three known NOS isozyme forms, two constitutive forms and one inducible form. Neuronal NOS (nNOS), predominantly expressed in neurons, produces NO for neurotransmission; endothelial NOS (eNOS), produces NO for the regulation of blood pressure and flow; and inducible NOS (iNOS) is induced by cytokines and pathogens to produce NO to combat infection and microorganisms. Each isozyme of NOS catalyzes the conversion of L-arginine to NO and L-citrulline.3 All three enzymes share similar domain architecture and are active as homodimers.4 Each monomer has a N-terminal domain consisting of the catalytic heme active site and a (6R)-5,6,7,8-tetrahydro-L-biopterin (H4B) binding site and a C-terminal domain, containing FMN, FAD, and NADPH binding sites, which serves as an electron donating domain. The linker between the two functional domains is a calmodulin binding motif. The binding of calmodulin enables electron flow from the flavins to heme.
To exert appropriate physiological functions, NO generation by the three different NOS isoforms is under tight regulation.5,6 The overproduction of NO by nNOS has been implicated in various neurodegenerative diseases,7 including Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease; as well as neuronal damage resulting from stroke,8 cerebral palsy,9 and migraine headaches.10 Inhibition of nNOS could have therapeutic benefit in these diseases. However, this inhibition must be achieved without effect on the other NOS isoforms.11 Inhibition of eNOS could cause hypertension, atherosclerosis, arterial thrombosis, and angina. 12 Macrophage NOS (iNOS) is an enzyme responsible for the generation of cytotoxic NO, playing an important role in the immune system.13 Additionally, the inhibition of iNOS might result in a higher probability of Alzheimer’s disease.14 Therefore, isoform-selective inhibitors are essential if nNOS is to be a viable therapeutic target.15,16 Herein lies the challenge, because the crystal structures of the catalytic domains of all three NOS isoforms show that the active sites are nearly identical, making structure-based design of isoform-selective inhibitors a difficult and challenging problem.17
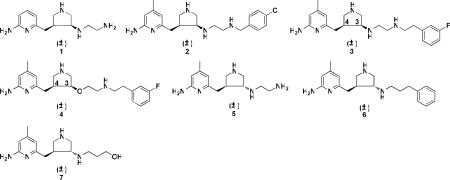
Previous structure-activity relationship studies in our laboratories on a series of Nω-nitroarginine containing dipeptide inhibitors have enabled us to identify a family of compounds that has high potency and selectivity for inhibition of nNOS over eNOS and iNOS.18,19 The selectivity of the dipeptide/peptidomimetic inhibitors for nNOS over eNOS and/or iNOS was investigated by crystallographic analysis20 and computer modeling (GRID/CPCA).21 Recently, we established a new fragment-based de novo design strategy called fragment hopping.22 Using this novel approach, together with what we learned from the dipeptide inhibitors, lead compound 1 (Table 1) with a pyrrolidinomethyl aminopyridine scaffold was designed and synthesized, which shows nanomolar nNOS inhibitory potency and more than 1000-fold nNOS selectivity over eNOS.22 Lead compound 1 was evolved into highly potent and selective inhibitors 2, 3, and 4.23,24 Compounds 2 and 3 were tested on a rabbit model for cerebral palsy and found to prevent hypoxia-ischemia induced death and to reduce the number of newborn kits exhibiting cerebral palsy phenotype without affecting eNOS-regulated blood pressure.25 In our previous study, although we found that the trans amine analogue (5) is a less potent and selective inhibitor for nNOS compared to 1,22 this structure still represents a new scaffold for inhibitor optimization. Two chiral centers (3′ and 4′ carbons) exist in the structure of 3, although our initial studies were carried out with the racemic mixtures. Recognizing the importance of chirality in molecular recognition and inhibitor binding, we synthesized four enantiomerically pure isomers of 3 in a previous study.26 The in vitro enzyme assay showed dramatic and exciting results. (3′R, 4′R)-3 is a more potent and selective inhibitor of nNOS over the other two NOS isozymes than (3′S, 4′S)-3 (Table 3). The Ki value for (3′R, 4′R)-3 is 5 nM, and the selectivities for nNOS over eNOS and iNOS are more than 3,800-fold and 700-fold, respectively. This compound is, to the best of our knowledge, the most potent and dual-selective nNOS inhibitor reported to date.26 Using a combination of crystallography, computation, and site-directed mutagenesis studies, it was found that nNOS undergoes an unanticipated structural change to accommodate the 2-amino-4-methylpyridine moiety of (3′R, 4′R)-3. A new “hot spot” was generated because of enzyme elasticity. The binding conformation of (3′R, 4′R)-3 in the crystal structure of nNOS is 180° flipped over compared to that of (3′S, 4′S)-3. We, therefore, synthesized the four enantiomerically pure isomers of 4 (Table 1), another very potent and highly selective inhibitor of nNOS with better drug-like properties, and determined their inhibitory potencies and isozyme selectivities. Here we describe the interactions of these enantiomers and of a series of compounds related to 3 and 4 with nNOS and eNOS, which reveal important structural requirements for induction of enzyme elasticity and generation of a new binding site.
Table 1.
Inhibition of NOS isozymes by 1–7.
| Compounds |
Ki (μM) |
Selectivitya |
|||
|---|---|---|---|---|---|
| nNOS | eNOS | iNOS | n/e | n/i | |
| (±)-1 | 0.388 | 434.5 | 58.4 | 1114 | 150 |
| (±)-2 | 0.085 | 85 | 8.95 | 1000 | 106 |
| (±)-3 | 0.014 | 28 | 4.06 | 2000 | 290 |
| (±)-4 | 0.015 | 31 | 9.5 | 2067 | 633 |
| (±)-5 | 8.76 | 866.2 | 77.4 | 99 | 9 |
| (±)-6 | 13.28 | 85.2 | 143.8 | 6 | 11 |
| (±)-7 | 28.54 | 683.7 | 233.8 | 24 | 8 |
n/e and n/i are the selectivity ratio of Ki (eNOS or iNOS) to Ki (nNOS).
Table 3.
Inhibition of NOS isozymes by the enantiomerically pure isomers of 3 and 4.
 | |||||
|---|---|---|---|---|---|
| Compounds |
Ki (nM)a |
Selectivityb |
|||
| nNOS | eNOS | iNOS | n/e | n/i | |
| (3′S, 4′S)-3c | 52.2 | 26,400 | 3,850 | 506 | 73.8 |
| (3′R, 4′R)-3c | 5.3 | 20,300 | 3,940 | 3830 | 743 |
| (3′R, 4′S)-3c | 18.9 | 57,100 | 16,100 | 3021 | 852 |
| (3′S, 4′R)-3c | 171.0 | 34,500 | 26,600 | 202 | 156 |
| (3′S, 4′S)-4 | 116.0 | 26,200 | 7,500 | 226 | 65 |
| (3′R, 4′R)-4 | 7.2 | 19,200 | 5,800 | 2667 | 806 |
| (3′R, 4′S)-4 | 63.0 | 36,190 | 18,710 | 574 | 297 |
| (3′S, 4′R)-4 | 193.5 | 18,780 | 18,060 | 97 | 93 |
The apparent Ki values are represented as the mean from two or more independent experiments preformed in duplicate with five or six data points each and correlation coefficients of 0.92–0.99. The experimental standard deviations were less than 10%.
n/e and n/i are the selectivity ratio of Ki (eNOS or iNOS) to Ki (nNOS).
The data were reported in the previous paper.26
Results and Discussion
Inhibitor Design
In our previous studies, starting with the nitroarginine-containing dipeptide inhibitors, fragment hopping was used to determine the minimal pharmacophoric elements for inhibitor potency and NOS isozyme selectivities.22 A series of non-peptide cis substituted amino pyrrolidinomethyl 2-aminopyridine derivatives was designed and synthesized to match the requirements of the minimal pharmacophoric elements (Figure 1). The crystal structures of nNOS in complex with 1 and 2 (Figure 2) show that the binding of the inhibitors exactly mimic the binding mode of the dipeptide nNOS inhibitors.27 As shown in Figure 1, two structural water molecules bridge the hydrogen bonding interactions of the pyrrolidino nitrogen atom and the side chain carboxylic acid group of Asp597. Therefore, there is some space between this nitrogen atom and the Asp597 carboxylate. Compounds 8–11 (Figure 3A) were designed to explore the space in this area and to determine whether or not the nNOS inhibitory potency and selectivities can be increased if a positively-charged amino group of the inhibitor is placed closer to the side chain carboxylic acid group of Asp597.
Figure 1.

Schematic drawing of the active site of nNOS. The binding mode of the cis pyrrolidinomethyl aminopyridine inhibitors and the amino acids and cofactors important for inhibitor binding are indicated. Numbering is for the sequence of nNOS if not indicated. The H-bonds observed in the crystal structure are indicated by dotted lines based on the crystal structures of nNOS in complexes with 1 and 2 (PDB id: 3B3N and 3B3O).27 Under physiological conditions, the nitrogen atoms of the pyridine ring, the pyrrolidine ring, and the terminal amino group of the ethylenediamine side chain in NOS are protonated, but the nitrogen atom attached to the pyrrolidine ring is in the neutral form.
Figure 2.

Crystallographic binding conformations of 1 (A) and 2 (B) complexed with nNOS (PDB id: 3B3N and 3B3O).27 The heme (orange), H4B (purple), and structural water molecules (green) involved in the binding of 1 and 2 to nNOS are shown. The active site residues and ligands are represented in an atom-type style (carbons in grey, nitrogens in blue, oxygens in red, and sulfur in yellow). The distances of some important H-bonds between the residues, structural water molecules, cofactors, and inhibitors are given in Angstroms (Å).
Figure 3.

Target molecules synthesized and tested in this study
Trans amine 5 represents a new lead compound for optimization. In the previous studies,23 it was found that there is more space at position 4 of the 2-aminopyridine ring that would favor hydrophobic and steric interactions (S pocket),28,23 and there is another hydrophobic cavity in the C1 pocket, as shown in Figure 4.23 Structural optimization of 5 is based on these hydrophobic pockets. One series of trans molecules, 13 through 20 in Figure 3B, were designed to introduce various conformational constraints to the ethylenediamine side chain to minimize the entropic penalty associated with flexibility of this side chain and also to extend the tail into the C1 pocket with a benzyl group. In the other series of trans molecules, 21 through 34 in Figure 3B, a methyl group was introduced at position 4 of the 2-aminopyridine ring to occupy the hydrophobic region in the S pocket, and the hydrophobic benzyl group or phenethyl group was extended from the terminal amino group of 5 to occupy the C1 pocket (Figure 4).
Figure 4.

Results of the GRID analyses of the substrate-binding site of nNOS (PDB id: 1P6I) illustrates the direction of lead optimization. The residues and inhibitor 5 are represented in an atom-type style (carbons in grey, nitrogens in blue, oxygen in red, and sulfur in yellow). Heme and H4B are colored magenta. The S, M, and C1 pockets are indicated. The GRID contours of the DRY probe (yellow) is set at an energy level of −0.75 kcal/mol. The GRID contours of the C3+ probe (green) is set at an energy level of −4.00 kcal/mol.
It was found in our previous study that the racemic mixture of the ether derivative (4) exhibits comparable nNOS inhibitory potency and selectivity over eNOS compared to 3.24 The difference between 4 and 3 is that compound 4 uses a hydrogen-bonding acceptor group (ether), and 3 uses a hydrogen-bonding donor group (secondary amine). Therefore, 4 exhibits better drug-like properties and improved bioavailability.24 Enzyme assays in the presence of the four enantiopure isomers of 3 revealed that (3′R, 4′R)-3 exhibits better nNOS inhibitory potency and selectivity over the other two NOS isozymes than (3′S, 4′S)-3. Crystallographic analysis shows that (3′R, 4′R)-3 induces enzyme elasticity, and a new “hot spot” is generated to accommodate the 2-amino-4-methylpyridine moiety of (3′R, 4′R)-3.26 Therefore, the four enantiomers of 4 were synthesized to determine whether or not (3′R, 4′R)-4 can similarly induce enzyme elasticity to generate a new ligand binding pocket for better nNOS inhibitory potency and selectivity over the other two NOSs. In the inhibitor design cycles, AutoDock 3.0.5 was routinely used to dock most of the inhibitors in the active site before the synthesis was carried out.29
Chemistry
Scheme 1 shows the synthetic route for target compounds 8–11. The synthesis of 37 was described in an earlier paper.22 Deprotection of the benzyl group by catalytic hydrogenation and then alkylation with N-Boc-2-bromoethylamine generated 39. Swern oxidation of 39 generates ketone intermediate 40 in high yields. The direct reductive amination of 40 with different amines generated the cis (41) and trans (42) amines. These two regioisomers can be completely separated by silica gel column chromatography; deprotection of the Boc groups generates the final products.
Scheme 1.

Characterization of the cis and trans isomers was studied in detail by NMR spectrometry, including 1H NMR, 13C NMR, 13C NMR-DEPT, 1H-1H COSY, 1H-13C HMQC and 1H-1H NOSEY spectrometry, as described in a previous paper.22 It was found that one prominent difference between the final products of the cis and trans isomers is that the 13C chemical shifts of the carbon atom attached to the pyridine ring is about 29.5 ppm for the cis compound, but 34.1 ppm for the trans isomer. This difference was further used in the rapid structural characterization of compounds 1–34. Typically, all of the trans isomers show a lower Rf value on TLC compared to the corresponding cis isomers.
The synthetic route for 12–20 is shown in Scheme 2. Deprotection of the Boc groups of 44 generated 12. The synthesis of intermediate 45 was described in earlier papers.22,23 To optimize the yield of the trans amine analogue, different conditions for the reductive amination reaction were tried. First, direct reductive amination reactions were tried. When molecular sieves were used as the desiccant, anhydrous MeOH was used as the solvent, one equivalent of acetic acid was used as the proton provider, and sodium cynoborohydride was used as the reducing agent, the yields of the amines were about 50–65%. The ratio of the cis to trans amines was about 45:55 for 46b–46d. As described in the previous study,23 when the catalyst for the hydrogenation reaction was changed to platinum IV oxide, the ratio of the cis to trans isomers changed to 55:45. When sodium triacetoxyborohydride was used as the reducing agent, the ratio of the cis to trans isomers was 95:5. Indirect reductive amination reactions also were attempted. The ketone was refluxed with the amine in benzene, and water was removed with a Dean-Stark trap. The imine compound was produced, which was then reduced with sodium borohydride in MeOH. However, the yield of the total amine was much lower (about 10–20%). The ratio of cis to trans isomers was 1:2. When molecular sieves were used as the desiccant, the reaction mixture was stirred at room temperature then reduced with sodium borohydride in MeOH, the yield of the total amines was increased to 30%, and the ratio of the cis to trans isomers was 1:2. When molecular sieves were used as the desiccant, and the catalytic hydrogenation reaction using Pd/C as catalyst was conducted, the ratio of the cis to trans isomers was changed to 3:1. Consequently, direct reductive amination reactions using sodium cynoborohydride as the reducing agent were carried out for the rest of the compounds. The yields of the total amines still remained about 50–65%. The ratio of the cis to trans isomers was about 30:70 for 46a, 46g, and 46h. For compounds 46b–46f in Scheme 2A and 50 in Scheme 2B, the ratio of cis to trans isomers was about 45:55.
Scheme 2.

In the early stage of the synthesis of this series of inhibitors we discovered that trans alcohols 44 or 48a could not be used in any SN2 reaction directly. The only product generated was a new cis five-membered ring (a quarternary ammonium) that occurred by intramolecular attack of the nitrogen atom of the 2-aminopyridine on the carbon atom of the pyrrolidino ring to which the hydroxyl group was attached. So 44 or 48a was oxidized to a ketone, and then reductive amination was carried out to generate the trans amines, as shown in Schemes 1 and 2.22,23 Later, we discovered that Mitsunobu reactions proceeded smoothly when the mono Boc protected 2-amino group of the 2-aminopyridine moiety of 44 was further protected with a benzyl group or an additional Boc group.30 In this study we continued to use a benzyl group as the protecting group to protect 48a, which allowed the SN2 reaction to proceed, and to synthesize the trans isomers, as shown in Scheme 3. Mitsunobu reaction of 51 and the subsequent hydrolysis of the acetate afforded cis alcohol 53, which could be converted to trans azide 54 by the application of another Mitsunobu reaction. Hydrogenation of azide 54 using Pd(OH)2/C as catalyst generated amine 55, which could be converted to 56 by reductive amination. Compounds 23, 32, and 34 were re-synthesized by this synthetic route.
Scheme 3.

The synthesis of the four enantiopure isomers of 4 is shown in Scheme 4. Different from the synthetic route reported in our previous study,26 compound 51 was directly subjected to the Mitsunobu reaction to generate ester 57 using 1S-(−)camphanic acid as a nucleophile (Scheme 4A). The synthesis from four optically pure alcohol isomers of 58 to four isomers of 4 is shown in Scheme 4B. The synthetic route is very similar to the synthesis of the racemic mixture of 4.24 Allylation of (3′R, 4′R)-58 with allyl bromide generated (3′R, 4′R)-60, which then underwent ozonolysis to generate aldehyde (3′R, 4′R)-61. Direct reductive amination of (3′R, 4′R)-61 with 3-fluorophenethylamine generated (3′R, 4′R)-62. Protection of the secondary amine of (3′R, 4′R)-62 with a Boc group and then deprotection of the benzyl group afforded (3′R, 4′R)-64. Deprotection of the Boc groups generated (3′R, 4′R)-4. The other three enantiopure isomers were synthesized by the same route.
Scheme 4.

NOS Inhibition and Structure-Activity Relationships of Compounds 8–34
Table 2 shows the results of the NOS enzyme inhibition assays. Trans amines 9 and 11 exhibited much better nNOS inhibitory potency and better nNOS selectivity over the other two NOS isozymes than the corresponding cis amine derivatives (8 and 10), respectively. This suggests that the aminoethyl group of the trans isomers is better accommodated than that of the cis isomers. Compared to the unsubstituted trans isomers, such as 6 and 7, compounds 9 and 11 exhibit two- to three-fold better inhibitory potency and five- to seven-fold better selectivities for nNOS over eNOS. These are not very significant improvements. Considering that the primary amine was reported to have a much larger polar surface area (PSA) than the secondary amine, which would be unfavorable for biomembrane permeability,31 the 2-aminoethyl side chain of 9 and 11 was not further pursued. Instead, other structural optimization of the inhibitors was based on the structure of 5.
Table 2.
Inhibition of NOS isozymes by synthetic compounds 8–34.
| Compounds |
Ki (μM)a |
Selectivityb |
|||
|---|---|---|---|---|---|
| nNOS | eNOS | iNOS | n/e | n/i | |
| L-NNAc | 0.57 | 0.75 | 4.55 | 1.3 | 8 |
| (±)-8 | 47.94 | 121.62 | 609.43 | 2.5 | 13 |
| (±)-9 | 3.98 | 172.91 | 437.76 | 43 | 110 |
| (±)-10 | 304.96 | 849.21 | 2,522.8 | 3 | 8 |
| (±)-11 | 13.47 | 1,696 | 651.85 | 126 | 48 |
| (±)-12 | 6.94 | 340.83 | 25.24 | 49 | 4 |
| (±)-13 | 15.68 | 241.7 | 308.1 | 15 | 20 |
| (±)-14 | 14.45 | 2,906 | 166.1 | 201 | 11.5 |
| (±)-15 | 13.86 | 1,453 | 298.55 | 105 | 21.5 |
| (±)-16 | 20.57 | 1,453 | 416.61 | 71 | 20 |
| (±)-17 | 12.85 | 625.42 | 234.06 | 49 | 18 |
| (±)-18 | 20.93 | 460.16 | 253.43 | 22 | 12 |
| (±)-19 | 17.92 | 108.69 | 209.01 | 6 | 12 |
| (±)-20 | 24.4 | 170.52 | 285.54 | 7 | 12 |
| (±)-21 | 0.61 | 262.74 | 32.03 | 431 | 52.5 |
| (±)-22 | 0.53 | 118.34 | 44.85 | 223 | 85 |
| (±)-23 | 0.25 | 95.18 | 44.11 | 380 | 176 |
| (±)-24 | 0.74 | 61.19 | 40.42 | 83 | 55 |
| (±)-25 | 0.87 | 78.75 | 27.35 | 90.5 | 31 |
| (±)-26 | 0.74 | 108.55 | 58.72 | 147 | 79 |
| (±)-27 | 0.66 | 201.61 | 83.9 | 305.5 | 127 |
| (±)-28 | 0.414 | 63.22 | 23.76 | 153 | 57 |
| (±)-29 | 0.483 | 76.13 | 44.23 | 158 | 92 |
| (±)-30 | 0.589 | 16.47 | 20.46 | 28 | 35 |
| (±)-31 | 1.891 | 58.14 | 75.36 | 31 | 40 |
| (±)-32 | 0.088 | 123.90 | 18.17 | 1408 | 206.5 |
| (±)-33 | 0.119 | 44.09 | 22.59 | 370.5 | 190 |
| (±)-34 | 0.048 | 49.07 | 26.59 | 1022 | 554 |
The apparent Ki values are represented as the mean of two or more independent experiments preformed in duplicate with five or six data points each and correlation coefficients of 0.88–0.99. The experimental standard deviations were less than 10%.
n/e and n/i are the selectivity ratio of Ki (eNOS or iNOS) to Ki (nNOS).
L-NNA: L-Nω-nitroarginine, a positive control.
Structural optimization of 5 leads to the generation of three interesting compounds: 23, 32, and 34. Compound 23 is the most potent nNOS inhibitor among the compounds with a benzyl moiety. Compound 34 is the most potent inhibitor among the compounds with a phenethyl moiety. The Ki value for 34 with nNOS is 48 nM, and the selectivity for nNOS over eNOS and iNOS is more than 1000-fold and 500-fold, respectively. Introduction of conformational constraints, such as a ring or a methyl group, to the ethylenediamine side chain of 5 generates 17–20, 30, and 31. The enzyme assay shows that these structural modifications do not work well (Table 2).
The crystal structures of 23 in a complex with nNOS and eNOS were reported previously (PDB code: 3B3P and 3DQT).27 The binding mode of the (3R, 4S)-isomer of 23 with nNOS is very similar to that of 2, except for the nitrogen atom attached to the pyrrolidine ring (Figures 2B and 5). In 2 this nitrogen atom makes a H-bond with heme propionate A, but the nitrogen atom in 23 points away from the propionate because of the trans configuration. Although 23 has a meta-chloro and 2 has a para-chloro substituent, both the meta-chlorobenzyl and para-chlorobenzyl groups point to the same surface hydrophobic pocket, and the active site is large enough to accommodate both. The location of the pyrrolidine ring of 23 is the same as that in 2. The ring nitrogen H-bonds to the side chain of Glu592 in both cases. Two structural waters were observed to bridge the H-bonding interactions between the side chain carboxylic acid group of Asp597 and the pyrrolidine nitrogen atom in both 2 and 23. Therefore, the principal difference between 2 and 23 derives from the chirality of the pyrrolidino ring. The trans configuration in 23 makes it impossible to have a H-bond between its nitrogen attached to the pyrrolidine and heme propionate A. In contrast, the additional H-bond in 2 gives the cis molecule its better inhibitory potency.
Figure 5.

Crystallographic binding conformations of 23 in complex with nNOS (PDB id: 3B3P).27 The heme (orange), H4B (purple), and structural water molecules (green) involved in the binding of 23 to nNOS are shown. The active site residues and ligands are represented in an atom-type style (carbons in grey, nitrogens in blue, oxygens in red, and sulfur in yellow). The distances of some important H-bonds between the residues, structural water molecules, cofactors, and inhibitors are given in Angstroms (Å).
The crystallographic binding conformations of 2 and 23 in nNOS and eNOS are very similar, which is drastically different from the binding mode of the L-Nω-nitroarginine-containing dipeptide inhibitors we developed before.19 The dipeptide inhibitors have multiple flexible bonds around the α-amino group of the L-Nω-nitroarginine moiety that generates nNOS selectivity over eNOS. Correspondingly, the binding conformation of L-Nω-nitroarginine-containing dipeptide inhibitors in nNOS is different from that in eNOS. The dipeptide inhibitor adopts a curled conformation in nNOS, but an extended conformation in eNOS.20 The introduction of the pyrrolidino ring in the pyrrolidinomethyl 2-aminopyridine derivatives (such as 2 and 23) potentially increases their nNOS selectivity over eNOS because this scaffold has a lower entropic penalty.
NOS Inhibition and Crystallographic Analysis of Four Enantiomers of 4
Identification of 32 and 34 as the most potent inhibitors among those compounds with a phenethyl moiety prompted us to synthesize and characterize the enantiopure compounds. The synthesis and enzyme assay results for the four enantiomers of 3 have been reported recently.25 It was found that (3′R, 4′R)-3 is a more potent inhibitor of nNOS and more selective over the other two NOS isozymes than (3′S, 4′S)-3. The Ki value for (3′R, 4′R)-3 with nNOS is 5 nM. The nNOS selectivities over eNOS and iNOS are more than 3800-fold and 700-fold, respectively. Trans (3′R, 4′S)-3 is also a very potent and selective nNOS inhibitor with a Ki value of 19 nM with selectivities over eNOS and iNOS of more than 3000 fold and 800 fold, respectively.
Similarly, enzyme assays show that (3′R, 4′R)-4 is a more potent and selective inhibitor of nNOS than (3′S, 4′S)-4 (Table 2). The Ki for nNOS is 7 nM, and the selectivities over eNOS and iNOS are more than 2,600-fold and 800-fold, respectively. To the best of our knowledge, this compound and (3′R, 4′R)-3 represent two of the most potent and selective nNOS inhibitors reported to date. Trans (3′R, 4′S)-4 also is a potent and selective nNOS inhibitor with a Ki value of 63 nM with selectivities over eNOS and iNOS of over 500-fold and 300-fold, respectively (Table 3).
Crystal structures of nNOS in a complex with the four enantiopure isomers of 4 were determined. The binding conformation of (3′S, 4′S)-4 in nNOS (Figure 6A) shows that the 2-amino-4-methylpyridine moiety interacts with active site Glu592. The pyrrolidine nitrogen is close to the selective residue (nNOS Asp597/eNOS Asn368). The nitrogen atom of the 3-fluorophenethylamino group forms one H-bond with heme propionate D, and the 3-fluorophenyl moiety is located in the surface hydrophobic pocket lined with Met336, Leu337, Tyr706, and Trp306 from the second monomer. This binding mode is termed the “normal” binding mode, which means it mimics the binding mode of the dipeptide inhibitors. (3′R, 4′R)-4, however, adopts the 180°-flipped orientation and induces nNOS enzyme elasticity to generate a new “hot spot” to accommodate the 2-amino-4-methylpyridine (Figure 6B). For this to occur, the side chain of Tyr706 must swing away to make room for π-π stacking interactions with the 2-amino-4-methylpyridine ring. The 3-fluorophenyl group is, therefore, positioned over the heme while the 2-amino-4-methylpyridine ring extends out of the substrate binding site. The pyrrolidine nitrogen is placed in between the heme propionate A (2.9 Å) and the carbonyl group of H4B (2.6 Å). One structural water molecule bridges three H-bonds between heme propionate D, the oxygen atom attached to the pyrrolidine ring, and the nitrogen atom of the phenethylamine side chain. The binding mode of (3′R, 4′R)-4 is identical with that of (3′R, 4′R)-3, which has been termed the “flipped” binding mode.
Figure 6.

Crystallographic binding conformations of four enantiomerically pure isomers of 4 [A: (3′S, 4′S)-4, B: (3′R, 4′R)-4, C: (3′R, 4′S)-4, D: (3′S, 4′R)-4] with rat nNOS. Shown also is the 2Fo – Fc electron density for the ligands contoured at 1 σ. The active site residues and ligands are represented in an atom-type style (carbons in green or cyan (chain B), nitrogens in blue, oxygen in red, and sulfur in yellow). The important H-bonds between the residues, structural water, cofactors, and inhibitors are depicted with dashed lines. The resolution and Rwork/Rfree values for the four structures shown are (A) 2.02 Å, 0.183/0.219; (B) 1.85 Å, 0.189/0.222; (C) 2.00 Å, 0.196/0.231; (D) 2.30 Å, 0.201/0.266. The 4-methyl-2-aminopyridine ring in (3′S, 4′S)-4, (3′R, 4′S)-4, and (3′S, 4′R)-4 and the m-fluoro-phenyl ring in (3′R, 4′R)-4 are roughly parallel to the heme plane with the closest distance to the heme iron at 4.0–4.1 Å. It is not clear if this is a π-π stacking interaction or a cation-π interaction.
Like the two trans isomers of 3, (3′R, 4′S)-4 and (3′S, 4′R)-4 only adopt the “normal” binding mode. The 2-aminopyridine moiety forms a bifurcated salt bridge with active site Glu592. The fluorophenyl group extends out of the substrate-catalytic site and is located in the surface hydrophobic pocket (Figures 6C and 6D). The pyrrolidine nitrogen atom of the (3′R, 4′S)-isomer forms one H-bond with Glu592 and another H-bond with a structural water, which in turn binds to Asp597. The positively charged nitrogen atom in the pyrrolidine ring interacts favorably with the side chain carboxylic acid group of Asp597. In the case of (3′S, 4′R)-4, although the binding of the 2-aminopyridine next to the Glu592 side chain is supported by good electron density, the density for the pyrrolidine ring of (3′S, 4′R)-4 is not well defined, and the 3-fluorophenethylamino group is almost fully disordered (Figure 6D). The model that fits the density has the orientation of the pyrrolidine ring of (3′S, 4′R)-4 opposite that of (3′R, 4′S)-4. From the binding conformations of these two isomers, it can be concluded that (3′R, 4′S)-4 can form better electrostatic interactions with active site residues than (3′S, 4′R)-4, which rationalizes why (3′R, 4′S)-4 exhibits better nNOS inhibitory potency and selectivity than (3′S, 4′R)-4.
The enzyme assays show that the trans isomers, (3′R, 4′S)-3 and (3′R, 4′S)-4, exhibit better nNOS inhibitory potency and selectivity than the cis isomers (3′S, 4′S)-3 and (3′S, 4′S)-4 (Table 3), although all of these compounds adopt the “normal” binding mode. The superimposition of (3′R, 4′S)-3 and (3′S, 4′S)-3 or (3′R, 4′S)-4 and (3′S, 4′S)-4 by the backbone atoms of the active site residues, as defined in our previous paper,21 shows that the 2-amino-4-methylpyridino and the 3-fluorophenyl groups overlap fairly well (Figure 7). The low-energy structures of the cis (3′S, 4′S)-isomers of 3 and 4 are typically longer than those of the trans (3′R, 4′S)-isomers if they have the same orientations. One conclusion that can be drawn on the basis of the enzyme assay results and the crystallographic analysis is that the length of the cis (3′S, 4′S)-isomers of 3 and 4 exceeds the requirement between the substrate-catalytic pocket (the S pocket in Figure 3) and the hydrophobic pocket (the C1 pocket in Figure 3) of nNOS. Therefore, the cis (3′S, 4′S)-isomers of 3 and 4 cannot be accommodated as well as the trans (3′R, 4′S)-isomers (Figure 7). The 2-aminopyridine ring in (3′R, 4′S)-3 or (3′R, 4′S)-4 is more parallel to the heme plane than that seen in the (3′S, 4′S)-structures, which strengthens the stacking interaction between the two aromatic rings in the trans structure. The trans configuration forces the pyrrolidine rings in the (3′R, 4′S)-isomers to be better aligned with the carboxylic acid side chain of Glu592, thus resulting in a stronger H-bond than that found in the cis (3′S, 4′S) nNOS structures. However, it is noteworthy that the different configurations of the pyrrolidino rings in the (3′R, 4′S) and (3′S, 4′S)-isomers lead to the formation of two extra H-bonds for the less favorable cis isomers, (3′S, 4′S)-3 and (3′S, 4′S)-4. One extra H-bond is from the nitrogen atom attached to the pyrrolidine ring in (3′S, 4′S)-3 with heme propionate A. Second, the positively charged nitrogen atom of the 3-fluorophenethylamino side chain can form a H-bond with heme propionate D in both (3′S, 4′S)-3 and (3′S, 4′S)-4. Overall enzyme-inhibitor interactions seen in the trans (3′R, 4′S)-3 and (3′R, 4′S)-4 structures, dominated by those involving the 2-aminopyridine and pyrrolidine rings, are slightly better than those in cis (3′S, 4′S)-3 and (3′S, 4′S)-4 structures, which accounts for a two to three-fold better inhibitory potency for the trans (3′R, 4′S)- over the cis (3′S, 4′S)-isomers (Table 3). From this study, it can be concluded that if the inhibitor adopts the “normal” binding mode, the nNOS active site can optimally accommodate the inhibitors with a length that matches those of the trans (3′R, 4′S)-isomers of 3 or 4.
Figure 7.

Superimposition of the crystallographic binding conformations of (3′R, 4′S)-4 and (3′S, 4′S)-4. The heme (orange), H4B (purple), and structural water (green) involved in the binding of inhibitor to nNOS are shown. The active site residues are represented in an atom-type style (carbons in grey, nitrogens in blue, oxygen in red, and sulfur in yellow). The carbons of (3′R, 4′S)-4 are colored green, and the carbons of (3′S, 4′S)-4 are colored yellow. The nitrogens and the fluorine are colored blue and green, respectively. The distances of the H-bonds between the residues, structural water, cofactors and inhibitors from the crystal structure of nNOS in a complex with (3′S, 4′S)-4 are indicated in Angstroms (Å). The corresponding distances from the crystal structure of nNOS in a complex with (3′R, 4′S)-4 are similar but not shown.
Similar to what we have described for the nNOS and eNOS structures bound with the four enantiopure isomers of 3,26 the binding conformations of the four enantiopure isomers of 4 to the eNOS active site (Figure S1) are generally the same as what we have just discussed for nNOS. The rigidity of the pyrrolidine ring brings its ring nitrogen to the vicinity of Glu592 (Glu363 in eNOS) and Asp597 (Asn368 in eNOS) when the configuration is (3′S, 4′S) or (3′R, 4′S), regardless of whether inhibitors are bound to nNOS or to eNOS. The electrostatic stabilization of this positively charged ring nitrogen is less optimal in eNOS than in nNOS because of the single amino acid difference (Asn368 in eNOS vs. Asp597 in nNOS). This is the structural basis for isoform selectivity of these inhibitors. The (3′S, 4′R) isomer of 4 behaves slightly differently when its complex structures of nNOS and eNOS are compared. While the pyrrolidine ring adopts an opposite orientation in the (3′S, 4′R)-4 nNOS structure, the electron density in the (3′S, 4′R)-4 eNOS structure supports an orientation that makes H-bonds to both heme propionates. Finally, the (3′R, 4′R) isomer of 4 binds to eNOS also in a “flipped” mode (Figure S1) exactly as observed in nNOS. Three residues, Met336, Asp597, and Tyr706, were found partially responsible for the selectivity for nNOS over eNOS, the same as discussed previously for (3′R, 4′R)-3.26 However, further studies are required to better understand 2600-fold nNOS selectivity of this compound over eNOS.
Conclusions
In this study we reported the design, synthesis, and structure-activity relationships of the racemic trans substituted amino pyrrolidinomethyl 2-aminopyridine derivatives 8–34, which leads to the discovery of a new series of inhibitors with high nNOS inhibitory potency and high nNOS selectivities over the other two NOS isozymes. The active site requirements for the trans 2-aminopyridine nNOS inhibitors were explored through rational inhibitor design and structure-activity relationship analyses. Low nanomolar trans amine nNOS inhibitors [(±)-32 and (±)-34] were discovered, having Ki values for nNOS of 88 and 48 nM, respectively. The nNOS selectivities of these two inhibitors over eNOS are more than 1000-fold. Four enantiomerically pure isomers of 4 also were synthesized, and structures of complexes with nNOS and eNOS were determined in this study. It was found that, similar to (3′R, 4′R)-3, (3′R, 4′R)-4 can also induce enzyme elasticity to generate a “hot spot” that accommodates the 2-amino-4-methylpyridine moiety. The binding mode of this isomer adopts a 180° “flipped” orientation compared with the expected “normal” binding mode. This compound and (3′R, 4′R)-3 represent the two most potent and selective nNOS inhibitors reported so far. The influence of the pyrrolidine configuration to enzyme-inhibitor interactions is also observed when the structures of nNOS in complex with the cis (3′S, 4′S)- and trans (3′R, 4′S)-isomers of 3 or 4 are compared. The critical structural determinants by which inhibitors can be simultaneously accommodated by the substrate-catalytic site and the surface hydrophobic pocket of nNOS have been revealed.
Our previous26 and current studies implicate a new avenue for the rational design of enzyme inhibitors, that is, to look for a potential hot spot in the target protein that is occluded by the present active site residues, for instance the side chains of the residues. This finding is particularly useful for those inhibitor design studies where the active sites are shallow and/or of highly charged nature, and a new hot spot for inhibitor potency is needed. The 2-amino-4-methylpyridine moiety is one of the most important pharmacophores for this series of inhibitors, as is evident by the fact that (±)-3 (Ki =14 nM for nNOS) is much more potent than the corresponding (±)-2-amino-6-methyl derivative (Ki = 326 nM for nNOS).23 The pocket of nNOS that accommodates this “new hot” is only generated when the enzyme undergoes an induced fit upon inhibitor binding.
Experimental Section
Computer Modeling
Protein Structures
The crystallographic coordinates for NOS from the Research Collaboratory for Structural Bioinformatics (RCSB) protein database are as follows: rat nNOS1: 1P6I (1.90 Å resolution, Rcryst = 0.228); 3B3N (1.98 Å resolution, Rcryst = 0.230). The amino acid sequences of NOS were retrieved from the PIR protein sequence database. All of the computational work was performed on Silicon Graphics Octane 2 Workstations with an IRIX 6.5 operating system. The molecular modeling was achieved with commercially available InsightII 200032 and SYBYL 6.833 software packages. The NOS model was prepared by first adding H-atoms using the Accelrys/InsightII 2000 software, and the protonation states of the residues were set to pH 7.0. The cvff force field of the Discover 98.0 program within Insight II was used to optimize the orientation of hydrogen atoms of the protein and of structural waters. The ligands and solvent molecules of the protein structures were removed, but the heme and H4B were retained near the active site.
GRID Calculations
The calculations were performed with version 22 of the GRID software.34 Hydrogens were added with the program GRIN. The GRID box dimensions were chosen to encompass all of the active site residues. This resulted in a box size of 31 × 28 × 31 Å. The grid spacings were set to 1 Å (directive NPLA = 1) and 0.5 Å (directive NPLA = 2), respectively. The amino acids in the active site were considered rigid (directive move = 0). The directives NETA and ALMD were set to 120 and 1, respectively, to include the atoms of heme and H4B in calculations and to interpret which atom(s) in the active site contribute(s) to the interaction with a specific probe atom. The following single atom probes were used in the calculation: DRY, C3, C1=, NM3, N1+, N2+, Cl and F.
Chemical Synthesis
General Methods, Reagents, and Materials
All reagents were purchased from Aldrich, Acros Organics, or Fisher Scientific and were used without further purification unless stated otherwise. NADPH, calmodulin, and human ferrous hemoglobin were obtained from Sigma Chemical Co. Tetrahydrobiopterin (H4B) was purchased from Alexis Biochemicals. 1H NMR and 13C NMR spectra were recorded on a Varian Mercury 400 MHz or a Varian Inova 500 MHz spectrometer (100.6, or 125.7 MHz, for 13C NMR spectra, respectively) in CDCl3, DMSO-d6, CD3OD, or D2O. Chemical shifts are reported as values in parts per million (ppm) and the reference resonance peaks set at 0 ppm [TMS(CDCl3)], 2.50 ppm [(CD2H)2SO], 3.31 ppm (CD2HOD), and 4.80 ppm (HOD) respectively for 1H NMR spectra, 77.23 ppm (CDCl3), 39.52 ppm (DMSO-d6), and 49.00 ppm (CD3OD) for 13C NMR spectra, and 0 ppm (CF3CO2H) for 19F NMR spectra. An Orion research model 701H pH meter with a general combination electrode was used for pH measurements. Mass spectra were performed on a Micromass Quattro II triple quadruple HPLC/MS mass spectrometer with an electrospray ionization (ESI) source or atmospheric pressure chemical ionization (APCI) source. High-resolution mass spectra were carried out using a VG70-250SE mass spectrometer. Chemical ionization (CI) or electron impact (EI) was used as the ion source. Elemental analyses were performed by Atlantic Microlab Inc., Norcross, GA. Thin-layer chromatography was carried out on E. Merck pre-coated silica gel 60 F254 plates with visualization accomplished with phosphomolybdic acid or ninhydrin spray reagent or with a UV–visible lamp. Column chromatography was performed with E. Merck silica gel 60 (230–400 mesh) or Sorbent Technologies 250 mesh silica gel. Tetrahydrofuran (THF) and ethyl ether were redistilled under nitrogen using sodium and benzophenone as the indicator. Dichloromethane was redistilled from CaH2 under nitrogen. Other dry solvents were directly purchased from the companies.
N-Boc-2-bromoethylamine, N1, N1-dimethylethane-1,2-diamine, (S)-(−)-1-Boc-3-aminopyrrolidine, (R)-(+)-1-Boc-3-aminopyrrolidine, and (S)-(+)-1-benzyl-3-aminopyrrolidine, (R)-(−)-1-benzyl-3-aminopyrrolidine were purchased from Aldrich. All compounds were characterized by TLC, 1H- and 13C NMR, and MS. All final products were also characterized by HRMS. The TLC, HPLC, NMR, and analytical data confirmed that the purity of all of the products was ≥ 95%. The purity of the most potent compounds was further confirmed by elemental analyses.
(±)-tert-Butyl {6-[trans-(4′-hydroxypyrrolidin-3′-yl)methyl]pyridin-2-yl}carbamate (38)
A suspension of 37 (0.766 g, 0.002 mol), which was prepared in the previous study,22 and 10% Pd/C (0.7 g) in MeOH (30 mL) was stirred at 45 °C under one hydrogen atmosphere overnight. The catalyst was removed by filtration and washed with MeOH (30 mL). The filtrate was concentrated by high-vacuum rotary evaporation to give the desired product in a quantitative yield (1.482 g). 1H NMR (CDCl3, 500 MHz): δ 9.206 (brs, 1H), 8.519 (brs, 1H), 7.798 (d, 1H, J=8Hz), 7.577 (t, 1H, J=7.5Hz), 6.8235 (d, 1H, J=7.5Hz), 4.242 (m, 1H), 3.542 (m, 1H), 3.399-3.302 (m, 2H), 3.102-3.090 (m, 1H), 2.868-2.832 (m, 1H), 2.713-2.641 (m, 2H), 1.985 (s, 6H), 1.495 (s, 9H). 13C NMR (CDCl3, 125.7 MHz): δ 177.392 (2C), 156.724 (1C), 152.699 (1C), 152.049 (1C), 139.141 (1C), 118.030 (1C), 110.605 (1C), 80.946 (1C), 74.146 (1C), 51.226 (1C), 48.148 (1C), 45.937 (1C), 38.160 (1C), 28.263 (3C), 22.410 (2C). MS (ESI, CH3OH): [C15H23N3O3] m/z 294.2([M+H]+).
(±)-tert-Butyl {6-[trans-1′-(2″-tert-butoxycarbonylamino-ethyl)-4′-hydroxy-pyrrolidin-3′-ylmethyl]-pyridi n-2-yl}carbamate (39)
A mixture of 38 (0.741 g, 0.001 mol), N-Boc-2-bromoethylamine (0.268 g, 0.0012 mol), anhydrous K2CO3 (0.552 g, 0.004 mol) in 10 mL anhydrous DMF was stirred at room temperature overnight. Solids were removed by filtration. The filtrate was evaporated under reduced pressure. The residue was partitioned between EtOAc and water. The organic layer was washed with brine, dried over anhydrous Na2SO4, and evaporated in vacuo. The obtained residue was purified by column chromatography (silica gel, CH2Cl2: MeOH = 9: 1) to afford a colorless oil (0.248 g, 57%). 1H NMR (CDCl3, 400 MHz): δ 7.758 (d, 1H, J=8Hz), 7.583 (t, 1H, J=8Hz), 7.357 (brs, 1H), 6.8295 (d, 1H, J=7.6Hz), 5.144 (brs, 1H), 4.099 (m, 1H), 3.300-3.100 (m, 2H),3.080-2.980 (m, 1H), 2.900-2.782 (m, 3H), 2.721-2.706 (m, 1H), 2.601-2.541 (m,3H), 2.189-2.146 (m, 1H), 1.510 (s, 9H), 1.442 (s, 9H). 13C NMR (CDCl3, 100.6 MHz): δ 158.514 (1C), 156.303 (1C), 152.251 (1C), 151.303 (1C), 138.891 (1C), 118.183 (1C), 110.024 (1C), 81.279 (2C), 76.800 (1C), 61.769 (1C), 59.176 (1C), 55.468 (1C), 51.033 (1C), 40.591 (1C), 39.052 (1C), 28.740 (3C), 28.558 (3C). MS (ESI, CH3CN): [C22H36N4O5] m/z 437.4 ([M+H]+).
(±)-tert-Butyl {6-{[1′-(2″-tert-butoxycarbonylamino-ethyl)-4′-oxo-pyrrolidin-3′-yl]methyl}-pyridin-2-yl}ca rbamate (40)
To a solution of dimethyl sulfoxide (DMSO, 0.313 g, 0.284 mL, 0.004 mol) in 30 mL of dry CH2Cl2 was added oxalyl chloride (0.381 g, 0.285 mL, 0.003 mol) dropwise. The mixture was stirred at −78 °C for 10 min. After this time a solution of 39 (0.873g, 0.002 mol) in 10 mL of anhydrous CH2Cl2 was added dropwise at a rate to keep the reaction temperature below −60 °C. Upon complete addition, the mixture was allowed to stir at −78 °C for 2 h. Anhydrous triethylamine (0.607 g, 0.836 mL, 0.006 mol) was added dropwise to the mixture; then the reaction mixture was allowed to warm to room temperature. The resulting solution was partitioned between 1 M NaOH (40 mL) and CH2Cl2, and the product was extracted with CH2Cl2 (30 mL × 2). All organic layers were combined, washed with brine, dried over Na2SO4, and concentrated in vacuo to yield the crude product, which was purified using silica gel column chromatography (CH2Cl2: EtOAc = 3: 2) to afford a pale-yellow oil (0.738 g, 85%). 1H NMR (CDCl3, 400 MHz): δ 7.763 (d, 1H, J=8.8Hz), 7.560 (t, 1H, J=8Hz), 7.279 (brs, 1H), 6.790 (d, 1H, J=7.2Hz), 4.997 (brs, 1H), 3.399-3.133 (m, 5H), 2.995-2.924 (m, 1H), 2.787-2.584 (m, 4H), 2.438-2.392 (m, 1H), 1.522 (s, 9H), 1.442 (s, 9H). 13C NMR (CDCl3, 100.6 MHz): δ 214.670 (1C), 157.347 (1C), 156.009 (1C), 152.388 (1C), 151.514 (1C), 138.739 (1C), 117.930 (1C), 109.941 (1C), 81.133 (1C), 79.450 (1C), 61.643 (1C), 57.185 (1C), 55.665 (1C), 48.550 (1C), 38.432 (1C), 35.921 (1C), 28.632 (3C), 28.496 (3C).
(±)-tert-Butyl {6-{[cis-1′-(2″-tert-butoxycarbonylaminoethyl)-4′-(3″-phenylpropylamino)- -pyrrolidin-3′-yl]methyl}pyridin-2-yl}-carbamate (41a) and (±)-tert-Butyl {6-{[trans-1′-(2″-tert-butoxycarbonylaminoethyl)-4′-(3″-phenylpropylamino)-pyrrolidin-3′-yl]methyl}pyridin-2-yl}-carbamate (42a)
To a solution of 40 (0.434 g, 0.001 mol), a substituted amine such as 3-phenyl-1-propylamine (0.203 g, 0.0015 mol), acetic acid (0.090 g, 0.086 mL, 0.0015 mol), and 3 Å molecular sieves (1 g) in dry MeOH (20 mL) was added NaBH3CN (0.126 g, 0.002 mol). The reaction was stirred at room temperature under a N2 atmosphere for 36 h. The reaction mixture was filtered, and the filtrate was concentrated in vacuo. The residue was diluted with 1M NaOH (30 mL) and extracted with CH2Cl2 (30 mL × 2). The organic layers were combined, washed with brine, dried over MgSO4 and concentrated in vacuo to give the crude product, which was purified by silica gel column chromatography (hexanes: CH2Cl2: EtOAc: Et3N = 5: 3: 2: 0.5) to afford a pale-yellow oil (0.360 g, 65%). The cis isomer (41a) and the trans isomer (41b) can be separated with the above eluent. The ratio of cis and trans isomers was 45: 55.
41a
(Rf = 0.25, pale-yellow oil, 0.162 g): 1H NMR (CDCl3, 500 MHz): δ 7.730 (d, 1H, J=8Hz), 7.542 (t, 1H, J=8Hz), 7.289-7.165 (m, 6H), 6.8025 (d, 1H, J=7.5Hz), 5.074 (brs, 1H), 3.235-3.171 (m, 3H), 2.891-2.861 (m, 1H), 2.645-2.382 (m, 12H), 1.820-1.781 (m, 2H), 1.512 (s, 9H), 1.446 (s, 9H). 13C NMR (CDCl3, 125.7 MHz): δ 159.881 (1C), 156.260 (1C), 152.534 (1C), 151.440 (1C), 142.294 (1C), 138.619 (1C), 128.577 (2C), 128.542 (2C), 125.985 (1C), 118.151 (1C), 109.536 (1C), 81.071 (1C), 79.269 (1C), 60.096 (1C), 59.327 (1C), 58.007 (1C), 55.427 (1C), 48.379 (1C), 41.586 (1C), 39.152 (1C), 36.518 (1C), 33.903 (1C), 32.104 (1C), 28.653 (3C), 28.483 (3C). MS (ESI, CH3OH): [C31H47N5O4] m/z 554.4 ([M+H]+)
42a
(Rf = 0.2, pale-yellow oil, 0.198 g): 1H NMR (CDCl3, 500 MHz): δ 7.740 (d, 1H, J=8Hz), 7.546 (t, 1H, J=8Hz), 7.284-7.138 (m, 6H), 6.7905 (d, 1H, J=7.5Hz), 5.040 (brs, 1H), 3.192 (m, 2H), 2.940-2.928 (m, 1H), 2.899-2.756 (m, 2H), 2.724-2.681 (m, 2H), 2.648-2.449 (m, 7H), 2.332-2.322 (m, 1H), 2.216-2.187 (m, 1H), 1.734-1.705 (m, 2H), 1.509 (s, 9H), 1.446 (s, 9H). 13C NMR (CDCl3, 125.7 MHz): δ 159.142 (1C), 156.236 (1C), 152.519 (1C), 151.517 (1C), 142.213 (1C), 138.670 (1C), 128.569 (2C), 128.526 (2C), 125.977 (1C), 118.132 (1C), 109.710 (1C), 81.099 (1C), 79.292 (1C), 63.0450 (1C), 60.905 (1C), 59.129 (1C), 55.241 (1C), 47.934 (1C), 45.373 (1C), 42.410 (1C), 38.978 (1C), 33.791 (1C), 31.907 (1C), 28.657 (3C), 28.468 (3C). MS (ESI, CH3OH): [C31H47N5O4] m/z 554.4 ([M+H]+).
(±)-6-{{trans-1′-(2″-aminoethyl)-4′-[(3″-phenylpropyl)amino]pyrrolidin-3′-yl}methyl}pyridi n-2-amine tetrahydrochloride (8)
A solution of 4M HCl in 1,4-dioxane (4 mL) was added to 42a (0.111 g, 0.2 mmol) at 0 °C under argon. The ice-water bath was removed after 3 h, and the reaction mixture was stirred at room temperature 48 h. After the completion of the reaction, liquids were evaporated under reduced pressure, and the residue was partitioned between water (10 mL) and ethyl acetate (10 mL). The aqueous layer was washed with ethyl acetate (5 mL × 2). After evaporation of water by high-vacuum rotary evaporation, the residue was dried with a lyophilizer to afford a hygroscopic white solid (0.100 g, quantitative yield). 1H NMR (D2O, 500 MHz): δ 7.795 (t, 1H, J=8.5Hz), 7.385-7.258 (m, 5H), 6.883 (d, 1H, J=9Hz), 6.764 (d, 1H, J=7Hz), 3.902-3.894 (m, 2H), 3.702-3.648 (m, 2H), 3.482-3.457 (m, 2H), 3.389-3.344 (m, 2H), 3.189-3.126 (m, 3H), 3.000-2.888 (m, 3H), 2.751-2.658 (m, 2H), 2.048-1.972 (m, 2H). 13C NMR (D2O, 125.7 MHz): δ 154.686 (1C), 144.740 (2C), 140.369 (1C), 128.976 (2C), 128.577 (2C), 126.717 (1C), 112.801 (1C), 112.511 (1C), 59.558 (1C), 57.469 (1C), 55.369 (1C), 51.384 (1C), 46.363 (1C), 39.875 (1C), 35.430 (1C), 34.343 (1C), 27.399 (1C), 27.016 (1C). MS (ESI, CH3CN-H2O): [C21H31N5] m/z 354.3 ([M+H]+). HRMS (CI+, CH3OH) Calc.: 354.2652, Found: 354.2648. Anal. (C21H35Cl4N5·1.75H2O), Calcd: C, 47.51; H, 7.31; N, 13.19; Found: C, 47.75; H, 7.42; N, 12.87.
(±)-trans-4-[(6′-aminopyridin-2′-yl)methyl]pyrrolidin-3-ol dihydrochloride (12)
The procedure to prepare 12 is the same as that to prepare 8 except using 44 (0.079 g, 0.2 mmol) instead of 42a, affording a hygroscopic white solid (0.053 g, quantitative yield). 1H NMR (D2O, 500 MHz): δ 7.840 (t, 1H, J=8.5Hz), 6.894 (d, 1H, J=9Hz), 6.7955 (d, 1H, J=7.5Hz), 4.375-4.350 (m, 1H), 3.674-3.634 (m, 1H), 3.600-3.563 (m, 1H), 3.302-3.270 (m, 1H), 3.166-3.128 (m, 1H), 2.982-2.936 (m, 1H), 2.884-2.837 (m, 1H), 2.725-2.672 (m, 1H). 13C NMR (D2O, 125.7 MHz): δ 154.675 (1C), 146.377 (1C), 144.802 (1C), 112.612 (1C), 111.846 (1C), 73.009 (1C), 50.873 (1C), 47.991 (1C), 44.359 (1C), 33.430 (1C). MS (ESI, CH3OH) : [C10H15N3O] m/z 194.3 ([M+H]+). HRMS (CI+, CH3OH) Calc. : 194.1288, Found : 194.1285. Anal. (C10H17Cl2N3O·0.1H2O), Calcd : C, 44.82; H, 6.47; N, 15.68; Found : C, 44.78; H, 6.45; N, 15.42.
(±)-trans-tert-Butyl 3-{{6′-[(tert-butoxycarbonyl)amino]-4′-methylpyridin-2′-yl}methyl}-4-{{2′-[(tert-butoxycarbonyl)amino]ethyl}amino}pyrrolidine-1-carboxylate (50a)
The procedure to prepare 50a is the same as that to prepare 42a except using 49 (0.203 g, 0.5 mmol)23 and N-Boc-1,2-diaminoethane (0.088 g, 0.55 mmol) instead of 40 (0.434 g, 0.001 mol) and 3-phenyl-1-propylamine (0.203 g, 0.0015 mol). The desired product was purified by column chromatography (silica gel, hexanes : EtOAc : Et3N = 6 : 4 : 0.5, the isomer with lower Rf value, Rf = 0.1) to afford a pale-yellow oil (0.083 g, 55%, diastereomer ratio : cis : trans = 45 : 55). 1H NMR (CDCl3, 500 MHz) : δ 7.624-7.609 (m, 1H), 7.554 (m, 1H), 6.611 (s, 1H), 5.215-5.172 (m, 1H), 3.723-3.702 (m, 0.5H), 3.689-3.628 (m, 0.5H), 3.561-3.526 (m, 0.5H), 3.477-3.444 (m, 0.5H), 3.181-2.983 (m, 4H), 2.936-2.906 (m, 1H), 2.892-2.801 (m, 1H), 2.707-2.699 (m, 2H), 2.584-2.562 (m, 1H), 2.410-2.290 (m, 4H), 1.519 (s, 9H), 1.444 (s, 18H). 13C NMR (CDCl3, 125.7 MHz) : δ (157.759+157.685) (1C), 156.231 (1C), 154.598 (1C), 152.594 (1C), 151.650 (1C), 149.983 (1C), 119.237 (1C), 110.436 (1C), 80.782 (1C), 79.308 (2C), (61.786+61.082) (1C), (51.940+51.417) (1C), (49.820+49.572) (1C), 47.363 (1C), (44.051+43.537) (1C), 40.569 (1C), (39.552+39.413) (1C), 28.568 (3C), 28.487 (3C), 28.344 (3C), 21.353 (1C). MS (ESI, CH3OH) : [C28H47N5O6] m/z 550.4 ([M+H]+).
(±)-trans-tert-Butyl 3-{{2′-[(tert-butoxycarbonyl)(3″-chlorobenzyl)amino]ethyl}amino}-4-{{6′-[(tert-butoxycarbonyl)amino]-4′-methylpyridin-2′-yl}methyl}pyrrolidine-1-carboxylate (50c)
The procedure to prepare 50c is the same as that to prepare 42a except using 49 (0.203 g, 0.5 mmol)23 and tert-butyl (2-aminoethyl)(3-chlorobenzyl)carbamate (0.156 g, 0.55 mmol)23 instead of 40 (0.434 g, 0.001 mol) and 3-phenyl-1-propylamine (0.203 g, 0.0015 mol). The desired product was purified by column chromatography (silica gel, hexanes : EtOAc : Et3N = 9.5 : 0.5 : 0.5, the isomer with lower Rf value, Rf = 0.1) to afford a pale-yellow oil (0.117 g, 63%, diastereomer ratio : cis : trans = 45 : 55). 1H NMR (CDCl3, 500 MHz) : δ 7.611-7.592 (m, 1H), 7.249-7.233 (m, 4H), 7.105 (s, 1H), 6.603 (s, 1H), 4.413 (s, 2H), 3.690-3.656 (m, 0.5H), 3.613-3.608 (m, 0.5H), 3.555-3.518 (m, 0.5H), 3.465-3.429 (m, 0.5H), 3.394-3.216 (m, 2H), 3.128-3.091 (m, 1H), 3.052-3.031 (m, 1H), 2.956-2.943 (m, 1H), 2.821-2.761 (m, 3H), 2.545-2.500 (m, 1H), 2.410-2.243 (m, 4H), 1.514-1.449 (m, 27H). 13C NMR (CDCl3, 125.7 MHz) : δ 157.876 (1C), 155.873 (1C), (154.755+154.664) (1C), 152.539 (1C), 151.513 (1C), 150.008 (1C), 140.828 (1C), 134.519 (1C), 129.936 (1C), 127.489 (2C), (125.825+125.242) (1C), (119.311+119.238) (1C), (110.368+110.295) (1C), 80.867 (1C), 80.320 (1C), 79.325 (1C), (62.179+61.347) (1C), (51.925+51.372) (1C), 50.826 (1C), (49.927+49.733) (1C), (47.456+47.213) (1C), 46.339 (1C), (44.165+43.631) (1C), (39.727+39.636) (1C), 28.641 (3C), 28.538 (3C), 28.392 (3C), 21.410 (1C). MS (ESI, CH3OH) : [C35H52ClN5O6] m/z 674.3 ([M+H]+).
(±)-trans-tert-Butyl 3-{{2′-[(tert-butoxycarbonyl)(phenethyl)amino]ethyl}amino}-4-{{6′-[(tert-butoxycarbonyl)amino]-4′-methylpyridin-2′-yl}methyl}pyrrolidine-1-carboxylate (50l)
The procedure to prepare 50l is the same as that to prepare 42a except using 49 (0.203 g, 0.5 mmol)23 and tert-butyl (2-aminoethyl)(phenethyl)carbamate (0.145 g, 0.55 mmol)23 instead of 40 (0.434 g, 0.001 mol) and 3-phenyl-1-propylamine (0.203 g, 0.0015 mol). The desired product was purified by column chromatography (silica gel, hexanes : EtOAc : Et3N = 9 : 1 : 0.5, the isomer with lower Rf value, Rf = 0.1) to afford a pale-yellow oil (0.101 g, 56%, diastereomer ratio : cis : trans = 45 : 55). 1H NMR (CDCl3, 500 MHz) : δ 7.602-7.586 (m, 1H), 7.297-7.170 (s, 6H), 6.597 (s, 1H), 3.699-3.665 (m, 0.5H), 3.626-3.604 (m, 0.5H), 3.556-3.520 (m, 0.5H), 3.467-3.392 (m, 2.5H), 3.227-2.967 (m, 5H), 2.807 (m, 3H), 2.681 (m, 2H), 2.544-2.500 (m, 1H), 2.407-2.272 (m, 4H), 1.511-1.444 (m, 27H). 13C NMR (CDCl3, 125.7 MHz) : δ 157.920 (1C), 155.783 (1C), (154.751+154.666) (1C), 152.541 (1C), 151.527 (1C), 149.991 (1C), 139.281 (1C), 128.990 (2C), 128.619 (2C), 126.427 (1C), (119.305+119.227) (1C), (110.368+110.289) (1C), 80.891 (1C), 79.677 (1C), 79.677 (1C), (62.239+61.371) (1C), (51.960+51.384) (1C), (50.060+49.933+49.750) (2C), 47.674 (1C), 46.478 (1C), (44.171+43.655) (1C), 39.660 (1C), 35.361 (1C), 28.634 (3C), 28.561 (3C), 28.385 (3C), 21.396 (1C). MS (ESI, CH3OH) : [C36H55N5O6] m/z 654.4 ([M+H]+).
(±)-trans-tert-Butyl 3-{{2′-[(tert-butoxycarbonyl)(3″-fluorophenethyl)amino]ethyl}amino}-4-{{6′-[(tert-butoxycarbonyl)amino]-4′-methylpyridin-2′-yl}methyl}pyrrolidine-1-carboxylate (50n)
The procedure to prepare 50n is the same as that to prepare 42a except using 49 (0.203 g, 0.5 mmol)23 and tert-butyl (2-aminoethyl)(3-fluorophenethyl)carbamate (0.l55 g, 0.55 mmol)23 instead of 40 (0.434 g, 0.001 mol) and 3-phenyl-1-propylamine (0.203 g, 0.0015 mol). The desired product was purified by column chromatography (silica gel, hexanes : EtOAc : Et3N = 9.5 : 0.5 : 0.5, the isomer with lower Rf value, Rf = 0.1) to afford a pale-yellow oil (0.120 g, 65%, diastereomer ratio : cis : trans = 45 : 55). 1H NMR (CDCl3, 500 MHz) : δ 7.611-7.595 (m, 1H), 7.355 (brs, 1H), 7.262-7.220 (m, 1H), 6.917-6.885 (m, 3H), 6.604 (s, 1H), 3.704-3.670 (m, 0.5H), 3.636-3.614 (m, 0.5H), 3.559-3.523 (m, 0.5H), 3.476-3.397 (m, 2.5H), 3.233-2.980 (m, 5H), 2.813 (m, 3H), 2.695 (m, 2H), 2.552-2.508 (m, 1H), 2.406-2.367 (m, 0.5H), 2.296-2.276 (m, 3.5H), 1.512-1.446 (m, 27H). 13C NMR (CDCl3, 125.7 MHz) : δ (163.889+161.934) (1C), 157.811 (1C), 155.564 (1C), (154.672+154.581) (1C), 152.486 (1C), 151.484 (1C), 149.924 (1C), 141.800 (1C), 129.955 (1C), (124.630+124.606) (1C), (119.227+119.148) (1C), (115.851+115.681) (1C), (113.343+113.167) (1C), (110.326+110.247) (1C), 80.745 (1C), 79.713 (1C), 79.234 (1C), (62.179+61.323) (1C), (51.869+51.293) (1C), (49.878+49.678) (2C), (48.293+47.589) (1C), 46.387 (1C), (44.098+43.582) (1C), (39.678+39.587) (1C), (35.039+34.377) (1C), 28.555 (3C), 28.458 (3C), 28.306 (3C), 21.324 (1C). 19F NMR (CDCl3, 376.5 MHZ) : δ −113.900. MS (ESI, CH3OH) : [C36H54FN5O6] m/z 672.5 ([M+H]+).
N1-{(±)-trans-4-[(6-amino-4-methylpyridin-2-yl)methyl]pyrrolidin-3-yl}ethane-1,2-diamine tetrahydrochloride (21)
The procedure to prepare 21 is the same as that to prepare 8 except using 50a (0.110 g, 0.2 mmol) instead of 42a, affording a hygroscopic white solid (0.079 g, quantitative yield). 1H NMR (D2O, 500 MHz) : δ 6.748 (s, 1H), 6.714 (s, 1H), 4.098-4.022 (m, 2H), 3.765-3.682 (m, 2H), 3.573-3.421 (m, 4H), 3.376-3.288 (m, 2H), 3.150-3.109 (m, 1H), 2.938-2.888 (m, 1H), 2.3549 (s, 3H). 13C NMR (D2O, 125.7 MHz) : δ 158.396 (1C), 154.306 (1C), 143.625 (1C), 114.988 (1C), 111.328 (1C), 60.713 (1C), 48.596 (1C), 46.507 (1C), 43.965 (1C), 40.525 (1C), 35.728 (1C), 33.453 (1C), 21.340 (1C). MS (ESI, CH3OH-H2O) : [C13H23N5] m/z 250.2 ([M+H]+). HRMS (CI+, CH3OH) Calc. : 250.2026, Found : 250.2026. Anal. (C13H27Cl4N5·1.25 H2O), Calcd : C, 37.38; H, 7.12; N, 16.77; Found : C, 37.48; H, 7.05; N, 16.56.
N1-{(±)-trans-4-[(6′-amino-4′-methylpyridin-2′-yl)methyl]pyrrolidin-3-yl}-N2-(3-chlorobenzyl)ethane-1,2-diamine tetrahydrochloride (23)
The procedure to prepare 23 is the same as that to prepare 8 except using 50c (0.135 g, 0.2 mmol) instead of 42a, affording a hygroscopic white solid (0.103 g, quantitative yield). 1H NMR (D2O, 500 MHz) : δ 7.523-7.418 (m, 4H), 6.699 (s, 1H), 6.680 (s, 1H), 4.316 (s, 2H), 4.038-4.002 (m, 2H), 3.731-3.654 (m, 2H), 3.538 (m, 4H), 3.342-3.243 (m, 2H), 3.097-3.090 (m, 1H), 2.902-2.853 (m, 1H), 2.316 (s, 3H). 13C NMR (D2O, 125.7 MHz) : δ 158.321 (1C), 154.211 (1C), 143.562 (1C), 134.461 (1C), 132.014 (1C), 130.964 (1C), 130.114 (1C), 129.883 (1C), 128.396 (1C), 114.960 (1C), 111.268 (1C), 60.742 (1C), 51.119 (1C), 48.545 (1C), 46.493 (1C), 42.881 (2C), 40.519 (1C), 33.403 (1C), 21.315 (1C). MS (ESI, CH3OH) : [C20H28ClN5] m/z 374.4 ([M+H]+). HRMS (CI+, CH3OH) Calc. : 374.2106, Found : 374.2103. Anal. (C20H32Cl5N5·1.25 H2O), Calcd : C, 44.30; H, 6.41; N, 12.91; Found : C, 44.93; H, 6.58; N, 12.51.
N1-{(±)-trans-4-[(6′-amino-4′-methylpyridin-2′-yl)methyl]pyrrolidin-3-yl}-N2-phenethylethane-1,2-diamine tetrahydrochloride (32)
The procedure to prepare 32 is the same as that to prepare 8 except using 50l (0.131 g, 0.2 mmol) instead of 42a, affording a hygroscopic white solid (0.094 g, quantitative yield). 1H NMR(D2O, 500 MHz) : δ 7.390-7.302 (m, 5H), 6.677 (s, 2H), 4.083-3.997 (m, 2H), 3.760-3.725 (m, 1H), 3.690-3.650 (m, 1H), 3.580-3.509 (m, 4H), 3.412-3.382 (m, 2H), 3.344-3.306 (m, 1H), 3.280-3.239 (m, 1H), 3.108-3.087 (m, 1H), 3.044-3.015 (m, 2H), 2.900-2.850 (m, 1H), 2.307 (s, 3H). 13C NMR(D2O, 125.7 MHz) : δ 158.347 (1C), 154.206 (1C), 143.514 (1C), 136.089 (1C), 129.301 (2C), 129.125 (2C), 127.692 (1C), 115.099 (1C), 111.353 (1C), 60.790 (1C), 48.641 (1C), 48.641 (1C), 46.480 (1C), 43.420 (1C), 42.964 (1C), 40.548 (1C), 33.468 (1C), 31.938 (1C) (21.465+21.423) (1C). MS (ESI, CH3OH) : [C21H31N5] m/z 354.4 ([M+H]+). HRMS (CI+, CH3OH) Calc. : 354.2652, Found : 354.2649. Anal. (C21H35Cl4N5·2.2H2O), Calcd : C, 46.80; H, 7.37; N, 12.99; Found : C, 47.01; H, 7.47; N, 12.67.
N1-{(±)-trans-4-[(6′-amino-4′-methylpyridin-2′-yl)methyl]pyrrolidin-3-yl}-N2-(3-fluorophenethyl)ethane-1,2-diamine tetrahydrochloride (34)
The procedure to prepare 34 is the same as that to prepare 8 except using 50n (0.134 g, 0.2 mmol) instead of 42a, affording a hygroscopic white solid (0.103 g, quantitative yield). 1H NMR (D2O, 500 MHz) : δ 7.377-7.333 (m, 1H), 7.117-7.008 (m, 3H), 6.678 (s, 2H), 4.094-4.003 (m, 2H), 3.766-3.730 (m, 1H), 3.698-3.657 (m, 1H), 3.597-3.520 (m, 4H), 3.425-3.396 (m, 2H), 3.351-3.312 (m, 1H), 3.290-3.249(m, 1H), 3.137-3.095(m, 1H), 3.059-3.030 (m, 2H), 2.907-2.856 (m, 1H), 2.303 (s, 3H). 13C NMR (D2O, 125.7 MHz) : δ (163.829+161.886) (1C), 158.328 (1C), 154.188 (1C), 143.472 (1C), (138.517+138.456) (1C), (130.952+130.885) (1C), (124.972+124.947) (1C), (115.895+115.725) (1C), 115.057 (1C), (114.486+114.316) (1C), 111.317 (1C), 60.760 (1C), 49.254 (1C), 48.611 (1C), 46.419 (1C), 43.407 (1C), 42.934 (1C), 40.487 (1C), 33.420 (1C), 31.617 (1C), 21.392 (1C). 19F NMR (D2O, 376.5 MHZ) : δ −113.580. MS (ESI, CH3OH) : [C21H30FN5] m/z 372.4 ([M+H]+). HRMS (CI+, CH3OH) Calc. : 372.2558, Found : 372.2549. Anal. (C21H34Cl4FN5·1.25H2O), Calcd : C, 46.72; H, 6.81; N, 12.97; Found : C, 46.99; H, 6.85; N, 12.67.
(±)-cis-tert-Butyl 3-acetoxy-4-{{6′-[benzyl(tert-butoxycarbonyl)amino]-4′-methylpyridin-2′-yl}methyl}pyrrolidine-1-carboxylate (52)
To an ice-cooled solution of triphenylphospine (Ph3P, 0.341 g, 0.0013 mol) in dry THF (5 mL) was added 51 (0.497 g, 0.001 mol), which was prepared in a previous study,23 in dry THF (15 mL) through a cannula under N2 atmosphere. Diisopropyl azodicarboxylate (DIAD, 0.263 g, 0.259 mL, 0.0013 mol) was then added dropwise, and the solution was stirred 20 min at 0 °C. Acetic acid (HOAc, 0.078 g, 0.074 mL, 0.0013 mol) was added at 0 °C, and the solution was stirred at room temperature for 48 h. The solution was concentrated, and the residue was purified directly by column chromatography (silica gel, hexanes : EtOAc = 8 : 2) to yield colorless oil (0.469 g, 87%). 1H NMR (CDCl3, 500MHz) : δ 7.446-7.426 (m, 1H), 7.293-7.179 (m, 5H), 6.638-6.628 (m, 1H), 5.163-5.091 (m, 3H), 3.506-3.361 (m, 3H), 3.127-3.086 (m, 1H), 2.876-2.835 (m, 1H), 2.700-2.614 (m, 2H), 2.334-2.239 (m, 3H), 2.051-2.039 (m, 3H), 1.448-1.445 (m, 9H), 1.413 (m, 9H). 13C NMR (CDCl3, 125.7 MHz) : δ (170.591+170.367) (1C), 156.657 (1C), (154.429+154.338) (1C), (154.271+154.229) (1C), 153.949 (1C), (148.679+148.625) (1C), (139.797+139.760) (1C), 128.048 (2C), (126.925+126.871) (2C), (126.543+126.440) (1C), (119.694+119.633) (1C), 117.229 (1C), (81.201+81.158) (1C), (79.440+79.422) (1C), (74.759+74.716+73.866+73.830) (1C), (52.792+52.343) (1C), 49.951 (1C), (49.289+48.846) (1C), (41.718+41.129) (1C), (34.730+34.669) (1C), (28.458+28.433) (3C), 28.124 (3C), (21.099+20.965) (1C). MS (ESI, CH3OH) : [C30H41N3O6] m/z 540.4 ([M+H]+); m/z 562.4 ([M+Na]+).
(±)-cis-tert-butyl 3-{{6′-[benzyl(tert-butoxycarbonyl)amino]-4′-methylpyridin-2′-yl}methyl}-4-hydroxypyrrolidine-1-carboxylate (53)
To a solution of 52 (1.079 g, 0.002 mol) in CH3OH (40 mL) was added at room temperature a solution of Na2CO3 (0.424 g, 0.004 mol) in H2O (10 mL). The solution was stirred at room temperature for 14 h. The solvent was evaporated in vacuo. The residue was dissolved in a mixture of EtOAc (10 mL) and H2O (10 mL). The aqueous layer was extracted with EtOAc (10 mL × 3). The combined organic layers were dried over Na2SO4, and concentrated in vacuo. The residue was purified by column chromatography (silica gel, hexanes : EtOAc = 6 : 4) to afford a colorless oil (0.995 g, quantitative yield). 1H NMR (CDCl3, 500MHz) : δ 7.369-7.348 (m, 1H), 7.272-7.185 (m, 5H), 6.748-6.730 (m, 1H), 5.087 (m, 2H), (4.078+3.863) (brs, 1H), 3.960-3.938 (m, 1H), 3.616-3.340 (m, 3H), 3.170-3.116 (m, 1H), 2.881-2.834 (m, 1H), 2.789-2.712 (m, 1H), 2.318 (m, 4H), 1.443 (s, 9H), 1.413 (s, 9H). 13C NMR (CDCl3, 125.7 MHz) : δ 157.592 (1C), (154.417+154.362) (1C), 154.241 (1C, 15), 153.889 (1C), (149.457+149.341) (1C), (139.020+138.983) (1C), 128.194 (2C), 126.731 (1C), 126.695 (2C), (120.404+120.319) (1C), (118.334+118.200) (1C), 81.340 (1C), 78.991 (1C), (70.988+70.193) (1C), (53.830+53.557) (1C), (50.309+50.266) (1C), (49.253+48.870) (1C), (44.735+44.177) (1C), (35.088+35.015) (1C), 28.470 (3C), 28.093 (3C), 21.075 (1C). MS (ESI, CH3OH) : [C28H39N3O5] m/z 520.3 ([M+Na]+).
(±)-trans-tert-Butyl 3-azido-4-{{6′-[benzyl(tert-butoxycarbonyl)amino]-4′-methylpyridin-2′-yl}methyl}pyrrolidine-1-carboxylate (54)
To Ph3P (0.328 g, 0.00125 mol) in a dry THF solution (5 mL) was added 53 (0.497 g, 0.001 mol) in dry THF (10 mL) at 0 °C under a N2 atmosphere via cannula. DIAD (0.262 g, 0.260 mL, 0.0013 mol) was added dropwise, and the solution was stirred at 0 °C for 20 min. Diphenylphosphoryl azide (DPPA, 0.358 g, 0.281 mL, 0.0013 mol) was added dropwise at 0 °C, and the reaction mixture was stirred for 22 h at room temperature. The solvent was concentrated in vacuo. The crude residue was directly purified by column chromatography (silica gel, hexanes : EtOAc = 8.5 : 1.5) to afford a colorless oil (0.480 g, 92%). 1H NMR (CDCl3, 500MHz) : δ 7.501-7.479 (m, 1H), 7.261-7.184 (m, 5H), 6.653 (m, 1H), 5.189 (m, 2H), 3.726-3.686 (m, 1H), 3.632-3.597 (m, 0.5H), 3.556-3.520 (m, 0.5H), 3.465-3.391 (m, 1H), 3.292-3.262 (m, 0.5H), 3.206-3.175 (m, 0.5H), 3.126-3.071 (m, 1H), 2.792-2.751 (m, 1H), 2.667-2.615 (m, 1H), 2.566-2.519 (m, 1H), 2.303-2.290 (m, 3H), 1.459-1.432 (m, 9H), 1.418 (s, 9H). 13C NMR (CDCl3, 125.7 MHz) : δ (156.074+155.989) (1C), (154.338+154.162) (2C), 153.968 (1C), 148.825 (1C), 139.675 (1C), 128.127 (2C), (126.895+126.871) (2C), (126.603+126.525) (1C), 119.998 (1C), (117.357+117.320) (1C), (81.280+81.231) (1C), 79.598 (1C), (63.496+62.950) (1C), 49.866 (1C), (49.617+49.441) (1C), (49.234+48.967) (1C), (43.752+42.999) (1C), (38.992+38.913) (1C), 28.439 (3C), 28.142 (3C), 21.148 (1C).
(±)-trans-tert-Butyl 3-amino-4-{[6′-(tert-butoxycarbonylamino)-4′-methylpyridin-2′-yl]methyl}pyrrolidine-1-carboxylate (55)
A solution of 54 (0.522 g, 0.001 mol) in EtOH (20 mL) was treated with 20% wt Pd(OH)2 on carbon (300 mg). The reaction mixture was stirred at 60 °C under a hydrogen atmosphere for 36 h. The catalyst was filtered through Celite. The Celite pad was washed with EtOH (10 mL × 2). The combined filtrate was concentrated in vacuo. The residue was purified by column chromatography (silica gel, CH2Cl2 : CH3OH = 9.5 : 0.5) to afford a pale-green oil (0.28 g, 85%). 1H NMR (CDCl3, 500MHz) : δ 7.625-7.583 (m, 2H), 6.629 (m, 1H), 3.727-3.693 (m, 0.5H), 3.669-3.634 (m, 0.5H), 3.619-3.581 (m, 0.5H), 3.529-3.492 (m, 0.5H), 3.167-3.154 (m, 1H), 3.107-3.068 (m, 0.5H), 3.035-2.986 (m, 1H), 2.966-2.929 (m, 0.5H), 2.889-2.821 (m, 1H), 2.595-2.552 (m, 1H), 2.312-2.290 (m, 3H), 2.262-2.251 (m, 0.5H), 2.184-2.142 (m, 0.5H), 1.510 (s, 9H), 1.441 (s, 9H). 13C NMR (CDCl3, 125.7 MHz) : δ (157.841+157.762) (1C), 154.551 (1C), 152.577 (1C), 151.648 (1C), 150.076 (1C), 119.166 (1C), (110.417+110.368) (1C), (80.849+80.764) (1C), 79.270 (1C), (56.180+55.397) (1C), (53.673+53.248) (1C), (50.224+49.975) (1C), (47.097+46.775) (1C), (39.289+39.083) (1C), 28.567 (3C), 28.336 (3C), 21.378 (1C). MS (ESI, CH3OH) : [C21H34N4O4] m/z 407.1([M+H]+); m/z 812.9([2M+H]+).
(±)-trans-tert-Butyl 3-{{2′-[(tert-butoxycarbonyl)(3″-chlorobenzyl)amino]ethyl}amino}-4-{{6′-[(tert-butoxycarbonyl)amino]-4′-methylpyridin-2′-yl}methyl}pyrrolidine-1-carboxylate (50c), (±)-trans-tert-Butyl 3-{{2′-[(tert-butoxycarbonyl)(phenethyl)amino]ethyl}amino}-4-{{6′-[(tert-butoxycarbonyl)amino]-4′-methylpyridin-2′-yl}methyl}pyrrolidine-1-carboxylate (50l), or (±)-trans-tert-Butyl 3-{{2′-[(tert-butoxycarbonyl)(3″-fluorophenethyl)amino]ethyl}amino}-4-{{6′-[(tert-butoxycarbonyl)amino]-4′-methylpyridin-2′-yl}methyl}pyrrolidine-1-carboxylate (50n)
To a mixture of 55 (0.203 g, 0.5 mmol), NaBH(OAc)3 (0.127 g, 0.6 mmol), and 3 Å molecular sieves (0.5 g) in dry 1,2-dichloroethane (10 mL) was added tert-butyl 3-fluorophenethyl(2-oxoethyl)carbamate (0.141 g, 0.5 mmol)23 in dry 1,2-dichloroethane (5 mL) via cannula under a N2 atmosphere. The reaction mixture was stirred at room temperature under a N2 atmosphere for 16 h and then was filtered through Celite, and the Celite pad was washed with CH2Cl2 (5 mL × 2). To the filtrate was then added 1 M aqueous NaOH (10 mL). The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (10 mL × 2). The combined organic layers were washed with brine (10 mL) and dried over MgSO4. The solvent was evaporated, and the residue was purified by column chromatography (silica gel, hexanes : EtOAc : Et3N = 8 : 2 : 0.25) to afford a colorless oil (50n, 0.306 g, 91%). 50c and 50l were prepared by the same procedure.
(3S,4S)-tert-Butyl 3-{{6′-[benzyl(tert-butoxycarbonyl)amino]-4′-methylpyridin-2′-yl}methyl}-4-{(1S,4R)-4′,7′,7 ′-trimethyl-3′-oxo-2′-oxabicyclo[2.2.1]heptane-1′-carbonyloxy}pyrrolidine-1-carboxylate (57a) and (3R,4R)-tert-Butyl 3-{{6′-[benzyl(tert-butoxycarbonyl)amino]-4′-methylpyridin-2′-yl}methyl}-4-{(1S,4R)-4′,7′,7′-trimethyl-3′-oxo-2′-oxabicyclo[2.2.1]heptane-1′-carbonyloxy}pyrrolidine-1-carboxylate (57b)
To Ph3P (0.328 g, 0.00125 mol) in a dry THF (5 mL) solution was added 51 (0.497 g, 0.001 mol) in dry THF (10 mL) at 0 °C under a N2 atmosphere via cannula. DIAD (0.262 g, 0.260 mL, 0.0013 mol) was added dropwise, and the solution was stirred at 0 °C for 20 min. (1S)-(−)-camphanic acid (0.258 g, 0.0013 mol) was added dropwise at 0 °C, and the reaction mixture was stirred for 14 h at room temperature. The solvent was concentrated in vacuo. The crude residue was purified by column chromatography (silica gel, hexanes : EtOAc = 8 : 2) to afford a white solid (0.644 g, 95%). Compounds 57a and 57b can be further separated by column chromatography (silica gel, hexane : EtOAc = 9.0 : 1.0. Rf : 57a : 0.19; 57b : 0.23).
57a, white solid (0.322 g). 1H NMR (CDCl3, 500MHz) : δ 7.508-7.442 (m, 1H), 7.298-7.185 (m, 5H), 6.633 (m, 1H), 5.233-5.080 (m, 3H), 3.549-3.394 (m, 3H), 3.151-3.121 (m, 1H), 2.861-2.837 (m, 1H), 2.728-2.703 (m, 2H), 2.401-2.380 (m, 1H), 2.285 (m, 3H), 2.028-2.024 (m, 1H), 1.958-1.933 (m, 1H), 1.699-1.680 (m, 1H), 1.448-1.418 (m, 18H), 1.108 (s, 3H), 1.014 (s, 3H), 0.921-0.904 (m, 3H). 13C NMR (CDCl3, 125.7 MHz) : δ (177.926+177.677) (1C), (166.815+166.736) (1C), (156.087+156.056) (1C), 154.253 (1C), (154.180+153.943) (2C), 148.643 (1C), (139.712+139.669) (1C), 128.018 (2C), (126.658+126.597) (2C), (126.464+126.367) (1C), (119.670+119.573) (1C), 117.108 (1C), (90.861+90.800) (1C), (81.189+81.164) (1C), (79.495+79.464) (1C), (76.143+75.330) (1C), 54.674 (1C), (54.116+54.079) (1C), (52.914+52.434) (1C), 49.890 (1C), (49.198+48.821) (1C), (41.366+40.819) (1C), (34.517+34.420) (1C), (30.674+30.601) (1C), 28.925 (1C), 28.373 (3C), 28.057 (3C), 21.063 (1C), (16.819+16.661+16.600) (2C), 9.606 (1C). MS (ESI, CH3OH) : [C38H51N3O8] m/z 678.5 ([M+H]+); m/z 700.5 ([M+Na]+); m/z 1355.1 ([2M+H+); m/z 1377.1 ([2M+Na]+).
57b, white solid (0.322 g). 1H NMR (CDCl3, 500MHz) : δ 7.467-7.446 (m, 1H), 7.272-7.183 (m, 5H), 6.664 (m, 1H), 5.198-5.112 (m, 3H), 3.544-3.382 (m, 3H), 3.154-3.112 (m, 1H), 2.926-2.884 (m, 1H), 2.744-2.680 (m, 2H), 2.443-2.378 (m, 1H), 2.291-2.280 (m, 3H), 1.971-1.884 (m, 2H), 1.696-1.646 (m, 1H), 1.440-1.419 (m, 18H), 1.115 (s, 3H), 1.031-1.012 (m, 3H), 0.933-0.910 (m, 3H). 13C NMR (CDCl3, 125.7 MHz) : δ (178.563+178.157) (1C), (167.191+167.082) (1C), (156.378+156.342) (1C), (154.441+154.405) (1C), (154.332+154.089) (1C), 154.010 (1C), 148.734 (1C), (139.870+139.821) (1C), 128.133 (2C), (126.889+126.822) (2C), (126.597+126.500) (1C), (119.931+119.876) (1C), (117.265+117.180) (1C), (91.146+91.061) (1C), (81.316+81.286) (1C), 79.580 (1C), (76.459+75.572) (1C), (54.935+54.881) (1C), (54.413+54.225) (1C), (52.914+52.495) (1C), 49.999 (1C), (49.277+48.870) (1C), (41.669+41.044) (1C), (34.596+34.523) (1C), 30.437 (1C), 28.834 (1C), 28.476 (3C), 28.215 (3C), 21.201 (1C), 16.749 (1C), 16.642 (1C), 9.782 (1C). MS (ESI, CH3OH) : [C38H51N3O8] m/z 678.4 ([M+H]+); m/z 1354.7 ([2M+H+).
(3′S,4′S)-tert-Butyl 3′-{{6″-[benzyl(tert-butoxycarbonyl)amino]-4″-methylpyridin-2″-yl}methyl}-4′-hydroxypyrrolidine-1′-carboxylate [(3′S,4′S)-58] and (3′R,4′R)-tert-butyl 3′-{{6″-[benzyl(tert-butoxycarbonyl)amino]-4″-methylpyridin-2″-yl}methyl}-4′-hydroxypyrrolidine-1′-carboxylate [(3′R,4′R)-58]
The procedure to prepare (3′S, 4′S)-58 or (3′R, 4′R)-58 is the same as that to prepare 53 except using 57a (1.355 g, 0.002 mol) or 57b (1.355 g, 0.002 mol) instead of 52 (1.079 g, 0.002 mol). The desired products were purified by column chromatography (silica gel, hexanes : EtOAc = 6 : 4).
(3′S,4′S)-58, colorless oil (0.995 g, quantitative yield). 1H NMR (CDCl3, 500MHz) : δ 7.379-7.357 (m, 1H), 7.275-7.168 (m, 5H), 6.734 (m, 1H), 5.116-5.109 (m, 2H), 4.197 (brs, 1H), 3.976-3.968 (m, 1H), 3.559-3.355 (m, 3H), 3.167-3.124 (m, 1H), 2.897-2.850 (m, 1H), 2.775-2.713 (m, 1H), 2.364-2.286 (m, 4H), 1.434 (s, 9H), 1.407 (s, 9H). 13C NMR (CDCl3, 125.7 MHz) : δ 157.477 (1C), (154.265+154.192) (1C), (154.077+154.047) (1C), 153.707 (1C), (149.153+149.044) (1C), (138.959+138.910) (1C), 128.012 (2C), 126.567 (3C), (120.210+120.143) (1C), (117.994+117.860) (1C), 81.098 (1C), 78.766 (1C), (70.849+70.053) (1C), (53.837+53.557) (1C), 50.109 (1C), (49.058+48.670) (1C), (44.407+43.873) (1C), (34.893+34.833) (1C), 28.312 (3C), (27.935+27.911) (3C), 20.905 (1C). MS (ESI, CH3OH) : [C28H39N3O5] m/z 498.4([M+H]+); m/z 520.3 ([M+Na]+). [α]25 = − 33.4° (c = 4, CH3OH).
(3′R,4′R)-58, colorless oil (0.995 g, quantitative yield), the NMR and MS are the same as those of (3′S,4′S)-isomer. [α]25 = + 33.4° (c = 4, CH3OH).
(3′R,4′S)-tert-Butyl 3′-acetoxy-4′-{{6″-[benzyl(tert-butoxycarbonyl)amino]-4″-methylpyridin-2″-yl}methyl}pyrrolidine-1′-carboxylate [(3′R,4′S)-59] and (3′S,4′R)-tert-butyl 3′-acetoxy-4′-{{6″-[benzyl(tert-butoxycarbonyl)amino]-4″-methylpyridin-2″-yl}methyl}pyrrolidine-1′-carboxylate [(3′S,4′R)-59]
The procedure to prepare (3′R,4′S)-59 or (3′S,4′R)-59 is the same as that to prepare 52 except using (3′S, 4′S)-58 (0.497 g, 0.001 mol) or (3′R, 4′R)-58 (0.497 g, 0.001 mol) instead of 51 (0.497 g, 0.001 mol). The desired products were purified by column chromatography (silica gel, hexanes : EtOAc = 7.5 : 2.5).
(3′R,4′S)-59, colorless oil (0.53 g, 98%), 1H NMR (CDCl3, 500MHz) : δ 7.446-7.427 (m, 1H), 7.262-7.174 (m, 5H), 6.672 (m, 1H), 5.176 (m, 2H), 4.980 (m, 1H), 3.715-3.663 (m, 1H), 3.493-3.413 (m, 1H), 3.557-3.332 (m, 2H), 2.765-2.589 (m, 3H), 2.288 (s, 3H), 1.952-1.946 (m, 3H), 1.449 (s, 9H), 1.441-1.406 (m, 9H). 13C NMR (CDCl3, 125.7 MHz) : δ (170.367+170.179) (1C), (156.214+156.117) (1C), (154.508+154.338) (1C), (154.229+154.198) (1C), 153.871 (1C), 148.607 (1C), 139.681 (1C), 127.970 (2C), 127.095 (2C), (126.470+126.427) (1C), 120.028 (1C), 117.363 (1C), (81.073+81.031) (1C), 79.428 (1C), (76.659+75.973) (1C), (50.358+49.945) (1C), 49.841 (1C), (49.040+48.603) (1C), (43.072+42.234) (1C), 38.797 (1C), 28.367 (3C), 28.063 (3C), 21.008(1C). MS (ESI, CH3OH) : [C30H41N3O6] m/z 562.2 ([M+Na]+).
(3′S,4′R)-59, colorless oil (0.40 g, 74%), the NMR and MS are the same as those of (3′R,4′S)-isomer.
(3′S,4′R)-tert-Butyl 4′-{{6″-[benzyl(tert-butoxycarbonyl)amino]-4″-methylpyridin-2″-yl}methyl}-3′-hydroxypyrrolidine-1′-carboxylate [(3′S,4′R)-58] and (3′R,4′S)-tert-butyl 4′-{{6″-[benzyl(tert-butoxycarbonyl)amino]-4″-methylpyridin-2″-yl}methyl}-3′-hydroxypyrrolidine-1′-carboxylate [(3′R,4′S)-58]
The procedure to prepare (3′S,4′R)-58 and (3′R,4′S)-58 is the same as that to prepare 53 except using (3′R,4′S)-59 (0.540 g, 0.001 mol) or (3′S,4′R)-59 (0.540 g, 0.001 mol) instead of 52 (1.079 g, 0.002 mol). The desired products were purified by column chromatography (silica gel, hexanes : EtOAc = 6 : 4).
(3′S,4′R)-58, colorless oil (0.5 g, quantitative yield). 1H NMR (CDCl3, 500 MHz) : δ 7.357-7.314 (m, 1H), 7.248-7.163 (m, 5H), 6.684 (m, 1H), 5.148-5.138 (m, 2H), (4.419+4.279) (brs, 1H), 4.007-3.996 (m, 1H), 3.661-3.489 (m, 2H), 3.209-3.156 (m, 1H), 3.092-3.058 (m, 1H), 2.855-2.786 (m, 1H), 2.656-2.563 (m, 1H), 2.462-2.396 (m, 1H), 2.270-2.257 (m, 3H), 1.443-1.415 (m, 9H), 1.405 (s, 9H). 13C NMR (CDCl3, 125.7 MHz) : δ (157.240+157.186) (1C), 154.447 (1C), (154.150+154.107) (1C), 153.658 (1C), (148.801+148.698) (1C), (139.323+139.244) (1C), 127.970 (2C), 126.974 (1C), 126.512 (1C), (120.301+120.162) (1C), (117.836+117.636) (1C), 81.025 (1C), 78.991 (1C), (74.474+73.763) (1C), (52.464+52.033) (1C), (50.103+49.987) (1C), (49.307+48.913) (1C), (45.367+44.650) (1C), (38.955+38.907) (1C), 28.294 (3C), (27.972+27.948) (3C), 20.874 (1C). MS (ESI, CH3OH) : [C28H39N3O5] m/z 498.4 ([M+H]+); m/z 520.4 ([M+Na]+); m/z 1017.2 ([2M+Na]+). [α]25 = − 35.6° (c = 2, MeOH).
(3′R,4′S)-58, colorless oil (0.5 g, quantitative yield). The NMR and MS are also the same as those of (3′S,4′R)-isomer. [α]25 = + 35.6° (c = 2, MeOH).
(3′R,4′R)-tert-Butyl 3′-(allyloxy)-4′-{{6″-[benzyl(tert-butoxycarbonyl)amino]-4″-methylpyridin-2″-yl}methyl}pyrrolidine-1′-carboxylate [(3′R,4′R)-60]
To an ice-cooled solution of NaH (0.048 g, 0.0012 mol, 60% dispersion in mineral oil) in anhydrous DMF (1 mL) was added dropwise (3′R,4′R)-58 (0.497 g, 0.001 mol) in anhydrous DMF (5 mL) under a N2 atmosphere. The suspension was then stirred vigorously for 30 min at room temperature. The color of the reaction mixture changed from colorless to pale-red. Allyl bromide (0.157 g, 0.113 mL, 0.0013 mol) was added dropwise at 0 °C, and the reaction mixture was stirred at room temperature for 3 h. The excess NaH was quenched with H2O (5 mL), and the solvent was evaporated in vacuo. The residue was further diluted with water (10 mL) and EtOAc (10 mL). The organic layer was separated, and the aqueous layer was extracted with EtOAc (10 mL × 3). The combined organic layers were washed with brine (10 mL × 2) and dried over Na2SO4. The solvent was evaporated, and the residue was purified by column chromatography (silica gel, hexanes : EtOAc = 8 : 2) to afford a colorless oil (0.505 g, 94%). 1H NMR (CDCl3, 500MHz) : δ 7.445-7.413 (m, 1H), 7.279-7.165 (m, 5H), 6.686-6.678 (m, 1H), 5.828-5.756 (m, 1H), 5.184(s, 2H), 5.229-5.091 (m, 2H), 3.980-3.930 (m, 1H), 3.751-3.635 (m, 2H), 3.555-3.379 (m, 2H), 3.210-3.099 (m, 2H), 2.950-2.907 (m, 1H), 2.747-2.705 (m, 1H), 2.601-2.549 (m, 1H), 2.287-2.272 (m, 3H), 1.457-1.439 (m, 9H), 1.410 (s, 9H). 13C NMR (CDCl3, 125.7 MHz) : δ (157.690+157.653) (1C), (154.642+154.405) (1C), (154.302+154.241) (1C), 153.767 (1C), 148.303 (1C), (139.803+139.760) (1C), (134.624+134.600) (1C), 127.982 (2C), (126.943+126.852) (2C), (126.482+126.403) (1C), 120.016 (1C), (116.901+116.840) (1C), (116.585+116.422) (1C), (81.019+80.958) (1C), (78.954+78.900) (1C), (78.554+77.691)(1C), (70.060+70.005) (1C), (50.916+50.364) (1C), 49.805 (1C), (49.289+48.870) (1C), (42.714+42.070) (1C), (34.517+34.462) (1C), 28.458 (3C), 28.087 (3C), 21.038 (1C). MS (ESI, CH3OH) : [C31H43N3O5] m/z 538.6 ([M+H]+); m/z 560.6 ([M+Na]+); m/z 1097.1 ([2M+Na]+).
(3′S, 4′S)-60 was synthesized as a colorless oil (0.521 g, 97%) by the same procedure as that to prepare the (3′R, 4′R)-isomer. The NMR and MS are the same as those for (3′R, 4′R)-isomer.
(3′R, 4′S)-60 was synthesized as a colorless oil (0.521 g, 97%) by the same procedure as that to prepare the (3′R, 4′R)-isomer. 1H NMR (CDCl3, 500MHz) : δ 7.453-7.432 (m, 1H), 7.273-7.173 (m, 5H), 6.659 (s, 1H), 5.809-5.764 (m, 1H), 5.192 (s, 2H), 5.167-5.082 (m, 2H), 3.840-3.787 (m, 2H), 3.715-3.698 (m, 1H), 3.613-3.579 (m, 0.5H), 3.544-3.510 (m, 0.5H), 3.485-3.417 (m, 1H), 3.338-3.317 (m, 0.5H), 3.276-3.253 (m, 0.5H), 3.175-3.153 (m, 0.5H), 3.132-3.104 (m, 0.5H), 2.766-2.702 (m, 1H), 2.633-2.549 (m, 2H), 2.295-2.284 (m, 3H), 1.459-1.429 (m, 9H), 1.407 (s, 9H). 13C NMR (CDCl3, 125.7 MHz) : δ (156.979+156.876) (1C), (154.696+154.532) (1C), (154.302+154.253) (1C), 153.871 (1C), 148.540 (1C), 139.724 (1C), (134.533+134.509) (1C), 128.042 (2C), 127.041 (2C), (126.555+126.506) (1C), 119.998 (1C), (117.229+117.187) (1C), (116.980+116.901) (1C), (81.201+81.091) (1C), (81.043+80.472) (1C), 79.100 (1C), (70.126+70.023) (1C), (50.400+49.726) (1C), 49.823 (1C), (49.192+48.767) (1C), (43.424+42.440) (1C), (39.295+39.241) (1C), 28.464 (3C), 28.112 (3C), 21.093 (1C). MS (ESI, CH3OH) : [C31H43N3O5] m/z 560.6 ([M+Na]+); m/z 1097.3 ([2M+Na]+).
(3′S, 4′R)-60 was synthesized as a colorless oil (0.510 g, 95%) by the same procedure as that to prepare the (3′R, 4′R)-isomer. The NMR and MS are the same as those for (3′R, 4′S)-isomer.
(3′R,4′R)-tert-Butyl 4′-{{6″-[benzyl(tert-butoxycarbonyl)amino]-4″-methylpyridin-2″-yl}methyl}-3′-(2″-oxoethoxy)pyrrolidine-1′-carboxylate [(3′R,4′R)-61]
Method A
(3′R,4′R)-60 (0.537 g, 0.001 mol) was dissolved in CH2Cl2 (30 mL) and cooled to −78 °C. Ozone was bubbled through the solution until a light blue color appeared. N2 was bubbled through the solution until the blue color disappeared (about 30 min). Zinc (0.098 g, 0.0015 mol) was added followed by 5 mL of 50% aqueous acetic acid solution. The suspension was allowed to warm slowly to 0 °C, stirred for 30 min, and then allowed to stir at room temperature for 30 min. The reaction mixture was diluted with CH2Cl2 (10 mL) and H2O (20 mL). The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (10 mL × 2). The combined organic layers were washed with H2O (10 mL), saturated aqueous NaHCO3 solution (10 mL × 2), and brine (10 mL), and then dried over MgSO4. The solvent was concentrated in vacuo, and the residue was purified by column chromatography (silica gel, hexanes : EtOAc = 5 : 5) to afford a pale-yellow oil (0.345 g, 64%).
Method B
(3′R,4′R)-60 (0.537 g, 0.001 mol) was dissolved in CH2Cl2 (30 mL) and cooled to −78 °C. Ozone was bubbled through the solution until a light blue color appeared. N2 was bubbled through the solution until the blue color disappeared (about 30 min). Ph3P (0.315 g, 0.0012 mol) was added to the reaction mixture. The reaction mixture was allowed to warm slowly to 0 °C, stirred for 15 min and then allowed to stir at room temperature for 30 min. The solvent was concentrated in vacuo, and the residue was purified by column chromatography (silica gel, hexanes : EtOAc = 5 : 5) to afford a pale-yellow oil (0.388 g, 72%).
1H NMR (CDCl3, 500MHz) : δ 9.552 (s, 1H), 7.457-7.429 (m, 1H), 7.228-7.176 (m, 5H), 6.728-6.691 (m, 1H), 5.165 (m, 2H), 4.017-3.938 (m, 1H), 3.805-3.635 (m, 2H), 3.535-3.382 (m, 2H), 3.220-3.107 (m, 2H), 3.021-2.958 (m, 1H), 2.788-2.576 (m, 2H), 2.296 (m, 3H), 1.454-1.413 (m, 18H). 13C NMR (CDCl3, 125.7 MHz) : δ 200.852-199.999 (1C), 157.501 (1C), (154.806+154.484+154.429) (2C), 153.974 (1C), 148.722 (1C), (139.930+139.876) (1C), 128.212 (2C), (126.895+126.810) (2C), (126.658+126.591) (1C), 120.089 (1C), (117.156+117.095) (1C), 81.371-80.211 (2C), 79.634-79.294 (1C), (74.807+74.583+74.540) (1C), (50.971+50.327) (1C), 49.975 (1C), (49.301+48.888) (1C), (42.786+42.155) (1C), (34.335+34.304) (1C), 28.597 (3C), 28.494 (3C), 21.239 (1C).
(3′S,4′S)-61 was synthesized as a pale-yellow oil (0.367 g, 68%) by the same procedure to prepare (3′R,4′R)-61 (method A). The NMR spectrum is the same as that for (3′R,4′R)-isomer.
(3′R,4′S)-61 was synthesized as a pale-yellow oil (0.399 g, 74%) by the same procedure to prepare (3′R,4′R)-61 (method A). 1H NMR (CDCl3, 500MHz) : δ (9.548+9.507) (s, 1H), 7.480-7.384 (m, 1H), 7.256-7.182 (m, 5H), 6.667 (s, 1H), 5.187-5.112 (m, 2H), 4.026-2.919 (m, 8H), 2.724-2.552 (m, 2H), 2.299-2.289 (m, 3H), 1.462-1.413 (m, 18H). 13C NMR (CDCl3, 125.7 MHz) : δ 199.965 (1C), (157.240+156.906+156.621+156.542) (1C), (154.747+154.544+154.368+154.320) (2C), (153.962+153.798) (1C), (148.922+148.825+148.716) (1C), 139.779 (1C), (128.170+128.133) (2C), (127.113+126.986+126.913+126.761) (2C), (126.713+126.634) (1C), (120.198+120.113) (1C), (117.338+117.278) (1C), 82.889-79.264 (3C), (75.554+74.431) (1C), (50.169-48.791) (3C), (43.205+42.197) (1C), 39.113 (1C), 28.500-28.160 (6C), 21.160 (1C).
(3′S,4′R)-61 was synthesized as a pale-yellow oil (0.469 g, 87%) by the same procedure to prepare (3′R,4′R)-61 (method A). The NMR spectrum is the same as that for (3′R,4′S)-isomer.
(3′R,4′R)-tert-Butyl 4′-{{6″-[benzyl(tert-butoxycarbonyl)amino]-4″-methylpyridin-2″-yl}methyl}-3′-[2″-(3‴-fluorophenethylamino)ethoxy]pyrrolidine-1′-carboxylate [(3′R,4′R)--62]
A mixture of (3′R,4′R)-61 (0.539 g, 0.001 mol), 3-fluorophenethylamine (0.153 g, 0.144 mL, 0.0011 mol), NaBH(OAc)3 (0.297 g, 0.0014 mol), and 3 Å molecular sieves (1 g) in dry 1, 2-dichloroethane (20 mL) was stirred at room temperature under a N2 atmosphere for 14 h. The reaction mixture was then filtered through Celite. The Celite pad was washed with CH2Cl2 (5 mL × 2). To the combined organic layers was added 1M NaOH (20 mL). The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (10 mL × 2). The combined organic layers were washed with brine (10 mL) and dried over MgSO4. The solvent was concentrated in vacuo, and the residue was purified by column chromatography (silica gel, hexanes : EtOAc : Et3N = 6 : 4 : 0.5) to afford a pale-yellow oil (0.623 g, 94%). 1H NMR (CDCl3, 500MHz) : δ 7.427-7.392 (m, 1H), 7.272-7.181 (m, 6H), 6.979-6.835 (m, 3H), 6.603 (m, 1H), 5.169 (s, 2H), 3.646-3.586 (m, 1H), 3.557-3.473 (m, 1H), 3.420-3.324 (m, 1H), 3.284-3.234 (m, 1H), 3.194-3.163 (m, 1H), 3.061-3.000 (m, 1H), 2.913-2.511 (m, 9H), 2.299-2.282 (m, 3H), 1.452-1.436 (m, 9H), 1.411 (s, 9H). 13C NMR (CDCl3, 125.7 MHz) : δ (164.034+162.079) (1C), (157.853+157.823) (1C), (154.909+154.708) (1C), (154.575+154.508) (1C), 154.022 (1C), 148.649 (1C), (142.802+142.748+142.669) (1C), (140.021+139.973) (1C), (130.107+130.052+129.991) (1C), 128.231 (2C), (127.174+127.077) (2C), (126.719+126.646) (1C), 124.539 (1C), 120.131 (1C), (117.253+117.187) (1C), (115.839+115.748+115.675+115.578) (1C), (113.313+113.270+113.149+113.106) (1C), (81.365+81.304) (1C), (79.646+79.306) (1C), (79.270+78.845) (1C), (68.687+68.536) (1C), 50.892 (1C), 50.746 (1C), 50.060 (1C), 49.374 (1C), (49.301+48.967) (1C), (42.877+42.270) (1C), 36.266 (1C), (35.567+34.602) (1C), 28.676 (3C), 28.324 (3C), 21.305 (1C). MS(ESI, CH3OH) : [C38H51FN4O5] m/z 663.7 ([M+H]+); m/z 1325.3 ([2M+H]+).
(3′S,4′S)-62 was synthesized as a pale-yellow oil (0.444 g, 67%) by the same procedure to prepare (3′R,4′R)-62. The NMR spectrum and MS are the same as those for (3′R,4′R)-isomer.
(3′R,4′S)-62 was synthesized as a pale-yellow oil (0.510 g, 77 %) by the same procedure to prepare (3′R,4′R)-62. 1H NMR (CDCl3, 500MHz) : δ 7.448-7.424 (m, 1H), 7.280-7.175 (m, 6H), 6.967-6.846 (m, 3H), 6.647 (s, 1H), 5.181 (s, 2H), 3.641-3.567 (m, 1.5H), 3.527-3.493 (m, 0.5H), 3.391-3.335 (m, 3H), 3.282-3.258 (m, 0.5H), 3.211-3.183 (m, 0.5H), 3.158-3.131 (m, 0.5H), 3.110-3.081 (m, 0.5H), 2.910-2.628 (m, 7H), 2.577-2.518 (m, 2H), 2.308-2.293 (m, 3H), 1.461-1.431 (m, 9H), 1.407 (s, 9H). 13C NMR (CDCl3, 125.7 MHz) : δ (163.961+162.006) (1C), (157.095+156.997) (1C), (154.860+154.702) (1C), (154.472+154.417) (1C), 154.010 (1C), 148.752 (1C), (142.705+142.669) (1C), 139.894 (1C), (129.979+129.912) (1C), 128.182 (2C), 127.198 (2C), (126.688+126.634) (1C), (124.472+124.448) (1C), 120.156 (1C), (117.429+117.393) (1C), (115.669+115.499) (1C), (113.204+113.034) (1C), (82.209+81.553) (1C), (81.310+81.261) (1C), 79.331 (1C), 68.523 (1C), (50.861+50.443) (1C), (50.801+49.866) (1C), 49.963 (1C), (49.222+48.840) (2C), (43.309+42.452) (1C), (39.392+39.344) (1C), 36.217 (1C), 28.603 (3C), 28.257 (3C), 21.257 (1C). MS (ESI, CH3OH) : [C38H51FN4O5] m/z 663.6 ([M+H]+); m/z 1325.5 ([2M+H]+).
(3′S,4′R)-62 was synthesized as a pale-yellow oil (0.325 g, 49%) by the same procedure to prepare (3′R,4′R)-62. The NMR and MS spectrum are the same as those for (3′S,4′R)-isomer.
(3′R,4′R)-tert-Butyl 4′-{{6″-[benzyl(tert-butoxycarbonyl)amino]-4″-methylpyridin-2″-yl}methyl}-3′-{2″-[tert-butoxycarbonyl(3‴-fluorophenethyl)amino]ethoxy}pyrrolidine-1′-carboxylate [(3′R,4′R)-63]
A solution of di-tert-butyl dicarbonate (0.219 g, 0.001 mol) in CH2Cl2 (5 mL) was added dropwise to a solution of (3′R,4′R)-62 (0.662 g, 0.001 mol) in CH2Cl2 (5 mL) and 1M NaOH (3 mL). The reaction mixture was then stirred at room temperature for 24 h. The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (10 mL × 2). The combined organic layers were washed with water (10 mL × 2) and dried over Na2SO4. The solvent was evaporated in vacuo, and the residue was purified by column chromatography (silica gel, hexanes : EtOAc = 7.5 : 2.5) to afford a colorless oil (0.572 g, 75 %). 1H NMR (CDCl3, 500 MHz) : δ 7.426-7.395 (m, 1H), 7.237-7.178 (m, 6H), 6.986-6.857 (m, 3H), 6.634-6.609 (m, 1H), 5.163 (s, 2H), 3.646-3.165 (m, 10H), 3.086-3.022 (m, 1H), 2.893-2.779 (m, 3H), 2.692-2.649 (m, 1H), 2.558 (m, 1H), 2.291-2.273(m, 3H), 1.445-1.409 (m, 27H). 13C NMR (CDCl3, 125.7 MHz) : δ (163.967+162.012) (1C), (157.756+157.665) (1C), (155.419+155.279) (1C), (154.812+154.623) (1C), (154.514+154.447) (1C), 153.974 (1C), 148.655 (1C), 142.013 (1C), 139.973 (1C), 129.997 (1C), 128.176 (2C), (127.120+127.035) (2C), (126.670+126.591) (1C), (124.685+124.667) (1C), 120.058 (1C), (117.174+117.126) (1C), (115.875+115.711) (1C), (113.337+113.246+113.173+113.076) (1C), (81.298+81.243) (1C), (79.725+79.203+78.881) (3C), (68.232+68.056) (1C), (50.934+50.430) (1C), (50.273+50.194) (1C), 50.024 (1C), (49.386+48.937) (1C), (47.971+47.468) (1C), (42.890+42.762+42.282) (1C), (35.003+34.711+34.341) (2C), 28.591 (3C), 28.488 (3C), 28.275 (3C), 21.239 (1C). MS (ESI, CH3OH) : [C43H59FN4O7] m/z 763.5 ([M+H]+); m/z 785.5 ([M+Na]+); m/z 1546.8 ([2M+Na]+).
(3′S,4′S)-63 was synthesized as a pale-yellow oil (0.595 g, 78%) by the same procedure to prepare (3′R,4′R)-63. The NMR spectrum and MS are the same as those for (3′R,4′R)-63.
(3′R,4′S)-63 was synthesized as a pale-yellow oil (0.473 g, 62%) by the same procedure to prepare (3′R,4′R)-63. 1H NMR(CDCl3, 500 MHz) : δ 7.446-7.423 (m, 1H), 7.244-7.171 (m, 6H), 6.967-6.837 (m, 3H), 6.636 (s, 1H), 5.170-5.161 (s, 2H), 3.639 (m, 1H), 3.580-3.556 (m, 0.5H), 3.510-3.495 (m, 0.5H), 3.396-3.313 (m, 6H), 3.216-3.095 (m, 3H), 2.812-2.691 (m, 3H), 2.574-2.538 (m, 2H), 2.291-2.279 (m, 3H), 1.448-1.407 (m, 27H). 13C NMR(CDCl3, 125.7 MHz) : δ (163.919+161.964) (1C), (157.028+156.937) (1C), (155.401+155.243) (1C), (154.854+154.702) (1C), (154.453+154.399) (1C), 153.974 (1C), 148.752 (1C), 142.025 (1C), 139.845 (1C), (130.016+129.955) (1C), 128.170 (2C), 127.192 (2C), (126.682+126.628) (1C), (124.673+124.654) (1C), 120.089 (1C), 117.344 (1C), (115.869+115.705) (1C), (113.283+113.191+113.119) (1C), (82.227+81.595) (1C), 81.292 (1C), 79.683 (1C), (79.373+79.313) (1C), (68.044+67.941) (1C), 50.327 (1C), 49.920 (1C), (50.078+49.756) (1C), (49.265+48.846) (1C), (48.117+47.613) (1C), (43.545+42.695) (1C), (39.392+39.344) (1C), (35.003+34.341) (1C), (28.555+28.452) (6C), 28.245 (3C), 21.233 (1C). MS (ESI, CH3OH) : [C43H59FN4O7] m/z 785.5 ([M+Na]+).
(3′S,4′R)-63 was synthesized as a pale-yellow oil (0.572 g, 75%) by the same procedure to prepare (3′R,4′R)-63. The NMR spectrum and MS are the same as those for (3′R,4′S)-63.
(3′R,4′R)-tert-butyl 3′-{2″-[tert-butoxycarbonyl(3‴-fluorophenethyl)amino]ethoxy}-4′-{[6″-(tert-butoxycarbonyl amino)-4″-methylpyridin-2″-yl]methyl}pyrrolidine-1′-carboxylate [(3′R,4′R)-64]
A solution of (3′R,4′R)-63 (0.381 g, 0.0005 mol) in EtOH (20 mL) was treated with 20% wt Pd(OH)2 on carbon (150 mg). The reaction mixture was stirred at 60 °C under a hydrogen atmosphere for 36 h. The catalyst was filtered through Celite. The Celite pad was washed with EtOH (10 mL × 2), and the combined filtrates were concentrated in vacuo. The residue was diluted with CH2Cl2 (10 mL) and 1M NaOH (10 mL), and the organic layer was separated. The aqueous layer was extracted with CH2Cl2 (10 mL × 2), and the combined organic layers were concentrated in vacuo. The residue was purified by column chromatography (silica gel, hexanes : EtOAc = 7 : 3) to afford a colorless oil (0.269 g, 80%). 1H NMR (CDCl3, 500 MHz) : δ 7.618-7.600 (m, 1H), 7.471-7.457 (m, 1H), 7.254-7.242 (m, 1H), 7.002-6.874 (m, 3H), 6.620-6.591 (m, 1H), 3.781-3.737 (m, 1H), 3.602-3.261 (m, 9H), 3.160-3.062 (m, 1H), 2.859-2.819 (m, 3H), 2.703-2.541 (m, 2H), 2.301-2.279 (m, 3H), 1.512 (s, 9H), 1.442 (m, 18H). 13C NMR (CDCl3, 125.7 MHz) : δ (163.870+161.915) (1C), (158.218+158.169) (1C), (155.382+155.334+155.170) (1C), (154.696+154.544) (1C), 152.492 (1C), (151.527+151.515) (1C), 149.894 (1C), (141.958+141.898) (1C), (129.979+129.906) (1C), (124.600+124.582) (1C), (119.117+119.063) (1C), (115.790+115.626) (1C), (113.240+113.149+113.070+112.979) (1C), (110.235+110.168) (1C), (80.721+80.660) (1C), (79.616+79.586) (1C), (79.750+79.221+79.149+78.651) (2C), (68.250+68.159+67.977) (1C), (50.922+50.485) (1C), (50.212+50.072+49.981) (1C), (49.216+48.864) (1C), (47.941+47.449) (1C), (43.333+43.175+42.707+42.556) (1C), (34.936+34.736+34.547+34.432+34.268) (2C), 28.482 (3C), 28.385 (3C), 28.263 (3C), 21.281 (1C). MS (ESI, CH3OH) : [C36H53FN4O7] m/z 673.8 ([M+H]+); m/z 695.6 ([M+Na]+); m/z 1367.5([2M+Na]+).
(3′S,4′S)-64 was synthesized as a colorless oil (0.286 g, 85%) by the same procedure to prepare (3′R,4′R)-64. The NMR spectrum and MS are the same as those for (3′R,4′R)-isomer.
(3′R,4′S)-64 was synthesized as a colorless oil (0.048 g, 48%) by the same procedure to prepare (3′R,4′R)-64 except using 0.114 g (0.0015 mol) of (3′R,4′S)-63. 1H NMR (CDCl3, 500 MHz) : δ 7.625-7.610 (m, 1H), 7.305-7.229 (m, 2H), 6.984-6.855 (m, 3H), 6.590-6.582 (s, 1H), 3.681-3.654 (m, 1H), 3.583-3.091 (m, 10H), 2.832-2.799 (m, 2H), 2.698-2.491 (m, 3H), 2.298-2.282 (m, 3H), 1.518 (s, 9H), 1.448-1.430 (m, 18H). 13C NMR(CDCl3, 125.7 MHz) : δ (164.004+162.049) (1C), 157.410 (1C), (155.534+155.316) (1C), (154.939+154.818) (1C), (152.644+152.559) (1C), 151.588 (1C), 150.155 (1C), 142.068 (1C), (130.101+130.040) (1C), (124.752+124.733) (1C), (119.397+119.324) (1C), (115.960+115.802) (1C), (113.392+113.228) (1C), 110.453 (1C), (82.439+81.699) (1C), 80.982 (1C), 79.798 (1C), 79.507 (1C), 68.153 (1C), 50.515 (1C), (50.315+49.963) (1C), (49.234+48.888) (1C), (48.287+47.880) (1C), (43.539+42.920) (1C), 39.320 (1C), (35.130+34.468) (1C), (28.652+28.549) (6C), 28.427 (3C), 21.457 (1C). MS (ESI, CH3OH) : [C36H53FN4O7] m/z 673.8 ([M+H]+); m/z 695.8 ([M+Na]+); m/z 1367.2 ([2M+Na]+).
(3′S,4′R)-64 was synthesized as a colorless oil (0.585 g, 58 %) by the same procedure to prepare (3′R,4′R)-64 except using 0.114 g (0.0015 mol) of (3′S,4′R)-63. The NMR and MS are the same as those of (3′R,4′S)-isomer.
6-{{(3′R,4′R)-3′-[2″-(3‴-fluorophenethylamino)ethoxy]pyrrolidin-4′-yl}methyl}-4-methylpyridin-2-amine trihydrochloride [(3′R,4′R)-4]
(3′R,4′R)-64 (0.067 g, 0.0001 mol) was cooled in an ice-water bath under argon. A solution of 4M HCl in 1,4-dioxane (4 mL) was then added slowly with stirring. The ice-water bath was removed after 3 h, and the reaction mixture was stirred at room temperature under an argon atmosphere for 34 h. The reaction mixture was concentrated in vacuo, and the residue was partitioned between water (10 mL) and ethyl acetate (10 mL). The aqueous layer was then washed with ethyl acetate (5 mL × 2). After evaporation of the water by high vacuum rotary evaporation, the residue was dried with a lyophilizer to afford a white solid (0.048 g, quantitative yield). 1H NMR (D2O, 500 MHz) : δ 7.336-7.293 (m, 1H), 7.111-7.096 (m, 1H), 7.063-7.044 (m, 1H), 6.972-6.937 (m, 1H), 6.631 (s, 1H), 6.560 (s, 1H), 4.189 (m, 1H), 3.871-3.850 (m, 1H), 3.684-3.620 (m, 2H), 3.500-3.466 (m, 1H), 3.354-3.309 (m, 5H), 3.195-3.154 (m, 1H), 3.065-3.040 (m, 2H), 2.964-2.930 (m, 1H), 2.807-2.771 (m, 2H), 2.289 (s, 3H). 13C NMR (D2O, 125.7 MHz) : δ (163.769+161.826) (1C), 158.328 (1C), 153.963 (1C), 145.882 (1C), (138.991+138.930) (1C), 130.849 (1C), (124.978+124.856) (1C), (115.852+115.682+115.543) (1C), (114.444+114.359) (1C), (114.201+114.060) (1C), 110.576 (1C), (78.422+78.361) (1C), 64.056 (1C), 49.558 (1C), 48.489 (1C), 47.141 (2C), 41.713 (1C), 31.434 (1C), 29.273 (1C), (21.386+21.344) (1C). 19F NMR (D2O, 376.5 MHZ) : δ −113.532 (F). MS (ESI, CH3OH) : [C21H29FN4O] m/z 373.5 ([M+H]+). HRMS (CI+, CH3OH) Calc. : 373.2404, Found : 373.2410. [α]25 +36.4°(c = 1, MeOH). Anal. (C21H33Cl3FN4O·2.5H2O·0.7CH3OH), Calcd : C, 47.45; H, 7.30; N, 10.20; Found : C, 47.56; H, 7.19; N, 9.97.
(3′S,4′S)-4 was synthesized as a white solid (0.048 g, quantitative) by the same procedure to prepare (3′R,4′R)-4. The NMR spectrum and MS are the same as those for (3′R,4′R)-isomer. [α]25 −36.4° (c = 1, MeOH). Anal. (C21H33Cl3FN4O·3.25H2O), Calcd : C, 46.67; H, 7.18; N, 10.37; Found : C, 46.70; H, 7.10; N, 10.13.
(3′R,4′S)-4 was synthesized as a white solid (0.048 g, quantitative) by the same procedure to prepare (3′R,4′R)-4. The NMR spectrum and MS are the same as those for the (3′R,4′R)-isomer. 1H NMR (D2O, 500 MHz) : δ 7.373-7.332 (m, 1H), 7.105-7.090 (m, 1H), 7.060-7.003 (m, 2H), 6.661 (s, 2H), 4.139 (m, 1H), 3.704 (m, 2H), 3.625-3.589 (m, 1H), 3.566-3.483 (m, 2H), 3.313-3.248 (m, 3H), 3.205-3.178 (m, 1H), 3.143-3.112 (m, 1H), 3.026-2.997 (m, 2H), 2.857-2.780 (m, 3H), 2.303 (s, 3H). 13C NMR (D2O, 125.7 MHz) : δ (163.811+161.874) (1C), 158.322 (1C), 154.084 (1C), 145.087 (1C), (138.948+138.888) (1C), (130.885+130.819) (1C), 124.747 (1C), (115.792+115.628+115.524) (1C), 114.796 (1C), 114.207 (1C), 110.752 (1C), (81.476+81.403) (1C), 63.941 (1C), 49.078 (1C), (48.228+48.058) (2C), 46.983 (1C) 42.460 (1C), 33.408 (1C), 31.337 (1C), (21.344+21.307) (1C). 19F NMR (D2O, 376.5 MHZ) : δ −113.558 (F). MS (ESI, CH3OH) : [C21H29FN4O] m/z 373.7 ([M+H]+). HRMS (CI+, CH3OH) Calc. : 373.2404, Found : 373.2403. [α]25 −33.6° (c = 1, MeOH). Anal. (C21H33Cl3FN4O·3H2O·0.3NaCl), Calcd : C, 45.57; H, 6.92; N, 10.12; Found : C, 45.43; H, 6.76; N, 9.82.
3′S,4′R)-4 was synthesized as a white solid (0.048 g, quantitative) by the same procedure to prepare (3′R,4′R)-4. The NMR spectrum and MS are the same as those for (3′R,4′R)-isomer. [α]25 +33.6° (c = 1, MeOH). Anal. (C21H33Cl3FN4O·3H2O·0.5NaCl), Calcd : C, 44.63; H, 6.78; N, 9.91; Found : C, 44.75; H, 6.76; N, 9.64
Enzyme and assay
All of the NOS isozymes used were recombinant enzymes overexpressed in E. coli. The murine macrophage iNOS was expressed and isolated according to the procedure of Hevel et al.35 The rat nNOS was expressed36 and purified as described.37 The bovine eNOS was isolated as reported.38 Nitric oxide formation from NOS was monitored by the hemoglobin capture assay at 30 °C as described.39 A typical assay mixture for nNOS and eNOS contained 10 μM L-arginine, 1.0 mM CaCl2, 600 unit/mL calmodulin (Sigma, P-2277), 100 μM NADPH, 0.125 mg/mL hemoglobin-A0 (ferrous form, Sigma, H0267), 10 μM H4B, in 100 mM HEPES (pH 7.5). A typical assay mixture for iNOS contained 10 μM L-arginine, 100 μM NADPH, 0.125 mg/mL hemoglobin-A0 (ferrous form), 10 μM H4B, in 100 mM HEPES (pH 7.5). All assays were in a final volume of 600 μL and were initiated by addition of enzyme. NOS assays were monitored at 401 nm on a Perkin–Elmer Lambda 10 UV–visible spectrophotometer.
Determination of Ki values
The apparent Ki values were obtained by measuring the percent enzyme inhibition in the presence of 10 μM L-arginine with at least five concentrations of inhibitor. The parameters of the following inhibition equation40 were fitted to the initial velocity data : % inhibition = 100[I]/{[I] + Ki(1+[S]/Km)}. Km values for L-arginine were 1.3 μM (nNOS), 8.2 μM (iNOS), and 1.7 μM (eNOS). The selectivity of an inhibitor was defined as the ratio of the respective Ki values.
Crystal preparation, x-ray data collection, and structure refinement
The nNOS or eNOS heme domain proteins, wild type or mutants, used for crystallographic studies were produced by limited trypsin digest from the corresponding full length enzymes and further purified through a Superdex 200 gel filtration column (GE Healthcare) as described previously.17d, 20a A batch of nNOS heme domain was also obtained by limited trypsin digest from the purified nNOS heme-FMN bidomain protein.41 This bidomain construct had its Arg349, a known sensitive trypsin cleavage site,42 mutated to Ala through the Quikchange protocol (Stratagene). The purified nNOS heme domain (48 kD) is therefore a R349A mutant, which is free of the 43 kD truncated heme domain. The nNOS heme domain at 7–9 mg/mL containing 20 mM histidine or the eNOS heme domain at 20 mg/mL with 2 mM imidazole were used for the sitting drop vapor diffusion crystallization setup under the conditions reported before.17b,17d Fresh crystals were first passed stepwise through cryo-protectant solutions as described17b,17d and then soaked with 10 mM inhibitor for 4–6 h at 4 °C before being flash cooled by plunging into liquid nitrogen. Crystals were stored in liquid nitrogen until data collection.
The cryogenic (100 K) X-ray diffraction data were collected at various beamlines at either Advanced Light Source (ALS, Berkeley, CA) or Stanford Synchrotron Radiation Lightsource (SSRL, Menlo Park, CA) using the data collection control software Blu-Ice.43 Raw data frames were indexed, integrated, and scaled using HKL2000.44 The binding of inhibitors was detected by the initial difference Fourier maps calculated with CNS.45 The inhibitor molecules were then modeled in O46 or COOT47 and refined using CNS. Water molecules were added and checked by COOT. Refinements at later stage were continued with REFMAC48 in order to implement the TLS49 protocol with each subunit as one TLS group. The refined structures were validated before deposition to RCSB protein data bank. The crystallographic data collection and structure refinement statistics for the four enantiopure isomers of 4 bound to the wild type eNOS and nNOS (including nNOS R349A mutant) are summarized in Supplementary Table S1; the statistics for the same four compounds bound to various eNOS and nNOS mutants are summarized in Table S2. Those eNOS or nNOS mutant structures in a complex with inhibitors provide no additional information on the enzyme-inhibitor interactions and thus are not discussed in the Results and Discussion section. Coordinates of all the crystal structures reported in this work have been deposited to RCSB protein data bank with their accession codes listed in both supplementary tables.
Supplementary Material
Acknowledgments
The authors are grateful for financial support from the National Institutes of Health, GM049725 to RBS, GM057353 to TLP, and GM052419 to Dr. Bettie Sue Masters, with whose lab PM and LJR are affiliated, and Grant No. AQ1192 from The Robert A. Welch Foundation to BSM. B.S.S.M. also is grateful to the Welch Foundation for a Robert A. Welch Foundation Distinguished Professorship in Chemistry (AQ0012). P.M. is supported by grants 0021620806 and 1M0520 from MSMT of the Czech Republic. We are grateful to Dr. Jinwen Huang for suggesting the use of a direct Mitsunobu inversion of 51 in Scheme 4. We thank the beamline support staff at ALS and SSRL for their assistance during X-ray data collections.
Footnotes
Abbreviations: NOS, nitric oxide synthase; nNOS, neuronal nitric oxide synthase; eNOS, endothelial nitric oxide synthase; iNOS, inducible nitric oxide synthase; H4B, (6R)-5,6,7,8-tetrahydro-L-biopterin. FMN, flavin mononucleotide; FAD, flavin adenine dinucleotide; NADPH, reduced nicotinamide adenine dinucleotide phosphate; CaM, calmodulin; CPCA, consensus principle component analysis; PSA, polar surface area; L-NNA, L-Nω-nitroarginine; Ph3P, triphenylphospine; DIAD, diisopropyl azodicarboxylate; DPPA, diphenylphosphoryl azide.
Supporting Information Available. The tables of crystallographic data collection and structure refinement (Tables S1 and S2). The figures for the wild type eNOS in complexes with four enatiopure isomers of 4 (Figure S1). The experimental details and data of 41b, 42b, 8, 10, 11, 46a–46h, 13–20, 50b, 50d–50k, 50m, 22, 24–31, 33; The 1H NMR and 13C NMR spectra of 8–34, and four enantiopure isomers of 4. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Bredt DS, Snyder SH. Nitric Oxide: A Physiologic Messenger Molecule. Annu Rev Biochem. 1994;63:175–195. doi: 10.1146/annurev.bi.63.070194.001135. [DOI] [PubMed] [Google Scholar]; (b) Kerwin JF, Jr, Lancaster JR, Jr, Feldman PL. Nitric Oxide: A New Paradigm for Second Messengers. J Med Chem. 1995;38(22):4343–4362. doi: 10.1021/jm00022a001. [DOI] [PubMed] [Google Scholar]; (b) Cary SPL, Winger JA, Derbyshire ER, Marletta MA. Nitric Oxide Signaling: No Longer Simply on or off. Trends Biochem Sci. 2006;31(4):231–239. doi: 10.1016/j.tibs.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 2.Griffith OW, Stuehr DJ. Nitric Oxide Synthases: Properties and Catalytic Mechanism. Annu Rev Physiol. 1995;57:707–736. doi: 10.1146/annurev.ph.57.030195.003423. [DOI] [PubMed] [Google Scholar]
- 3.Rosen GM, Tsai P, Pou S. Mechanism of Free-Radical Generation by Nitric Oxide Synthase. Chem Rev. 2002;102(4):1191–1199. doi: 10.1021/cr010187s. [DOI] [PubMed] [Google Scholar]
- 4.(a) Alderton WK, Cooper CE, Knowles RG. Nitric Oxide Synthases: Structure, Function and Inhibition. Biochem J. 2001;357(Pt 3):593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Knowles RG, Moncada S. Nitric Oxide Synthases in Mammals. Biochem J. 1994;298(2):249–258. doi: 10.1042/bj2980249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roman LJ, Martásek P, Masters BSS. Intrinsic and Extrinsic Modulation of Nitric Oxide Synthase Activity. Chem Rev. 2002;102(4):1179–1190. doi: 10.1021/cr000661e. [DOI] [PubMed] [Google Scholar]
- 6.(a) Moncada S, Palmer RM, Higgs EA. Nitric Oxide: Physiology, Pathophysiology, and Pharmacology. Pharmacol Rev. 1991;43(2):109–142. [PubMed] [Google Scholar]; (b) Domenico R. Pharmacology of Nitric Oxide: Molecular Mechanisms and Therapeutic Strategies. Curr Pharm Des. 2004;10(14):1667–1676. doi: 10.2174/1381612043384709. [DOI] [PubMed] [Google Scholar]
- 7.(a) Uehara T, Nakamura T, Yao D, Shi ZQ, Gu Z, Ma Y, Masliah E, Nomura Y, Lipton SA. S-Nitrosylated Protein-Disulphide Isomerase Links Protein Misfolding to Neurodegeneration. Nature. 2006;441(7092):513–517. doi: 10.1038/nature04782. [DOI] [PubMed] [Google Scholar]; (b) Chung KK, Thomas B, Li X, Pletnikova O, Troncoso JC, Marsh L, Dawson VL, Dawson TM. S-Nitrosylation of Parkin Regulates Ubiquitination and Compromises Parkin’s Protective Function. Science. 2004;304:1328–1331. doi: 10.1126/science.1093891. [DOI] [PubMed] [Google Scholar]; (c) Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, Lipton SA. S-Nitrosylation of Drp1 Mediates β-Amyloid-Related Mitochondrial Fission and Neuronal Injury. Science. 2009;324(5923):102–105. doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hantraye P, Brouillet E, Ferrante R, Palfi S, Dolan R, Matthews RT, Beal MF. Inhibition of Neuronal Nitric Oxide Synthase prevents MPTP-induced Parkinsonism in Baboons. Nat Med. 1996;2(9):1017–1021. doi: 10.1038/nm0996-1017. [DOI] [PubMed] [Google Scholar]
- 8.Huang Z, Huang PL, Panahian N, Dalkara T, Fishman MC, Moskowitz MA. Effects of Cerebral Ischemia in Mice Deficient in Neuronal Nitric Oxide Synthase. Science. 1994;265(5180):1883–1885. doi: 10.1126/science.7522345. [DOI] [PubMed] [Google Scholar]
- 9.(a) Ferriero DM, Holtzman DM, Black SM, Sheldon RA. Neonatal Mice Lacking Neuronal Nitric Oxide Synthase Are Less Vulnerable to Hypoxic-Ischemic Injury. Neurobiol Dis. 1996;3(1):64–71. doi: 10.1006/nbdi.1996.0006. [DOI] [PubMed] [Google Scholar]; (b) Tan S, Bose R, Derrick M. Hypoxia-Ischemia in Fetal Rabbit Brain Increases Reactive Nitrogen Species Production: Quantitative Estimation of Nitrotyrosine. Free Radic Biol Med. 2001;30(9):1045–1051. doi: 10.1016/s0891-5849(01)00499-3. [DOI] [PubMed] [Google Scholar]; (c) Derrick M, Drobyshevsky A, Ji X, Tan S. A Model of Cerebral Palsy from Fetal Hypoxia-Ischemia. Stroke. 2007;38(2 Suppl):731–735. doi: 10.1161/01.STR.0000251445.94697.64. [DOI] [PubMed] [Google Scholar]; (d) Derrick M, Luo NL, Bregman JC, Jilling T, Ji X, Fisher K, Gladson CL, Beardsley DJ, Murdoch G, Back SA, Tan S. Preterm Fetal Hypoxia-Ischemia Causes Hypertonia and Motor Deficits in the Neonatal Rabbit: A Model for Human Cerebral Palsy? J Neurosci. 2004;24(1):24–34. doi: 10.1523/JNEUROSCI.2816-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Calabrese V, Mancuso C, Calvani M, Rizzarelli E, Butterfield DA, Stella AM. Nitric Oxide in the Central Nervous System: Neuroprotection versus Neurotoxicity. Nat Rev Neurosci. 2007;8(10):766–775. doi: 10.1038/nrn2214. [DOI] [PubMed] [Google Scholar]; (b) Contestabile A, Monti B, Contestabile A, Ciani E. Brain Nitric Oxide and Its Dual Role in Neurodegeneration/Neuroprotection: Understanding Molecular Mechanisms to Devise Drug Approaches. Curr Med Chem. 2003;10(20):2147–2174. doi: 10.2174/0929867033456792. [DOI] [PubMed] [Google Scholar]; (c) Estevez AG, Jordan J. Nitric Oxide and Superoxide, A Deadly Cocktail. Ann New York Acad Sci. 2002;962:207–211. doi: 10.1111/j.1749-6632.2002.tb04069.x. [DOI] [PubMed] [Google Scholar]
- 11.(a) Marletta MA. Approaches toward Selective Inhibition of Nitric Oxide Synthase. J Med Chem. 1994;37(13):1899–1907. doi: 10.1021/jm00039a001. [DOI] [PubMed] [Google Scholar]; (b) Vallance P, Leiper J. Blocking NO Synthesis: How, Where and Why? Nat Rev Drug Discov. 2002;1(12):939–950. doi: 10.1038/nrd960. [DOI] [PubMed] [Google Scholar]
- 12.(a) Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, Fishman MC. Hypertension in Mice Lacking the Gene for Endothelial Nitric Oxide Synthase. Nature. 1995;377(6546):239–242. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]; (b) Endres M, Laufs U, Liao JK, Moskowitz MA. Targeting eNOS for Stroke Protection. Trends Neurosci. 2004;27(5):283–289. doi: 10.1016/j.tins.2004.03.009. [DOI] [PubMed] [Google Scholar]; (c) Braam B, Verhaar MC. Understanding eNOS for Pharmacological Modulation of Endothelial Function: A Translational View. Curr Pharm Des. 2007;13(17):1727–1740. doi: 10.2174/138161207780831275. [DOI] [PubMed] [Google Scholar]
- 13.Kim SF, Huri DA, Snyder SH. Inducible Nitric Oxide Synthase Binds, S-Nitrosylates, and Activates Cyclooxygenase-2. Science. 2005;310(5756):1966–1970. doi: 10.1126/science.1119407. [DOI] [PubMed] [Google Scholar]
- 14.Wilcock DM, Lewis MR, Van Nostrand WE, Davis J, Previti ML, Gharkholonarehe N, Vitek MP, Colton CA. Progression of Amyloid Pathology to Alzheimer’s Disease Pathology in an Amyloid Precursor Protein Transgenic Mouse Model by Removal of Nitric Oxide Synthase 2. J Neurosci. 2008;28(7):1537–1545. doi: 10.1523/JNEUROSCI.5066-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Salerno L, Sorrenti V, Di Giacomo C, Romeo G, Siracusa MA. Progress in the Development of Selective Nitric Oxide Synthase (NOS) Inhibitors. Curr Pharm Des. 2002;8(3):177–200. doi: 10.2174/1381612023396375. [DOI] [PubMed] [Google Scholar]; (b) Erdal EP, Litzinger EA, Seo J, Zhu Y, Ji HB, SR Selective Neuronal Nitric Oxide Synthase Inhibitors. Curr Top Med Chem. 2005;5(7):603–624. doi: 10.2174/1568026054679317. [DOI] [PubMed] [Google Scholar]; (c) Tafi A, Angeli L, Venturini G, Travagli M, Corelli F, Botta M. Computational Studies of Competitive Inhibitors of Nitric Oxide Synthase (NOS) Enzymes: Towards the Development of Powerful and Isoform-Selective Inhibitors. Curr Med Chem. 2006;13(16):1929–1946. doi: 10.2174/092986706777585031. [DOI] [PubMed] [Google Scholar]
- 16.Garcin ED, Arvai AS, Rosenfeld RJ, Kroeger MD, Crane BR, Andersson G, Andrews G, Hamley PJ, Mallinder PR, Nicholls DJ, St-Gallay SA, Tinker AC, Gensmantel NP, Mete A, Cheshire DR, Connolly S, Stuehr DJ, Åberg A, Wallace AV, Tainer JA, Getzoff ED. Anchored Plasticity Opens Doors for Selective Inhibitor Design in Nitric Oxide Synthase. Nat Chem Biol. 2008;4(11):700–707. doi: 10.1038/nchembio.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Crane BR, Arvai AS, Ghosh DK, Wu C, Getzoff ED, Stuehr DJ, Tainer JA. Structure of Nitric Oxide Synthase Oxygenase Dimer with Pterin and Substrate. Science. 1998;279(5359):2121–2126. doi: 10.1126/science.279.5359.2121. [DOI] [PubMed] [Google Scholar]; (b) Raman CS, Li H, Martásek P, Kral V, Masters BS, Poulos TL. Crystal Structure of Constitutive Endothelial Nitric Oxide Synthase: A Paradigm for Pterin Function Involving a Novel Metal Center. Cell. 1998;95(7):939–950. doi: 10.1016/s0092-8674(00)81718-3. [DOI] [PubMed] [Google Scholar]; (c) Fischmann TO, Hruza A, Niu XD, Fossetta JD, Lunn CA, Dolphin E, Prongay AJ, Reichert P, Lundell DJ, Narula SK, Weber PC. Structural Characterization of Nitric Oxide Synthase Isoforms Reveals Striking Active-Site Conservation. Nat Struct Biol. 1999;6(3):233–242. doi: 10.1038/6675. [DOI] [PubMed] [Google Scholar]; (d) Li H, Shimizu H, Flinspach M, Jamal J, Yang W, Xian M, Cai T, Wen EZ, Jia Q, Wang PG, Poulos TL. The Novel Binding Mode of N-Alkyl-N′-Hydroxyguanidine to Neuronal Nitric Oxide Synthase Provides Mechanistic Insights into NO Biosynthesis. Biochemistry. 2002;41(47):13868–13875. doi: 10.1021/bi020417c. [DOI] [PubMed] [Google Scholar]
- 18.Silverman RB. Design of Selective Neuronal Nitric Oxide Synthase Inhibitors for the Prevention and Treatment of Neurodegenerative Diseases. Acc Chem Res. 2009;42(3):439–451. doi: 10.1021/ar800201v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Huang H, Martásek P, Roman LJ, Masters BS, Silverman RBN. ω-Nitroarginine-Containing Dipeptide Amides. Potent and Highly Selective Inhibitors of Neuronal Nitric Oxide Synthase. J Med Chem. 1999;42(16):3147–3153. doi: 10.1021/jm990111c. [DOI] [PubMed] [Google Scholar]; (b) Hah J-M, Roman LJ, Martásek P, Silverman RB. Reduced Amide Bond Peptidomimetics. (4S)-N-(4-Amino-5-[Aminoakyl]Aminopentyl)-N′-Nitroguanidines, Potent and Highly Selective Inhibitors of Neuronal Nitric Oxide Synthase. J Med Chem. 2001;44(16):2667–2670. doi: 10.1021/jm0101491. [DOI] [PubMed] [Google Scholar]; (c) Hah JM, Martásek P, Roman LJ, Silverman RB. Aromatic Reduced Amide Bond Peptidomimetics as Selective Inhibitors of Neuronal Nitric Oxide Synthase. J Med Chem. 2003;46(9):1661–1669. doi: 10.1021/jm0202932. [DOI] [PubMed] [Google Scholar]; (d) Gómez-Vidal JA, Martásek P, Roman LJ, Silverman RB. Potent and Selective Conformationally Restricted Neuronal Nitric Oxide Synthase Inhibitors. J Med Chem. 2004;47(3):703–710. doi: 10.1021/jm030297m. [DOI] [PubMed] [Google Scholar]; (e) Ji H, Gómez-Vidal JA, Martásek P, Roman LJ, Silverman RB. Confomationally Restricted Dipeptide Amide as Potent and Selective Neuronal Nitric Oxide Synthase Inhibitors. J Med Chem. 2006;49(21):6254–6263. doi: 10.1021/jm0604124. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Seo J, Igarashi J, Li H, Martasek P, Roman LJ, Poulos TL, Silverman RB. Structure-Based Design and Synthesis of Nω-Nitro-L-Arginine-Containing Peptidomimetics as Selective Inhibitors of Neuronal Nitric Oxide Synthase. Displacement of the Heme Structural Water. J Med Chem. 2007;50(9):2089–2099. doi: 10.1021/jm061305c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Flinspach ML, Li H, Jamal J, Yang W, Huang H, Hah JM, Gómez-Vidal JA, Litzinger EA, Silverman RB, Poulos TL. Structural Basis for Dipeptide Amide Isoform-Selective Inhibition of Neuronal Nitric Oxide Synthase. Nat Struct Mol Biol. 2004;11(1):54–59. doi: 10.1038/nsmb704. [DOI] [PubMed] [Google Scholar]; (b) Flinspach M, Li H, Jamal J, Yang W, Huang H, Silverman RB, Poulos TL. Structures of the Neuronal and Endothelial Nitric Oxide Synthase Heme Domain with D-Nitroarginine-Containing Dipeptide Inhibitors Bound. Biochemistry. 2004;43(18):5181–5187. doi: 10.1021/bi0361867. [DOI] [PubMed] [Google Scholar]; (c) Li H, Flinspach ML, Igarashi J, Jamal J, Yang W, Gómez-Vidal JA, Litzinger EA, Huang H, Erdal EP, Silverman RB, Poulos TL. Exploring the Binding Conformations of Bulkier Dipeptide Amide Inhibitors in Constitutive Nitric Oxide Synthases. Biochemistry. 2005;44(46):15222–15229. doi: 10.1021/bi0513610. [DOI] [PubMed] [Google Scholar]
- 21.Ji H, Li H, Flinspach M, Poulos TL, Silverman RB. Computer Modeling of Selective Regions in the Active Site of Nitric Oxide Synthases: Implication for the Design of Isoform-Selective Inhibitors. J Med Chem. 2003;46(26):5700–5711. doi: 10.1021/jm030301u. [DOI] [PubMed] [Google Scholar]
- 22.Ji H, Stanton BZ, Igarashi J, Li H, Martásek P, Roman LJ, Poulos TL, Silverman RB. Minimal Pharmacophoric Elements and Fragment Hopping, An Approach Directed at Molecular Diversity and Isozyme Selectivity. Design of Selective Neuronal Nitric Oxide Synthase Inhibitors. J Am Chem Soc. 2008;130(12):3900–3914. doi: 10.1021/ja0772041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ji H, Li H, Martásek P, Roman LJ, Poulos TL, Silverman RB. Discovery of Highly Potent and Selective Inhibitors of Neuronal Nitric Oxide Synthase by Fragment Hopping. J Med Chem. 2009;52(3):779–797. doi: 10.1021/jm801220a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lawton GR, Ralay Ranaivo H, Chico LK, Ji H, Xue F, Martásek P, Roman LJ, Watterson DM, Silverman RB. Analogues of 2-Aminopyridine-Based Selective Inhibitors of Neuronal Nitric Oxide Synthase with Increased Bioavailability. Bioorg Med Chem. 2009;17(6):2371–2380. doi: 10.1016/j.bmc.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ji H, Tan S, Igarashi J, Li H, Derrick M, Martásek P, Roman LJ, Vásquez-Vivar J, Poulos TL, Silverman RB. Selective Neuronal Nitric Oxide Synthase Inhibitors and the Prevention of Cerebral Palsy. Ann Neurol. 2009;65(2):209–217. doi: 10.1002/ana.21555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Delker SL, Ji H, Li H, Jamal J, Fang J, Xue F, Silverman RB, Poulos TL. Unexpected Binding Modes of Nitric Oxide Synthase Inhibitors Effective in the Prevention of a Cerebral Palsy Phenotype in an Animal Model. J Am Chem Soc. 2010;132(15):5437–5442. doi: 10.1021/ja910228a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Igarashi J, Li H, Jamal J, Ji H, Fang J, Lawton GR, Silverman RB, Poulos TL. Crystal Structures of Constitutive Nitric Oxide Synthases in Complex with De Novo Designed Inhibitors. J Med Chem. 2009;52(7):2060–2066. doi: 10.1021/jm900007a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Faraci WS, Nagel AA, Verdries KA, Vincent LA, Xu H, Nichols LE, Labasi JM, Salter ED, Pettipher ER. 2-Amino-4-methylpyridine as a Potent Inhibitor of Inducible NO Synthase Activity in vitro and in vivo. Br J Pharmacol. 1996;119(6):1101–1108. doi: 10.1111/j.1476-5381.1996.tb16010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated Docking using a Lamarckian Genetic Algorithm and an Empirical Binding Free Energy Function. J Comp Chem. 1998;19(14):1639–1662. [Google Scholar]
- 30.Lawton GR, Ji H, Silverman RB. Remote Protection Prevents Unwanted Cyclizations with 2-Aminopyridines. Tetrahedron Lett. 2006;47(34):6113–6115. [Google Scholar]
- 31.Ertl P, Rohde B, Selzer P. Fast Calculation of Molecular Polar Surface Area as a Sum of Fragment-Based Contributions and Its Application to the Prediction of Drug Transport Properties. J Med Chem. 2002;43(20):3714–3717. doi: 10.1021/jm000942e. [DOI] [PubMed] [Google Scholar]
- 32.Insight II 2000. Accelrys Inc; 10188 Telesis Court, Suite 100, San Diego, CA 92121, Phone: (858) 799–5000 Fax: (858)799–5100: http://www.accelrys.com/ [Google Scholar]
- 33.SYBYL 6.8. Tripos, Inc; 1699 South Hanley Road St. Louis, MO 63144–2319 Phone: (800) 323–2960. Fax: (314) 647–9241: http://www.tripos.com/ [Google Scholar]
- 34.GRID 22. Molecular Discovery; Via Stoppani, 38, 06087 – Ponte San Giovanni – PG, ITALY. Telephone and fax: +39–075–397411: ( http://www.moldiscovery.com/) [Google Scholar]
- 35.Hevel JM, White KA, Marletta M. A Purification of the Inducible Murine Macrophage Nitric Oxide Synthase. J Biol Chem. 1991;266(34):22789–22791. [PubMed] [Google Scholar]
- 36.Roman LJ, Sheta EA, Martásek P, Gross SS, Liu Q, Masters BSS. High-Level Expression of Functional Rat Neuronal Nitric Oxide Synthase in Escherichia coli. Proc Natl Acad Sci USA. 1995;92(18):8428–8432. doi: 10.1073/pnas.92.18.8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gerber NC, Ortiz de Montellano PR. Neuronal Nitric Oxide Synthase: Expression in Escherichia coli, Irreversible Inhibition by Phenyldiazene, and Active Site Topology. J Biol Chem. 1995;270(30):17791–17796. doi: 10.1074/jbc.270.30.17791. [DOI] [PubMed] [Google Scholar]
- 38.Martasek P, Liu Q, Roman LJ, Gross SS, Sessa WC, Masters BSS. Characterization of Bovine Endothelial Nitric Oxide Synthase Expressed in Escherichia coli. Biochem Biophys Res Commun. 1996;219(2):359–365. doi: 10.1006/bbrc.1996.0238. [DOI] [PubMed] [Google Scholar]
- 39.Hevel JM, Marletta MA. Nitric Oxide Synthase Assays. Methods Enzymol. 1994;233:250–258. doi: 10.1016/s0076-6879(94)33028-x. [DOI] [PubMed] [Google Scholar]
- 40.Segel IH. Enzyme Kinetics. John Wiley and Sons; NewYork: 1975. p. 105. [Google Scholar]
- 41.Li H, Igarashi J, Jamal J, Yang W, Poulos TL. Structural Studies of Constitutive Nitric Oxide Synthases with Diatomic Ligands Bound. J Biol Inorg Chem. 2006;11(6):753–768. doi: 10.1007/s00775-006-0123-8. [DOI] [PubMed] [Google Scholar]
- 42.Lowe PN, Smith D, Stammers DK, Riveros-Moreno V, Moncada S, Charles I, Boyhan A. Identification of the Domains of Neuronal Nitric Oxide Synthase by Limited Proteolysis. Biochem J. 1996;314(Pt 1):55–62. doi: 10.1042/bj3140055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McPhillips TM, McPhillips SE, Chiu HJ, Cohen AE, Deacon AM, Ellis PJ, Garman E, Gonzalez A, Sauter NK, Phizackerley RP, Soltis SM, Kuhn P. Blu-Ice and the Distributed Control System: Software for Data Acquisition and Instrument Control at Macromolecular Crystallography Beamlines. J Synchrotron Radiat. 2002;9(Pt 6):401–406. doi: 10.1107/s0909049502015170. [DOI] [PubMed] [Google Scholar]
- 44.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 45.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR System: A New Software Suite for Macromolecular Structure Determination. Acta Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 46.Jones TA, Zou JY, Cowan SW, Kjeldgaarrd M. Improved Methods for Building Models in Electron Density and the Location of Errors in These Models. Acta Cryst. 1991;A47(2):110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 47.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 48.Murshudov GN, Vagin AA, Dodson EJ. Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Cryst. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 49.Winn MD, Isupov MN, Murshudov GN. Use of TLS Parameters to Model Anisotropic Displacements in Macromolecular Refinement. Acta Cryst. 2001;D57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.