

Abstract
Aromatic interactions are well-known players in molecular recognition but their catalytic role in biological systems is less documented. Here, we report that a conserved aromatic stacking interaction between dUTPase and its nucleotide substrate largely contributes to the stabilization of the associative type transition state of the nucleotide hydrolysis reaction. The effect of the aromatic stacking on catalysis is peculiar in that uracil, the aromatic moiety influenced by the aromatic interaction is relatively distant from the site of hydrolysis at the alpha-phosphate group. Using crystallographic, kinetics, optical spectroscopy and thermodynamics calculation approaches we delineate a possible mechanism by which rate acceleration is achieved through the remote π–π interaction. The abundance of similarly positioned aromatic interactions in various nucleotide hydrolyzing enzymes (e.g. most families of ATPases) raises the possibility of the reported phenomenon being a general component of the enzymatic catalysis of phosphate ester hydrolysis.
INTRODUCTION
The prevalence of aromatic interactions in the conformation control of macromolecules is widely accepted (1) and studied in a number of biological systems [e.g. DNA double helix (2), ribonucleoproteins (3), protein folding (4)]. In the reported cases, the contribution of intra- or inter-molecular aromatic stacking (also termed as π–π interactions) to structural stabilization is comparable in strength to hydrogen bonding (3). The role of π–π interactions in enzymatic catalysis, however, receives less attention mainly restricted to studies of flavoenzymes (5) and of the N-glycosidic bond cleavage by a nucleoside hydrolase (6). These two examples are comparable in that the interacting aromatic ring is bonded (covalently or non-covalently) to the chemical reaction center. In redox reactions catalyzed by flavoenzymes, the aromatic ring of the electron acceptor flavin cofactor is the catalytic reaction center that is directly affected by a stacked aromatic amino acid residue resulting in a decreased reduction potential (7). In the second example, the investigated nucleoside hydrolase uses aromatic stacking for efficient protonation of the leaving purine base (6).
By examining the structures of nucleotide hydrolyzing enzymes in the PDB database, it appears that aromatic residues frequently reside in their active sites (8) (Figure 1A) with reported roles mostly in substrate binding [ABC transporters (9), kinesins (10), kinases (11)]. The role of these interactions in catalysis has not been addressed and may be counterintuitive given the relatively large physical and chemical distances between the site of hydrolysis and the aromatic ring of the nucleotide. We present data on the catalytic role of a π–π interaction between the enzyme dUTPase and its nucleotide substrate in a hydrolysis reaction that occurs between the α and β phosphate groups. dUTPase is a ubiquitous enzyme that hydrolyzes dUTP into the dTTP precursor dUMP and pyrophosphate, thus preventing potentially fatal uracil incorporation into DNA (12). Homotrimeric dUTPases contain a conserved aromatic residue in their active sites which is stacked over the uracil ring in all substrate-containing complete dUTPase crystal structures (12). This aromatic interaction was attributed a role in substrate binding and possibly product release purely on the basis of structural considerations (13).
Figure 1.

Structural aspects of the enzyme–substrate π−π interaction in dUTPase and in other nucleotide hydrolases. (A) The π−π interactions between enzyme and substrate in representatives of various nucleotide hydrolase families [PDB IDs 2HQU (13), 2NCD (29) and 1XEF (30)]. (B) Structural superimposition of dUTPase active sites conferring different aromatic amino acids [Phe, His, Trp in PDB IDs 2HQU (13), 2PY4 (16), 3HZA, respectively]. (C) Superimposition of the newly acquired mutant structures with wild-type mtDUT. Grey and black arrows point to water molecules found in both mtDUT and mtDUTH145A but not in mtDUTH145W or only in mtDUTH145A, respectively. D83 is the catalytic residue that polarizes the nucleophile attacking water molecule (Wcat) (25). (D) Same view of the mtDUTH145A active site colored by B factors. Note the relatively low mobility of the Trp-replacing waters.
To pursue this hypothesis, we created mutations at the conserved aromatic site in the human and Mycobacterium tuberculosis (MT) dUTPase (hDUTF158W, hDUTF158A, mtDUTH145W, mtDUTH145A) and performed crystallographic, kinetic and spectroscopic experiments using these mutants to reveal the effect of the change/loss of the aromatic interaction on the enzymatic cycle. Contrary to the expectations, we show that elimination of the aromatic interaction only slightly affected substrate binding while it specifically decreased the rate constant of the chemical step resulting in an overall 100-fold decrease in the catalytic efficiency.
MATERIALS AND METHODS
Proteins were expressed and purified as described previously [human dUTPase (hDUT) (14), MT dUTPase (mtDUT) (15)]. Site-directed mutagenesis was performed by the QuikChange method (Stratagene) and verified by sequencing of both strands. Mutagen forward and reverse primers were 5′-ggggttcaggaggtgctggttccactgg-3′ and 5′-ccagtggaaccagcacctcctgaacccc-3′ for hDUTF158A and 5′-ggcgacggtggcgcgggttcctccggc-3′ and 5′-ggcggaggaacccgcgccaccgtcgcc-3′ for mtDUTH145A. Protein concentration was measured using the Bradford method (Bio-Rad Protein Assay) or by UV absorbance (λ280 = 10555 M−1cm−1 for hDUT and for hDUTF158A, 16055 M−1cm−1 for hDUTF158W, 8480 M−1cm−1 for mtDUTH145W and 2980 M−1cm−1 for mtDUTH145A) and is given in monomers. Proteins were dialyzed into 20 mM HEPES pH 7.5 buffer, also containing 100 mM NaCl, 5 mM MgCl2 and 1 mM DTT. This dialysis buffer was used for further measurements (unless otherwise stated). Electrophoresis and chromatography reagents were from Bio-Rad and Qiagen, molecular biology products were from New England Biolabs or Fermentas. Other chemicals were from Sigma-Aldrich.
Crystallization
The mtDUTH145W and mtDUTH145A mutants were crystallized as described for the wild-type enzyme using the hanging drop method (16). The 3 mg ml−1 dUTPase and 1.25 mM α,β-imido-dUTP was mixed with the reservoir solution containing 50 mM Tris–HCl pH 7.5, 10 mM MgCl2, 1.20–1.75 M ammonium sulphate and 10% glycerol in a 1:1 ratio.
Data collection and structure determination
Complete high resolution (1.2Å for mtDUTH145W and 1.25Å for mtDUTH145A) crystallographic data sets from well diffracting crystals were recorded at the EMBL-beamline X12 of DESY (Hamburg) at λ = 0.9786Å or 0.9769Å wavelength. Data reduction was performed using the XDS and XSCALE (17) programs and the CCP4 program suite (18). Both structures of the mutant: α,β-imido-dUTP:Mg2+ complexes were determined by molecular replacement [MOLREP (19)] using the truncated wild-type mtDUT structure (PDB ID: 2PY4) as a model without ligands. Model building was carried out using the Coot program (20). For atomic resolution refinement, Shelxl from the Shelx-97 program package was used (21). A 5% random subset of the data set was computed for cross-validation throughout the refinement, resulting in Rfree-values. Ramachandran statistics (percentage of all residues): most favored/additionally allowed/generously allowed = 89.9/10.1/0 for mtDUTH145W and = 92.1/7.9/0 for mtDUTH145A. Atomic coordinates and structure factor data have been deposited in the Protein Data Bank with the accession codes 3HZA and 3LOJ.
Steady-state colorimetric dUTPase assay
Protons released in the dUTPase reaction were detected by a phenol red indicator assay described in Vertessy et al. (22) in 1mM Hepes, pH 7.5 buffer containing 100 mM KCl, 40 μM phenol red (Merck) and 5 mM MgCl2. A Specord 200 (Analytic Jena, Germany) spectrophotometer and 10-mm path length thermostatted cuvettes were used at 20°C. Absorbance was recorded at 559 nm. The Michaelis–Menten equation was fitted to the steady-state curves using Origin 7.5 (OriginLab Corp., Northampton, MA, USA).
Stopped-flow experiments
Measurements were done using an SX-20 (Applied Photophysics, UK) stopped-flow apparatus. In the absorbance stopped-flow setup, an assay buffer containing 100 µM phenol red indicator and 1 mM HEPES pH 7.5 provided optimal monitoring of dUTP hydrolysis. To avoid mixing artifacts, the enzyme was dialyzed in the assay buffer prior to the measurements. Active site titrations were carried out to assess the active protein fraction (>90% in each protein preparation). The presented single turnover (STO) curves were measured upon mixing 10 µM dUTP with 35 µM enzyme at 20°C. The measurement in the fluorescence setup was carried out exactly as described in (23). Time courses were analyzed using the curve fitting software provided with the stopped-flow apparatus or by Origin 7.5.
‘Quench flow’ experiments were carried out using an RQF-3 (KinTek Corp., Austin, TX, USA) quench-flow apparatus as described in Toth et al. (23). To obtain the presented curve, the reaction of 25-µM protein with 17.5-µM γ32P-dUTP was monitored at 20°C. The resulting 32PPi product was quantified in water using a Wallac 1409 Liquid Scintillation Counter (PerkinElmer, Inc.).
Circular dichroism intensity titrations
CD spectra were recorded at 20°C on a JASCO 720 spectropolarimeter using a 1-mm path length cuvette. The 50-µM protein was titrated by step-wise addition of the non-hydrolysable substrate analogue α,β-imido-dUTP (dUPNPP, purchased from Jena Bioscience, Germany) in a buffer containing 10 mM potassium-phosphate pH 7.5, and 1 mM MgCl2. A spectrum between λ = 250–290 nm was recorded at each nucleotide concentration. Differential curves were obtained by subtracting the signal of dUPNPP alone from that of the corresponding complex. Differential ellipticity at λmax = 269 nm was plotted against the dUPNPP concentration to obtain the binding curves. The following quadratic equation was fitted to the experimental curves:
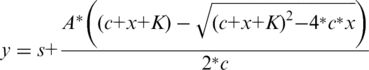 |
s = y at c = 0; A = amplitude; c = protein concentration; K = Kd.
Fluorescence spectroscopy
Maximal fluorescence quenches of the various enzyme–ligand complexes were measured in a Jobin Yvon Spex Fluoromax-3 spectrofluorometer at 20°C, with excitation at 295 nm (slit 1 nm) and emission at 347 (slit 5 nm). The saturating concentrations of ligands (4 mM dUTP, 100 µM dUPNPP, 500 µM dUMP) were added to 4 μM protein.
Statistical analysis
All measurements were carried out at least three times. Error bars represent standard deviations. In case no error bars are shown, a representative curve is displayed while the summary table (Table 1) shows the relevant standard deviations of a certain parameter obtained from several different measurements. In the stopped-flow experiments, typically 5–8 traces were collected and averaged.
Table 1.
Kinetic and substrate binding parameters of human dUTPase mutants
| kcat (s−1) | kH (s−1) | kP (s−1) | KM (µM) | Kd,dUPNPP (µM) | kcat/KM (M−1*s−1) | |
|---|---|---|---|---|---|---|
| hDUTWT | 5.8 ± 0.5 | 8.0 ± 2 | 1.0 ± 0.4 | 2.0 ± 1 | 5.8*106 | |
| hDUTF158W | 6.8 ± 2a | 5.5 ± 2.5a | 6.5 ± 0.1a | 3.6 ± 2a | 1.5 ± 1 | 1.9*106 |
| hDUTF158A | 0.32 ± 0.1 | 0.13 ± 0.01 | 0.25 ± 0.05 | 5.2 ± 0.4 | 4.8 ± 2 | 6.2*104 |
kH: observed rate constant of the hydrolysis step.
kP: observed rate constant of proton release.
Errors represent standard deviations of three independent measurements. In case fitting was carried out to data points with appended standard deviations, error propagation was calculated by the fitting software.
aToth et al. (23).
RESULTS
Full conservation of the aromatic residue within the active site of dUTPases
To assess the degree of conservation of the aromatic residue located in the C-terminal segment of the dUTPase subunit, we performed a database search (p-blast) using the human enzyme as query. Following an alignment of the first 500 dUTPase sequences (Supplementary Figure S1), we analyzed the amino acid distribution in the position that engages in a non-parallel face-to-face offset stack with the uracil ring of the substrate (Figure 1B and C) in all available complete crystal structures. As a result, we counted 96.4% Phe, 1.8% His, 1.6% Tyr and 0.2% Trp. It is of interest that the most frequent Phe is non-polar and relatively small.
The crystal structure of mtDUTH145W suggests a high degree of conformational conservation of the stacking interaction
We chose two enzymes from two different species for our investigations for the following reasons (i) they represent two different aromatic residues at the critical position (Phe in human and His in MT dUTPase); (ii) they share a high degree of sequence and structural similarity (Supplementary Figure S2A); (iii) a detailed enzymatic mechanism of the human enzyme is available (23); and (iv) the MT dUTPase as well as its mutants are readily crystallizable (16,24). Accordingly, we could obtain well diffracting crystals of the mtDUTH145W mutant in complex with the slowly hydrolysable (25) substrate analog α,β-imido-dUTP (dUPNPP). We determined its structure to 1.20 Å resolution (deposited in the PDB as 3HZA, Supplementary Table S1). Superimposition of the hDUT, mtDUT and the novel mtDUTH145W structures reveals that not only the aromatic residue is conserved but so is its conformation relative to the substrate (Figure 1B). Furthermore, three atoms of each aromatic ring (γ, δ1, ε1) can be superimposed despite the different dimensions of the rings (Figure 1B). The perfect conservation of the aromatic planes and the inter-planar angle among the three different structures implies that fine-tuning of the geometry and thus the potential energy of the π–π interaction is of significance (26).
The loss of the aromatic interaction does not perturb the overall structure and active site architecture of dUTPase
We aimed to investigate the consequences of abolishing the stacking interaction between uracil and Phe/His/Trp on the catalytic properties of dUTPase, we therefore engineered the hDUTF158A and mtDUTH145A mutants. Such a radical amino acid replacement is generally seen as the potential source of unspecific and unwanted effects on protein folding and/or activity. We therefore tried to crystallize the alanine mutants and succeeded to do so in the case of mtDUTH145A. The collected data set allowed us to solve the structure of the mutant in complex with dUPNPP at 1.25 Å resolution (deposited in the PDB as 3LOJ, Supplementary Table S1). Superimposition of the mtDUTH145A with the wild-type and mtDUTH145W dUTPase structures indicates that loss of the aromatic residue did not perturb the overall structure or the mode of substrate binding (Supplementary Figure S2B). The considerably high resolution of our dUTPase 3D structures (mtDUT, mtDUTH145W and mtDUTH145A) allowed an in-depth analysis of the water network within the active site. Figure 1C shows water molecules appearing upon loss of the aromatic elements. The replacing well-defined water network (c.f. B factors in Figure 1D) occupies approximately the space of an indole ring and integrates well into the H-bonding network observed in the wild-type crystal. The catalytic water molecule (25) as well as the rest of the conserved residues and metal cofactor are equally positioned in all mutants (Figure 1C) which suggests that any activity loss in the mtDUTH145A is not a result of disordering the interaction network of the catalytic site (Supplementary Figure S2). The modularity of the water network occupying the space of the aromatic rings explains the exchangeability of Phe/Trp/His/Tyr residues appearing in various dUTPase sequences (Supplementary Figure S1).
The loss of the aromatic interaction results in a decreased steady-state dUTPase activity
We have previously shown that introduction of a Trp residue to the aromatic site results in a wild-type enzymatic behavior (14,23). To evaluate the consequences of the loss of the aromatic residue to the enzymatic cycle, we measured the activity of hDUTF158A (Figure 2A) and mtDUTH145A (Figure 2B) taking advantage of the protons released in the hydrolysis reaction (‘Materials and Methods’ section). The maximal steady-state activity of the hDUTF158A mutant decreased to 0.3 s−1 compared to the wild-type (5.8 s−1), while the Michaelis constant (KM) increased from around 1 µM to 5 µM (Table 1). For mtDUT, we obtained a similar 20-fold decrease in kcat (from 3.1 s−1 to 0.16 s−1) and a small increase in the KM (from 0.46 µM to 0.56 µM, Figure 2B).
Figure 2.

Loss of the aromatic interaction brings about the same reduction in kcat in the mycobacterial as in the human enzyme. (A and B) Michaelis–Menten curves of hDUT versus hDUTF158A (steady-state parameters in Table 1) and of mtDUT versus mtDUTH145A, respectively. (B) We evaluated the enzymatic activity of the mycobacterial dUTPase mutant in addition to the human one since our structural data was obtained using this readily crystallizable isoform. The rectangular hyperbolic fit to the Michaelis–Menten curves shown yielded kcat = 3.1 ± 0.06, KM = 0.46 ± 0.2 µM and kcat = 0.16 ± 0.001, KM = 0.56 ± 0.1 µM for mtDUT and mtDUTH145A, respectively.
A hindered chemical step is responsible for the decreased activity in the hDUTF158A mutant
In previous kinetic investigations of the dUTPase enzymatic cycle we established several distinguishable kinetic reaction steps of which the hydrolysis event (or chemical step) proved to be rate-limiting (23). To measure the rate constant of hydrolysis, time courses of STO dUTPase reactions were recorded using stopped-flow (Figure 3A) and quench-flow (Figure 3B) techniques. The enzyme concentration was set high (‘Materials and Methods’ section) so that substrate binding is not rate-limiting and is quantitative. In such conditions, single exponential fit to the proton release STO curves (black line in Figure 3A) selectively reported on the proton release and any preceding conformational event coupled to it. Curve fitting yielded kP = 0.25 s−1, comparable to the maximal steady-state rate (Table 1). In order to directly observe the chemical step within the reaction cycle, we used rapid chemical quench and observed the hydrolysis of radioactively labeled dUTP. Single exponential fits to the time points (black lines in Figure 3B) yielded kH = 0.13 s−1 to hDUTF158A and kH = 5.5 s−1 to the wild-type enzyme. Compared with the steady-state rates, it is apparent that the stalled chemical step is responsible for the overall loss of enzymatic activity in the hDUTF158A mutant (Table 1). Also, consistently with our previous observations (23), proton release occurs concomitantly with the process observed in the chemical quench experiments as suggested by the similar rate constants obtained in the proton release and chemical quench experiments (kP ≈ kH, Table 1). Based on the above observations we may argue that loss of the aromatic interaction did not alter the basic enzymatic mechanism but it directly affected the chemical step.
Figure 3.

Proton release and hydrolysis events limit the observed steady-state rates in the hDUTF158A mutant. (A) Time courses of proton release upon mixing 15 µM hDUTF158A with buffer or with sub-stoichiometric (10 µM) dUTP in the stopped-flow. Single exponential fit to the curve yielded kobs = 0.23 ± 0.007 s−1. (B) Time courses of STO quench-flow experiments upon mixing hDUT or hDUTF158A with sub-stoichiometric radioactive dUTP. The inset shows the same on a logarithmic time scale. Calculated rate constants are summarized in Table 1.
Substrate binding is only slightly affected by the abolition of enzyme-substrate stacking
As was mentioned previously, the most acknowledged role of aromatic interactions lies in molecular recognition and thus in modulating the affinity of interacting partners. We investigated the substrate binding equilibrium of hDUTF158W and hDUTF158A using the circular dichroism signal of the enzyme–substrate complex (Supplementary Figure S3). Saturation curves of dUPNPP binding to hDUT, hDUTF158W and hDUTF158A (Figure 4) yielded dissociation constants of 2, 1.5 and 4.8 µM for hDUT, hDUTF158W and hDUTF158A, respectively (Table 1). This implies that changing one aromatic side chain to another does not affect the substrate binding affinity of dUTPase, while loss of the aromatic residue results in a 3-fold affinity decrease only. This phenomenon may be explained by earlier suggestions that the high selectivity and substrate specificity of dUTPase is provided by accommodation of the uridine moiety into a conserved β-hairpin via H-bonds (13). This binding site is apparently independent of the protein segment conferring the aromatic residue involved in the π–π interaction with the uracil ring.
Figure 4.

The effect of the loss of the aromatic interaction on substrate binding was investigated by the titration of 50 µM protein with the substrate-analog dUPNPP using the CD signal of the enzyme–substrate complex. Smooth lines represent quadratic fits to the data which yielded Kd values indicated in Table 1.
DISCUSSION
As a summary of the above presented results, we found that (i) the geometry of the face-to-face offset stacking interaction between the substrate nucleotide and dUTPase is conserved; (ii) the aromatic residue involved in this interaction is interchangeable with another aromatic residue without structural and/or enzymatic alterations; (iii) loss of the π–π interaction does not affect the ground-state active-site conformation in the alanine mutant; and (iv) results in a significant 20–30-fold decrease in the rate constant of the chemical step. Of the known kinetic steps, the present paper focuses on the chemical step as we obtained proof of its rate-limitation by performing STO experiments in increasing concentration regimes until the rates were unchanged. As to products release, we could not measure the rate constant of dUMP dissociation from the enzyme–products complex in lack of a detectable signal in hDUTF158A. However, even if the loss of aromatic interaction affects the otherwise fast [≈1000 s−1 (23)] dUMP dissociation, it is probably not manifested in the steady-state rate.
The relative energy states of the π−π interaction during the course of the reaction is of significance in order to understand the contribution it makes to the acceleration of the chemical event. We have three independent sets of evidences that help compiling a comprehensive picture of the behavior of the stacking interaction along the reaction trajectory.
First, we have to consider the energy contribution of the aromatic interaction to the ground state enzyme–substrate complex. The small increase in Kd, dUPNPP (Table 1) in the hDUTF158A mutant indicates that the aromatic interaction slightly stabilizes the enzyme–substrate complex. The geometry of the Phe relative to uracil would suggest a weakly repulsive interaction in a Phe–Phe model system (26) nevertheless, uracil is a heterocycle thus it may induce electrostatic polarization [dipole magnitude = 5.4 D (27)] in its interacting partner and such an effect may favor the observed geometry in the Phe–uracil interaction.
A second piece of relevant information arises from the calculation of the incremental activation energy (ΔΔG‡) upon loss of the aromatic interaction. Using the Eyring–Polanyi equation [Equation (1)] and the measured rate constants for the chemical step, we calculated the ΔΔG‡ to be 1.9 kcal mol−1 for hDUTF158A. This value corresponds to the energy of a typical aromatic interaction (11) and considering that no structural changes could be detected in the mutants, we may argue that the calculated ΔΔG‡ in the largest part is specifically attributable to the loss of the aromatic interaction. If we calculate the incremental Gibbs free energy [ΔΔGb, Equation (2)] we obtain a higher 2.6 kcal mol−1 value due to the fact that KM is also somewhat affected.
The third line of evidence is yielded by the intrinsic fluorescence change observed during the course of the reaction. The aromatic stacking between the uracil ring of the ligand and the single Trp residue in hDUTF158W and mtDUTH145W reports on the ligand-bound state of the active site with a characteristic fluorescence quench [Figure 5A and (23)]. The fluorescence quench is relatively small when the product dUMP is bound while it is much larger upon binding to the slowly hydrolysable substrate analog dUPNPP (note that the extent of dUPNPP hydrolysis is negligible during such an experiment). The largest quench is observed in an actively cycling enzyme (i.e. in the presence of dUTP). Since the rate-limiting step of the enzymatic cycle is the hydrolysis step itself and the subsequent product release is fast, in the presence of large amounts of dUTP most of the enzyme population will be in a hydrolysis-competent (close-to-transition or pre-hydrolysis) state. Therefore, the signal difference between dUPNPP and dUTP may lie in the fact that dUPNPP cannot efficiently induce the hydrolysis-competent conformation. It also suggests that going through the transition state (TS) results in further fluorescence quench probably owing to an enhanced aromatic interaction between the substrate and the enzyme. In a representative STO measurement shown in Figure 5B [c.f. Toth et al. (23)], the rate constant of fluorescence recovery, i.e. the exit from the low fluorescence conformational state, is identical with that measured for the hydrolysis step in the chemical quench experiments (Figure 3B). This also implies that the most highly quenched fluorescence enzyme–ligand conformation is related to the hydrolysis event.
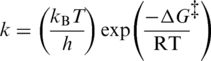 |
(1) |
| (2) |
 |
Figure 5.

The Trp fluorescence of hDUTF158W and mtDUTH145W reflects the relative energy changes of the π–π interaction during the enzymatic cycle. (A) Maximal fluorescence changes upon substrate or product binding to the Trp-bearing active site of mtDUTH145W (white bars) and of hDUTF158W (grey bars). The aromatic stacking between the uracil ring of the ligand and the Trp residue conveys a characteristic fluorescence quench with similar ligand-dependent tendency in both enzymes [human dUTPase data from (23)]. The largest quench is observed in an actively cycling enzyme (i.e. in the presence of dUTP). (B) Fluorescence changes on a rapid logarithmic time base in a STO stopped-flow experiment presented above. Similar fluorescence behavior has been reported for the human dUTPase (23), hence we only show that of the mtDUTH145W here. Upon mixing dUTPase with dUTP, a large and rapid fluorescence quench is observed which reflect substrate binding and subsequent conformational changes of the active site. Fluorescence then recovers to its near starting level (depending on the concentration of dUMP present in the solution). The rate constant of fluorescence recovery, i.e. the exit from the low fluorescence conformational state, is identical in a STO case with that of the hydrolysis step (23).
As schematically presented in Figure 6, the hydrolysis reaction most probably occurs through an associative mechanism (25) in which the TS complex is electron rich, particularly in the proximity of the α-phosphate. Based upon the above supports, weak stabilization of the reactants complex and increased stabilization of the TS seems to offer a plausible explanation for the rate acceleration brought about by the aromatic stacking interaction. As dUTPase does not go through large conformational changes during its enzymatic cycle, its catalytic effect is likely due to long-range electrostatic stabilization and/or by geometry optimization of the TS. This report shows that one of the important contributions to this electrostatic effect comes from the investigated aromatic interaction.
Figure 6.

Schematic representation of the associative-type hydrolysis reaction catalyzed by dUTPase is shown in this figure. Stick models are based on the structures 2HQU and 1SEH conferring the ligands dUPNPP and dUMP, respectively. The TS is most probably pentacovalent and electron rich (14,25).
Our results are in good agreement with a paper by Kaukinen et al. (28) that investigated the effect of stacking on self-catalyzed phosphodiester bond hydrolysis in linear single-stranded nucleic acid polymers. Their associative-type reaction mechanism and the TS geometry are comparable to the one we investigated. They found that enhancement of base stacking during the course of the reaction was an important rate accelerating and TS stabilizing factor. The stabilization energy was calculated to be 0.72–2.8 kcal mol−1. Importantly, they also found that strong base–base interactions in the initial state considerably retarded the reaction due to ground state stabilization. The slightly unfavorable geometry of the Phe–uracil interaction in the initial substrate-bound state of our system may efficiently be tuned up by increasing the lateral offset of the two aromatic rings (26). This can be brought about by movement of the aromatic residue in its plane to the direction of the δ1 atom (c.f. Figure 1B) without steric hindrance. The concept of stacking enhancement during the course of the dUTPase reaction is consistent with all of our results and calculations. This could, in fact, be a rather general mode of rate acceleration by electrostatic TS stabilization.
ACCESSION NUMBERS
3HZA, 3LOJ.
FUNDING
US National Institutes of Health (grant number 1R01TW008130-01); Howard Hughes Medical Institutes (grant number 55000342); Hungarian Scientific Research Funds (grant numbers PD72008, CK-78646, K68229, K72973, NI68466); National Office for Research and Technology, Hungary (grant number JÁP_TSZ_071128_TB_INTER); EU FP6 (grant numbers: SPINE2c LSHG-CT-2006-031220, TEACH-SG LSSG-CT-2007-037198). Funding for open access charge: National Institutes of Health (grant number 1R01TW008130-01).
Conflict of interest statement. None declared.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We thank Anna Lopata and Imre Zagyva for protein production and crystallization. We appreciate the support from Paul Tucker and Santosh Panjikar beamline scientists at the synchrotron beamline X12 at DESY/EMBL-Hamburg during diffraction data collection.
REFERENCES
- 1.Meyer EA, Castellano RK, Diederich F. Interactions with aromatic rings in chemical and biological recognition. Angew. Chem. Int. Ed. Engl. 2003;42:1210–1250. doi: 10.1002/anie.200390319. [DOI] [PubMed] [Google Scholar]
- 2.Hobza P, Sponer J. Structure, energetics, and dynamics of the nucleic Acid base pairs: nonempirical ab initio calculations. Chem. Rev. 1999;99:3247–3276. doi: 10.1021/cr9800255. [DOI] [PubMed] [Google Scholar]
- 3.Blakaj DM, McConnell KJ, Beveridge DL, Baranger AM. Molecular dynamics and thermodynamics of protein-RNA interactions: mutation of a conserved aromatic residue modifies stacking interactions and structural adaptation in the U1A-stem loop 2 RNA complex. J. Am. Chem. Soc. 2001;123:2548–2551. doi: 10.1021/ja005538j. [DOI] [PubMed] [Google Scholar]
- 4.Hughes RM, Waters ML. Model systems for beta-hairpins and beta-sheets. Curr. Opin. Struct. Biol. 2006;16:514–524. doi: 10.1016/j.sbi.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 5.Zhou Z, Swenson RP. The cumulative electrostatic effect of aromatic stacking interactions and the negative electrostatic environment of the flavin mononucleotide binding site is a major determinant of the reduction potential for the flavodoxin from Desulfovibrio vulgaris [Hildenborough] Biochemistry. 1996;35:15980–15988. doi: 10.1021/bi962124n. [DOI] [PubMed] [Google Scholar]
- 6.Versees W, Loverix S, Vandemeulebroucke A, Geerlings P, Steyaert J. Leaving group activation by aromatic stacking: an alternative to general acid catalysis. J. Mol. Biol. 2004;338:1–6. doi: 10.1016/j.jmb.2004.02.049. [DOI] [PubMed] [Google Scholar]
- 7.Swenson RP, Krey GD. Site-directed mutagenesis of tyrosine-98 in the flavodoxin from Desulfovibrio vulgaris (Hildenborough): regulation of oxidation-reduction properties of the bound FMN cofactor by aromatic, solvent, and electrostatic interactions. Biochemistry. 1994;33:8505–8514. doi: 10.1021/bi00194a015. [DOI] [PubMed] [Google Scholar]
- 8.Mao L, Wang Y, Liu Y, Hu X. Molecular determinants for ATP-binding in proteins: a data mining and quantum chemical analysis. J. Mol. Biol. 2004;336:787–807. doi: 10.1016/j.jmb.2003.12.056. [DOI] [PubMed] [Google Scholar]
- 9.Guo X, Chen X, Weber IT, Harrison RW, Tai PC. Molecular basis for differential nucleotide binding of the nucleotide-binding domain of ABC-transporter CvaB. Biochemistry. 2006;45:14473–14480. doi: 10.1021/bi061506i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rickert KW, Schaber M, Torrent M, Neilson LA, Tasber ES, Garbaccio R, Coleman PJ, Harvey D, Zhang Y, Yang Y, et al. Discovery and biochemical characterization of selective ATP competitive inhibitors of the human mitotic kinesin KSP. Arch. Biochem. Biophys. 2008;469:220–231. doi: 10.1016/j.abb.2007.10.016. [DOI] [PubMed] [Google Scholar]
- 11.Boehr DD, Farley AR, Wright GD, Cox JR. Analysis of the pi-pi stacking interactions between the aminoglycoside antibiotic kinase APH(3′)-IIIa and its nucleotide ligands. Chem. Biol. 2002;9:1209–1217. doi: 10.1016/s1074-5521(02)00245-4. [DOI] [PubMed] [Google Scholar]
- 12.Vertessy BG, Toth J. Keeping uracil out of DNA: physiological role, structure and catalytic mechanism of dUTPases. Acc. Chem. Res. 2009;42:97–106. doi: 10.1021/ar800114w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mol CD, Harris JM, McIntosh EM, Tainer JA. Human dUTP pyrophosphatase: uracil recognition by a beta hairpin and active sites formed by three separate subunits. Structure. 1996;4:1077–1092. doi: 10.1016/s0969-2126(96)00114-1. [DOI] [PubMed] [Google Scholar]
- 14.Varga B, Barabas O, Kovari J, Toth J, Hunyadi-Gulyas E, Klement E, Medzihradszky KF, Tolgyesi F, Fidy J, Vertessy BG. Active site closure facilitates juxtaposition of reactant atoms for initiation of catalysis by human dUTPase. FEBS Lett. 2007;581:4783–4788. doi: 10.1016/j.febslet.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 15.Varga B, Migliardo F, Takacs E, Vertessy B, Magazù S. Experimental study on dUTPase-inhibitor candidate and dUTPase/disaccharide mixtures by PCS and ENS. J. Mol. Struct. 2008;886:128–135. [Google Scholar]
- 16.Varga B, Barabas O, Takacs E, Nagy N, Nagy P, Vertessy BG. Active site of mycobacterial dUTPase: structural characteristics and a built-in sensor. Biochem. Biophys. Res. Commun. 2008;373:8–13. doi: 10.1016/j.bbrc.2008.05.130. [DOI] [PubMed] [Google Scholar]
- 17.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Cryst. 1993;26:795–800. [Google Scholar]
- 18.CCP4. The CCP4 suite. Programs for protein crystallography. Acta. Crystallogr. D Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 19.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 1997;30:1022–1025. [Google Scholar]
- 20.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta. Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 21.Sheldrick GM. A short history of SHELX. Acta. Crystallogr. A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 22.Vertessy BG, Persson R, Rosengren AM, Zeppezauer M, Nyman PO. Specific derivatization of the active site tyrosine in dUTPase perturbs ligand binding to the active site. Biochem. Biophys. Res. Commun. 1996;219:294–300. doi: 10.1006/bbrc.1996.0226. [DOI] [PubMed] [Google Scholar]
- 23.Toth J, Varga B, Kovacs M, Malnasi-Csizmadia A, Vertessy BG. Kinetic mechanism of human dUTPase, an essential nucleotide pyrophosphatase enzyme. J. Biol. Chem. 2007;282:33572–33582. doi: 10.1074/jbc.M706230200. [DOI] [PubMed] [Google Scholar]
- 24.Chan S, Segelke B, Lekin T, Krupka H, Cho US, Kim MY, So M, Kim CY, Naranjo CM, Rogers YC, et al. Crystal structure of the Mycobacterium tuberculosis dUTPase: insights into the catalytic mechanism. J. Mol. Biol. 2004;341:503–517. doi: 10.1016/j.jmb.2004.06.028. [DOI] [PubMed] [Google Scholar]
- 25.Barabas O, Pongracz V, Kovari J, Wilmanns M, Vertessy BG. Structural insights into the catalytic mechanism of phosphate ester hydrolysis by dUTPase. J. Biol. Chem. 2004;279:42907–42915. doi: 10.1074/jbc.M406135200. [DOI] [PubMed] [Google Scholar]
- 26.Hunter CA, Singh J, Thornton JM. Pi-pi interactions: the geometry and energetics of phenylalanine-phenylalanine interactions in proteins. J. Mol. Biol. 1991;218:837–846. doi: 10.1016/0022-2836(91)90271-7. [DOI] [PubMed] [Google Scholar]
- 27.Kulakowska I, Geller M, Lesyng B, Wierzchowski KL. Dipole moments of 2,4-diketopyrimidines. II. Uracil, thymine and their derivatives. Biochim. Biophys. Acta. 1974;361:119–130. [PubMed] [Google Scholar]
- 28.Kaukinen U, Lonnberg H, Perakyla M. Stabilisation of the transition state of phosphodiester bond cleavage within linear single-stranded oligoribonucleotides. Org. Biomol. Chem. 2004;2:66–73. doi: 10.1039/b309828a. [DOI] [PubMed] [Google Scholar]
- 29.Sablin EP, Case RB, Dai SC, Hart CL, Ruby A, Vale RD, Fletterick RJ. Direction determination in the minus-end-directed kinesin motor ncd. Nature. 1998;395:813–816. doi: 10.1038/27463. [DOI] [PubMed] [Google Scholar]
- 30.Zaitseva J, Jenewein S, Jumpertz T, Holland IB, Schmitt L. H662 is the linchpin of ATP hydrolysis in the nucleotide-binding domain of the ABC transporter HlyB. EMBO J. 2005;24:1901–1910. doi: 10.1038/sj.emboj.7600657. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.