


Abstract
Duplicated pseudogenes in the human genome are disabled copies of functioning parent genes. They result from block duplication events occurring throughout evolutionary history. Relatively recent duplications (with sequence similarity ≥90% and length ≥1 kb) are termed segmental duplications (SDs); here, we analyze the interrelationship of SDs and pseudogenes. We present a decision-tree approach to classify pseudogenes based on their (and their parents’) characteristics in relation to SDs. The classification identifies 140 novel pseudogenes and makes possible improved annotation for the 3172 pseudogenes located in SDs. In particular, it reveals that many pseudogenes in SDs likely did not arise directly from parent genes, but are the result of a multi-step process. In these cases, the initial duplication or retrotransposition of a parent gene gives rise to a ‘parent pseudogene’, followed by further duplication creating duplicated–duplicated or duplicated–processed pseudogenes, respectively. Moreover, we can precisely identify these parent pseudogenes by overlap with ancestral SD loci. Finally, a comparison of nucleotide substitutions per site in a pseudogene with its surrounding SD region allows us to estimate the time difference between duplication and disablement events, and this suggests that most duplicated pseudogenes in SDs were likely disabled around the time of the original duplication.
INTRODUCTION
It is known that genomic duplications provide opportunities for functional divergence of genes and contribute to the complexity of genomes. In fact, Ohno proposed that gene duplication is the driving force for the generation of new genes (1). Relatively new duplications in the genome with nucleotide sequence similarity of at least 90% and at least 1 kb in length cover ∼5% of the genome and are termed segmental duplications (SDs) (2). Assuming neutral rate of divergence, they are associated with duplications that likely happened in the last ∼40 million years of human evolution which corresponds roughly to the divergence between New and Old World monkeys (3). Several processes such as non-allelic homologous recombination (NAHR) and non-homologous end joining (NHEJ) are thought to be involved in the origin and propagation of SDs, and a strong association of Alu repeat elements and SDs has been reported (4,5). Genomic structural variations in humans such as insertions, deletions and inversions exhibit a 4- to 12-fold greater frequency near SD sites (4) and most SDs themselves exhibit copy number variations within humans although their duplication status seems fixed, i.e. they are duplicated at least once in all individuals resulting in a minimum copy number of two (6). SDs form the cores of genetic diversity which can potentially lead to evolution of new gene functions and diseased states (4). Moreover, many of the genes found in SDs exhibit strong signatures of positive selection and are enriched for functional categories related to primate and human adaptive evolution (4).
If a duplicating region of the genome contains a gene, the new sequence may contain a paralog performing the same function as the original gene or a new function. Duplicated pseudogenes are formed when the new sequence undergoes mutations that result in the loss of original function. In contrast, processed pseudogenes arise from retrotransposition events [typically indicated by the lack of introns and presence of a 3′-poly(A) tail] (7,8). They usually lack promoter sequences, and hence are considered dead-on-arrival. It has been estimated that a processed pseudogene loses about half of its DNA in ∼400 million years (9). The slow deletion rate ensures that the human genome contains many pseudogenes which can be used to study the evolution of the genome. Although pseudogenes are assumed to have lost the original coding functions of their parent genes due to the presence of disablements such as premature stop codons or frameshift mutations, recent studies indicate that they might have some regulatory roles (10). It has been reported that there is a higher enrichment of pseudogenes than genes in SDs since SDs comprise ∼5% of the genome and contain ∼36.8% of human pseudogenes and ∼17.8% of human genes (2,11). It has also been reported that the number of duplicated pseudogenes in pseudogene families shows a strong positive correlation to the number of pseudogenes located in SDs (12). However, a detailed comprehensive analysis dealing with the integration of these genomic elements has not been reported so far.
In an attempt to obtain insight about various genomic processes and genome evolution, we have performed an integrative analysis of SDs, paralogs and pseudogenes. We present a rigorous scheme of classification of pseudogenes based on their presence in SDs, and then discuss the biological implications of this classification, which leads to an improved annotation of pseudogenes in SDs. For instance, the presence of processed pseudogenes arising from same parent genes in SDs indicates that they might be the result of duplication events of other processed pseudogenes preceded by an initial retrotransposition event, and hence we call them ‘duplicated–processed’ pseudogenes. Another example where the integration of SDs and pseudogenes provides insight about genome evolution is the presence of pseudogenes and their parent genes in SD pairs (duplicated segments). For this category of pseudogenes, we compare the number of nucleotide substitutions in pseudogenes (relative to the parent genes) with the number of substitutions in the larger SD regions that contain them. We find that most pseudogenes exhibit similar number of substitutions per site as the larger SD regions containing them, likely indicating duplication immediately followed by disablement and then a neutral rate of nucleotide substitution. Hence, the classification of pseudogenes discussed here sheds light on the various processes that led to their formation thereby enhancing our understanding of the evolution of the genome. The categories of human duplicated and processed pseudogenes based on their presence in SDs are provided in the Supplementary Data and also made available at http://pseudogene.org/sdpgenes. The scheme of classification of human pseudogenes presented here can be extended to pseudogenes of other species as well, in order to compare formation and evolution of pseudogenes amongst different species.
MATERIALS AND METHODS
The SD pairs for NCBI Build 36 of the human genome were obtained from the Human Segmental Duplication Database at http://humanparalogy.gs.washington.edu/build36/build36.html. Pseudogenes were obtained by running PseudoPipe on proteins from Ensembl version 48 (13). Global alignments of the translated pseudogenes and their parent proteins were then obtained using the MSA program (14,15). These alignments were then converted to global nucleotide alignments based on their aligned coordinates and corresponding exonic sequences. Hence, the nucleotide alignments were for coding sequences only and UTRs were not included. The number of nucleotide substitutions per site, K2m, were computed using Kimura’s two parameter model (Equation 1) (16).
| (1) |
where, ti stands for the fraction of transitions, while tv stands for the fraction of transversions.
The gaps in the alignments were ignored for K2m calculation. This model assumes that all the sites in a sequence evolve at the same rate, while taking into account the fact that transitions are generally more frequent than transversions (3). It has also been used previously as a surrogate for evolutionary age of SDs (17) and hence is ideal to compare the substitutions in pseudogenes and SDs.
The coordinates for paralogs for Ensembl version 48 were obtained using BioMart (http://www.ensembl.org/index.html). The exonic sequence alignments for paralogs were obtained in a similar way as for pseudogenes using the MSA program. The ancestral loci for SDs were obtained from Supplementary Data provided by Jiang et al. (17). Since the loci were for hg17, the corresponding coordinates for hg18 were obtained using the liftOver tool at UCSC (http://genome.ucsc.edu/cgi-bin/hgLiftOver). Genes and pseudogenes in unassembled contigs and in mitochondrial DNA were not included in the analysis. Hence, only the genes and pseudogenes on chromosomes 1–22, X and Y were used.
RESULTS
We present the scheme of classification that emerges from the presence of pseudogenes and their parent genes in SDs (Figures 1 and 5), along with a discussion of the biological implications of this classification. The pseudogenes used for the analysis in this article were obtained using the pseudogene identification pipeline PseudoPipe (13). Out of a total of 21 315 pseudogenes (which includes duplicated, processed and fragmented pseudogenes, as well as pseudogenes tagged as possible false positives) we focus on 12 481 duplicated and processed pseudogenes in this article. We find that ∼25.4% of these pseudogenes (3172/12 481) are located in SDs, which enables us to improve their annotation as discussed below. We also discuss the presence of pseudogenes and parent genes on a set of ancestral loci in the genome that gave rise to many SDs. The set of loci identified previously by Jiang et al. (17) were used for this analysis.
Figure 1.

The scheme of classification based on the presence of pseudogenes (ψgenes) and their parent genes in SDs. SDs are shown by grey boxes and rest of the color coding is shown in (A). The various cases are named based on the question at each step where capital letters are used for positive answers and small letters for negative answers. Numbers of pseudogenes under each case are depicted in bold. Case SP from (A) is continued in (B). For question K: Q refers to log10 [K2m(ψgene)/K2m(SD)], with μ ≊ 0 and 2σ ≊ 0.4 from the distribution shown in Figure 2.
Figure 5.

Schematic showing the classification of processed pseudogenes based on their presence in SDs. Symbols used are the same as in Figure 1. Case SP from (A) is continued in (B).
Presence of pseudogenes and their parent genes in SDs
In order to integrate SD data with pseudogenes and infer meaningful relationships, we have developed a decision-tree like approach (Figures 1 and 5). This approach summarizes the various cases of occurrence of pseudogenes and their parent genes in SDs. Each category derives its name from the question asked at each step: capital letters are used for naming cases with positive answers, while small letters are used for negative answers. Whenever the biological implication of the category can be inferred, it is mentioned at the end of the branch. The numbers of events under each category are also shown in Figures 1 and 5 for duplicated and processed pseudogenes, respectively. The list of pseudogenes under each category is provided at http://pseudogene.org/sdpgenes as well as in Supplementary Tables S1–S10 along with a description of each Table (Tables_README.txt).
Broadly speaking, under the decision-tree approach we first check the presence of a pseudogene in SD, followed by queries to check the detailed structure of the set of regions where that segment is duplicated—including the presence of parent gene and ‘sister’ pseudogenes (i.e. pseudogenes from the same parent gene) in that set. This is followed by a comparison of the substitutions in the pseudogene (with respect to the parent) and the SD region that contains it. More specifically, the first question asked is whether a pseudogene is located in an SD (Figures 1A and 5A: question S). If a pseudogene is present in an SD (case S), we check if its parent gene is located in any SD (question P). The cases where a duplicated pseudogene and its parent gene are not in SD pairs [either due to the pseudogene not being in SD (case s) or the parent gene not being in SD (case Sp)] can also be sub-divided based on the sequence identity of exonic (and pseudo-exonic) sequences and length of the sequence (Figure 1A: question E) (Supplementary Tables S1–S4). A sequence identity cut-off of 90% and length cut-off of 1 kb provide hints about selection pressure acting on duplicated pseudogenes as discussed in the following section. For processed pseudogenes, the absence of parent in any SD (case Sp) aids in the identification of ‘duplicated–processed’ pseudogenes, discussed in detail in the sections below (Supplementary Table S8).
Continuing with the case SP (parent is in some SD), we then check if the parent gene and pseudogene are in an SD pair (in other words, if the parent gene is in any of the duplicates of the segment containing the pseudogene) (Figures 1B and 5B: question D). Case SPD branches further into SPDI (Supplementary Tables S5 and S9) and SPDi (Supplementary Tables S6 and S10) depending on the presence or absence of sister pseudogenes in the SD pair (question I). For duplicated pseudogenes (Figure 1B), such instances (SPDI) occur due to the complex architecture of SDs where tandem duplications are often followed by further duplications of the entire segment (2). Similarly, for processed pseudogenes (Figure 5B), case SPDI is the result of ‘tandem’ retrotransposition events (i.e. retrotransposition next to the parent gene) followed by duplication. In such instances, the pseudogene might be the direct result of duplication of a sister pseudogene rather than the parent gene itself, and hence may be called a duplicated–duplicated (Figure 1B) or duplicated–processed (Figure 5B) pseudogene (Supplementary Figure S1). Existing algorithms for whole-genome identification of pseudogenes, including PseudoPipe, rely on the sequence identity of pseudogenes with their parent genes, and hence can not differentiate between the two types of pseudogenes: ones that arise directly from the duplication of parent genes and those that arise from subsequent duplication events of pseudogenes themselves (13,18,19). Pseudogenes arising from the duplication of pseudogenes rather than the parent gene itself can only be identified by aligning the larger segmentally duplicated regions surrounding them. We are especially interested in the differentiation of duplicated–processed pseudogenes from processed pseudogenes since they are the results of different processes: duplication and retrotransposition, respectively. The list of duplicated–processed pseudogenes identified from case Sp as well as case SPDI is provided at http://pseudogene.org/sdpgenes (Supplementary Tables S8 and S9). We discuss case SPDI in detail in the following section entitled ‘Processed pseudogenes’.
We have applied the rigorous scheme of classification discussed above to all duplicated and processed pseudogenes and detailed examples of biological insight obtained from the classification are discussed in the following sections. It is noted that since the case sensitive nomenclature can be a problem for database searching, we have also assigned a node number to each category shown in Supplementary Figure S2 [e.g. Sp(6)].
Duplicated pseudogenes
We find that ∼44.4% of duplicated pseudogenes (corresponding to ∼43.6% of the duplicated-pseudogene sequence) are located completely within SDs. Since SDs contain ∼5% of the genomic sequence (2) but ∼43.6% of duplicated-pseudogene sequence, there is roughly a 9-fold enrichment of duplicated pseudogenes in SDs (P-value <1e−100). Using the scheme of classification correlating pseudogenes and SDs discussed above, insights about the formation and evolution of duplicated pseudogenes are obtained as discussed below. The number of events under each category is mentioned in Figure 1.
Comparison of nucleotide substitutions per site
Pseudogenes are assumed to evolve under neutral selection pressure after they lose their coding ability due to disablements (20). The number of nucleotide substitutions per site in a pseudogene computed from parent-gene–pseudogene alignment provides an estimate of the time of divergence of the parent gene and the pseudogene. The presence of pseudogenes and parent genes in SD pairs provides an opportunity to compare the evolution of pseudogene sequence with respect to the entire SD region that contains it. The number of nucleotide substitutions per site, K2m, were computed using Kimura’s two parameter model (16) (‘Materials and Methods’ section). An equal number of substitutions per site in the pseudogene and the SD region indicate that the time of divergence of parent gene and pseudogene sequence is roughly the same as the time of divergence of the two larger SD regions (Figure 1B: question K). This, in turn, implies that pseudogenization (or disablement) likely occurred at the same time as the duplication of the entire segment, and both the pseudogene and the larger region containing it are likely evolving neutrally. We note that the disablement time of pseudogene can be same as or less than the divergence time of pseudogene (from parent). If the disablement time is less than the divergence time, we expect lower divergence of pseudogene (from parent) than the divergence of the two larger SD regions (assuming a lower than neutral rate of nucleotide substitution in pseudogene before disablement). On the other hand, if the divergence times of pseudogene (from parent) and the two larger SD regions are similar, the disablement time is likely the same as divergence time.
When the pseudogene and parent gene align with each other within the two larger SD regions (case SPDi) (likely corresponding to events where the pseudogene is the direct result of duplication of parent gene) we compare the number of substitutions per site in pseudogenes and SDs. We find that the distribution of Q = log[K2m (ψgene)/K2m (SD)] roughly fits a Gaussian (µ ≊ 0 and σ ≊ 0.2) and most pseudogenes indeed show similar number of substitutions per site as the SD region containing them [Figure 1B: case SPDiK and Figure 2: (−0.4 < Q < 0.4)], indicating that most pseudogenes under this category were disabled around the time of duplication followed by neutral evolution. We note that K2m (ψgene) corresponds to the divergence between the parent gene sequence under purifying selection and pseudogene sequence under neutral selection; while K2m (SD) corresponds to the divergence between the two SD sequences. If both the SD sequences were accumulating mutations and diverging away from each other at neutral rates, we would expect K2m (SD) to be larger than K2m (ψgene) and hence the distribution of Q would be centered at a negative value. However, we find that the distribution is centered close to zero (though it is noted that the mode of the distribution is at a slightly negative value ≊ −0.02 in Figure 2). A possible explanation is that the entire SD region containing the parent gene is also under purifying selection (perhaps due to the presence of promoters and other regulatory sequences), while the pseudogene and SD sequence containing it are evolving at neutral rates. Zhang et al. (21) have shown that SDs containing genes show higher sequence similarity than those without genes and they also postulated that purifying selection acting on the SDs containing genes could be a possible explanation.
Figure 2.

Ratio of nucleotide substitutions per site in pseudogenes and the larger SD regions computed using Kimura’s two-parameter model. The distribution roughly fits a Gaussian, 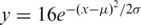 with µ ≊ 0 and σ ≊ 0.2.
with µ ≊ 0 and σ ≊ 0.2.
There are a few discrepant cases where pseudogenes exhibit much more [Figure 1: case SPDikM and Figure 2: (Q > 0.4)] or less [Figure 1: case SPDikm and Figure 2: (Q < −0.4)] substitutions per site than the surrounding SD region. More nucleotide substitutions per site in the pseudogene than the SD region likely indicate either (i) perhaps there is positive selection acting on the pseudogene and hence it shows a higher mutation rate than neutral or (ii) there is negative selection on the larger SD region and hence it shows a lower mutation rate than neutral. Similarly, a lower number of substitutions per site in the pseudogene could indicate either (i) pseudogenes that remained functional and hence under negative selection for some time after the duplication event, therefore accumulating lower substitutions per site than the surrounding SD region or (ii) pseudogenes that are currently under negative selection. Unfortunately, we are unable to distinguish between cases (i) and (ii) with the available data. These cases might be interesting for further experimental studies for a better understanding of evolution of these genomic locations. The list of pseudogenes under these categories is provided at http://pseudogenes.org/sdpgenes, as well as in Supplementary Table S6.
The estimation of time since divergence of two paralogous sequences can be complicated by gene conversion events. Such events shuffle DNA between paralogous regions leading to a reduction in the divergence of the two regions (17,22). In case of gene conversion events between parent gene and pseudogene, the situation can be even more complicated since transfer of DNA from pseudogene to parent gene could be harmful. Indeed, several genetic diseases are known to be caused by gene conversion from pseudogenes (23). Hence, while on one hand, gene conversion between parent gene and pseudogene might have reduced the sequence divergence; on the other hand, the sequence divergence might have been increased in order to avoid gene conversion (22). Thus, gene conversion events could offer another possible explanation for cases where we observe discrepancy between the number of substitutions per site in the pseudogene and the SD region containing it and pseudogene and parent gene are located on the same chromosome.
We note that the events where the pseudogene and parent gene are not in SD pairs even though sequence identity of exonic (and pseudo-exonic or exonic sequence of the pseudogene) sequences is ≥90% as well as the length of the sequence is ≥1kb (Figure 1A: cases sE and SpE) also indicate cases where the pseudogenic sequence exhibits lower substitutions than the surrounding region (Supplementary Tables S1 and S3). In all these cases, inclusion of the intronic sequence in the parent-gene–pseudogene alignment yields a sequence identity <90%, hence indicating that the pseudo-intronic sequence contains more substitutions than the pseudo-exonic sequence (relative to parent gene intronic and exonic sequences, respectively).
In Figures 3 and 4, we show examples of two pseudogenes within larger SDs that likely arose by duplication of a parent gene and a pseudogene, respectively.
Figure 3.

Duplicated pseudogene is the result of duplication of parent gene. Snapshots from UCSC genome browser (32) are shown. Segment on chr6 (top) is duplicated at the segment on chr1(bottom); the pair is marked in both top and bottom snapshots. Orange (seq id>99%), yellow (98%>seq id>99%) and grey (90%>seq id>98%) segments show the segmental duplications track; dark red shows the gene track and light red shows the duplicated pseudogene track. Multiple SD tracks show the complexity of the duplications where different fragments of one region are duplicated at various locations in the genome (some duplicated regions have been removed from the snapshots for clarity).
Figure 4.

Duplicated pseudogene is the result of duplication of another duplicated pseudogene. Intrachromosomal SD pair on chr4 is shown; color scheme is the same as used in Figure 3.
Processed pseudogenes
Retrotransposed or processed pseudogenes are identified by the absence of introns and presence of 3′-poly(A) tail, hence they are likely the result of retrotransposition events rather than duplication events (7,13). However, as discussed earlier, processed pseudogenes could also be the result of duplication of other processed pseudogenes and therefore show absence of introns (termed as duplicated–processed pseudogenes) (24). SDs contain ∼5% of genomic sequence but ∼25.1% processed pseudogenes sequence (corresponding to ∼21.1% processed pseudogenes by number), hence there is roughly 5-fold enrichment of processed pseudogenes in SDs relative to genomic background (P-value <1e−100). Unlike the enrichment of duplicated pseudogenes in SDs, the enrichment of processed pseudogenes in SDs is surprising—the possible reasons for this enrichment are discussed later.
The scheme of classification of processed pseudogenes based on their presence in SDs, analogous to the scheme for duplicated pseudogenes (Figure 1) is shown in Figure 5. The branches corresponding to question E in Figure 1A are eliminated in Figure 5A since unlike duplicated pseudogenes, processed pseudogenes do not contain introns. Case Sp (Figure 5A) and case SPDI (Figure 5B) aid identification of duplicated–processed pseudogenes as discussed earlier. At a first glance, case SPDi in Figure 5B where the SD pair contains only the parent gene and processed pseudogene seems hard to explain, since it would imply retrotransposition and duplication of the parent gene to the same genomic location (where pseudogene is present). However, closer examination indicates that the pseudogenes under this category are indeed duplicated pseudogenes and not processed pseudogenes. We find that most parent genes under this category consist of single exons. Hence, a lack of introns in the pseudogene alignments possibly led to the misannotation of these pseudogenes as processed by the pseudogene identification pipeline (PseudoPipe) (13). A comparison of the nucleotide substitutions per site in these pseudogenes with the entire SD region yields similar results as for duplicated pseudogenes (Supplementary Figure S3).
The ability to distinguish between processed pseudogenes and duplicated–processed pseudogenes is one of the most interesting consequences of the integration of SDs and pseudogenes as discussed above. Among all the processed pseudogenes we are able to identify 307 sets of pseudogenes with each set containing a parent gene, a parent-processed pseudogene and multiple duplicated-processed pseudogenes (Figure 6). The sub-categorization of processed pseudogenes into these two categories should aid correct understanding of pseudogene formation and avoid misinterpretation of pseudogene analyses such as overestimation of the number of retrotransposition events of a parent gene due to the abundance of its processed pseudogenes or underestimation of duplication events of a certain gene family (24,25).
Figure 6.

Schematic showing the formation of a processed pseudogene and subsequent duplicated–processed pseudogenes. The ancestral loci were identified using outgroup mammalian species (macaque, mouse, rat and dog) by Jiang et al. (17). Blue, green and yellow indicate sequence around the SD regions. The ancestral locus (surrounded by blue) shares a larger synteny block with the outgroup species. 959 duplicated-processed pseudogenes corresponding to 307 parent genes are identified. Asterisk denotes that unique parent-processed pseudogenes could be identified only for 158/307 sets.
Identification of parent-processed and duplicated–processed pseudogenes
A schematic for the formation of duplicated-processed pseudogenes, where a retrotransposition event is followed by multiple duplication events (of grey regions) is shown in Figure 6. The SD architecture is very complex since duplicated segments often correspond to multiple prior duplication events (17). The SD data by itself does not offer any information about the directionality of duplication. In an SD pair either of the two regions could be the original segment that duplicated to form the second segment, or they could both be the result of duplication of another paralogous segment which may or may not have been deleted from the genome. Hence, it is hard to distinguish the parent-processed and the subsequent duplicated–processed pseudogenes discussed above. However, a set of ancestral loci have been identified using outgroup mammalian genome sequence data (macaque, mouse, rat and dog) with the assumption that an ancestral locus should share a larger homologous synteny block with an outgroup species than the derived duplicated region (Figure 6) (17). It is noted that this syntenic approach limits the identification of ancestral loci to duplications occurring after the speciation events separating humans and outgroup mammalian genomes used. Based on the overlap of the sister-processed pseudogenes found in SDs (Figure 6) with the ancestral loci obtained by Jiang et al. (17), we are able to identify unique parent-processed pseudogenes for the 158 sets out of the 307 sets discussed above (Figure 6). Hence, we are able to successfully differentiate parent-processed and duplicated-processed pseudogenes for the 158 sets (Supplementary Table S8). For the remaining 149 sets of pseudogenes, the parent-processed pseudogenes cannot be distinguished from the subsequent duplicated–processed pseudogenes due to the lack of information about the ancestral segments and the derived segments.
Identification of novel pseudogenes
In the preceding paragraphs we have focused on the correlation of SDs and pseudogenes based on the presence or absence of pseudogenes in SDs. Similarly, we analyzed all the cases (1174) where coding genes are located in SDs while a paralog or pseudogene is not annotated in the corresponding duplicated segment. It is noted that all the pseudogenes identified by pseudogene identification pipeline PseudoPipe (including duplicated, processed and fragmented pseudogenes, as well as possible false positives) were used for this analysis (13). In these cases, we can identify sequence similar to the parent gene in the duplicated segment using tfasty (26). This step is followed by additional filters including amino acid sequence identity of at least 50%, at least two disablements in the aligned sequence, and zero overlap with any exons. Using these relatively stringent criteria we are able to identify 140 novel pseudogenes that were missed previously by PseudoPipe (Supplementary Table S11).
PseudoPipe finds BLAST hits in the entire genome which go through various filters, for example sufficiently low E-value, to be included or rejected as pseudogene candidates (13). Thus, it is not very surprising that we are able to identify more pseudogenes which were not identified by PseudoPipe, since due to the complexity of the SD regions where smaller duplications can be located within larger ones, a crude method involving BLAST search on the entire genome is likely to miss a few regions. Here, we demonstrate that knowledge of the large duplications in the genome should provide additional information for whole genome pseudogene detection, by increasing confidence in the hits obtained in SD pairs, thereby leading to more sensitive detection of pseudogenes.
Pseudogenes and parent genes on ancestral loci
As discussed in the preceding section and also depicted in Figure 6, a set of ancestral loci have been identified by Jiang et al. using outgroup mammalian genome sequence data. Based on the analysis of all duplicated and processed pseudogenes, as well as their parent genes that lie in SDs and overlap these loci, we find a higher percentage of parent genes of duplicated pseudogenes on these loci compared to both duplicated and processed pseudogenes (Table 1). 158 out of 326 processed pseudogenes on these loci are the parent-processed pseudogenes for the 307 sets of duplicated–processed pseudogenes as discussed above and shown in Figure 6; the remaining 168 pseudogenes could not be assigned uniquely to sets of duplicated–processed pseudogenes. Interestingly, none of the parent genes of processed pseudogenes that are found in SDs lie on ancestral loci. Since duplicated and processed pseudogenes arise from duplication and retrotransposition events, respectively, it is not surprising that the SD loci that underwent multiple duplication events contain a high percentage of parent genes of duplicated pseudogenes (31.3%) compared to the parent genes of processed pseudogenes (0%). The lower percentage of pseudogenes on ancestral loci could indicate that the original segments are more likely to retain the functional genes than the derived segments. We note that since the ancestral loci for the entire SD sequence are not available [they could be identified only for 102.4/152.2 Mb (67.3%) of SD sequence] (17), we are unable to compute a rigorous enrichment score for genes and pseudogenes on these loci relative to the entire SD sequence. However, our observations are in general agreement with those of Zheng who noted that parent loci of SDs contain more genes but fewer pseudogenes compared to the derived loci, although his analysis was restricted to a small set of post-macaque SDs (1646/25 914 pairs of SDs) (11).
Table 1.
Pseudogenes and parent genes that overlap ancestral loci
| Number in SDs | Overlap ancestral loci (%) | |
|---|---|---|
| Duplicated ψgenes | 1019 | 115/1019 (11.3) |
| Parent genes of duplicated ψgenes | 246 | 77/246 (31.3) |
| Processed ψgenes | 2153 | 326/2153 (15.1) |
| Parent genes of processed ψgenes | 312 | 0/312 (0) |
Paralogs located in SDs
Paralogs are duplicated copies of genes that retain their coding functions, although they may display similar or diverged functions as the original genes. The set of paralogs obtained using EnsemblCompara gene trees (27) was used for the analysis discussed here. ∼57.7% of Ensembl genes have paralogs (13 170/22 817) and we find that ∼12% of those (1579/13 170) are located in SDs. Both paralogs and duplicated pseudogenes are the result of duplication events. Hence, it is interesting that SDs contain a higher percentage of disabled copies of genes (∼44.4% of duplicated pseudogenes) than coding copies (∼12% of paralogs). We note that this is likely a consequence of the sequence identity distribution of duplicated pseudogenes (obtained from parent-gene–pseudogene alignments) and paralog pairs, with the mode for pseudogenes located at ∼95% exonic sequence identity and for paralogs at ∼60% (Supplementary Figure S4). However, considering the fact that SDs are highly prone to genomic structural variation (4,6,28), it is still notable that they contain a higher percentage of disabled copies than coding copies.
DISCUSSION
The genomic elements of SDs, paralogs and pseudogenes are the result of duplication events in the genome and hence are closely related. We have performed an integrative analysis of these elements, which provides additional information about the formation of pseudogenes and sheds light on the underlying genomic processes. We present a rigorous scheme of classification based on the presence of pseudogenes in SDs.
It is known that genes are enriched in SDs with ∼17.8% genes located in SDs (11). We find that ∼44.4% duplicated pseudogenes and ∼21.1% processed pseudogenes are located in SDs. The alignments of the SD regions that contain them reveal the ‘true parents’ of the pseudogenes and allow annotation of duplicated–processed pseudogenes: pseudogenes that result from the duplication of other processed pseudogenes. It was reported by Kim et al. (5) that processed pseudogenes show a significant association with SDs and from the presence of highly similar processed pseudogenes at SD junctions, they concluded that processed pseudogenes may have contributed to SD formation in some cases. This might partly explain the observed enrichment of processed pseudogenes in SDs in the current analysis. However, a high enrichment of processed pseudogenes in SDs confirms our view that most processed pseudogenes located in SDs are indeed a result of duplication events—mostly of other processed pseudogenes (duplicated–processed) and in some instances (case SPDi—Figure 5B) of parent genes (misassigned previously as processed pseudogenes).
When the pseudogene and parent gene align with each other within the two larger SD regions (case SPDi) we compare the number of nucleotide substitutions per site in pseudogene and the SD region containing it. This comparison indicates that most pseudogenes were likely disabled at roughly the same time as original duplication and have been evolving under neutral rates of selection since then. We note that this conclusion applies for pseudogenes under this category (case SPDi) even if the pseudogene is a direct result of duplication of another pseudogene. In such cases, the first pseudogene formed by duplication of segment containing parent gene likely started evolving with a neutral rate of nucleotide substitution after the disablement event; followed by a second SD event to give a second pseudogene that continued to evolve neutrally. Hence, similar number of nucleotide substitutions per site in the second pseudogene (relative to the parent) as the larger SD region containing it (relative to the SD region containing parent) indicate that the initial disablement (in the first pseudogene) likely occurred at the same time as the initial duplication event.
We find that even though the enrichment of pseudogenes in SDs is not due to the presence of olfactory receptor (OR) pseudogenes, 98 out of 300 (∼32.7%) OR pseudogenes that were classified as processed pseudogenes by PseudoPipe are found in SDs. ORs form the largest mammalian gene superfamily and it is estimated that ∼63% of them are actually non-functional duplicated pseudogenes (29). OR genes consist of single protein-coding exons and hence the classification of OR pseudogenes into processed and duplicated by PseudoPipe can be tricky. We think that most OR pseudogenes previously classified as processed by PseudoPipe are indeed the result of duplication events and we have now changed their annotation to duplicated pseudogenes. Indeed, 62 out of 98 OR pseudogenes in SDs fall under the case SPDi (Figure 5B) where parent gene and pseudogene align with each other in SD pairs.
We note that although the SD data provides pair-wise information, we extract the entire set of regions where a particular segment is duplicated from this data for our analysis. Additionally, since the SD pair-wise data does not provide information about the directionality of duplications, we use a set of ancestral loci obtained previously in a separate study by Jiang et al. (17). We find that amongst parent genes and pseudogenes that are found in SDs, a higher percentage of parent genes (of duplicated pseudogenes) than pseudogenes are located on these ancestral loci of SDs.
A limitation of our current analysis is its dependence on annotated SDs which correspond to relatively new duplications in the genome (≥90% sequence identity). For instance, some processed pseudogenes which are not located in SDs could be the result of older duplication events of other processed pseudogenes, but we are unable to label them as duplicated–processed based on current analysis. However, we note that the classification scheme presented in this article does not fundamentally rely on SD definition and can be applied with a different set of SDs obtained using a lower sequence similarity criteria. We have demonstrated that this classification scheme enables integration of the knowledge of the entire length of the sequence that was copied during the duplication events (SD regions) with the pseudogene data and helps gain significant additional insight which can not be obtained solely from the sequence homology between the parent genes and pseudogenes.
It is interesting to note that while SDs are hotspots for various kinds of structural variations such as insertions, deletions and inversions (4), both genes and pseudogenes are enriched in these regions. It is possible that the genes and pseudogenes located in SDs are strongly correlated with the variations between individuals. Hence, in the search to find polymorphic genes or polymorphic pseudogenes (genes that are functional in certain populations and rendered non-functional in others) (30), perhaps the genes and pseudogenes located in SD regions would be the best candidates for further investigation. With individual genomics data becoming available at an unprecedented rate, the variability of these pseudogenes in different populations would be the focus of future studies (31).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health and the A.L. William’s Professorships funds. Funding for open access charge: National Institutes of Health.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Gang Fang and Suganthi Balasubramanian for discussion.
REFERENCES
- 1.Ohno S. Evolution by Gene Duplication. Springer, Berlin; 1970. [Google Scholar]
- 2.Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, Adams MD, Myers EW, Li PW, Eichler EE. Recent segmental duplications in the human genome. Science. 2002;297:1003–1007. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- 3.Li W. Molecular Evolution. Sinauer Associates, MA, USA; 1997. [Google Scholar]
- 4.Bailey JA, Eichler EE. Primate segmental duplications: crucibles of evolution, diversity and disease. Nat. Rev. Genet. 2006;7:552–564. doi: 10.1038/nrg1895. [DOI] [PubMed] [Google Scholar]
- 5.Kim PM, Lam HY, Urban AE, Korbel JO, Affourtit J, Grubert F, Chen X, Weissman S, Snyder M, Gerstein MB. Analysis of copy number variants and segmental duplications in the human genome: evidence for a change in the process of formation in recent evolutionary history. Genome Res. 2008;18:1865–1874. doi: 10.1101/gr.081422.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marques-Bonet T, Kidd JM, Ventura M, Graves TA, Cheng Z, Hillier LW, Jiang Z, Baker C, Malfavon-Borja R, Fulton LA, et al. A burst of segmental duplications in the genome of the African great ape ancestor. Nature. 2009;457:877–881. doi: 10.1038/nature07744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mighell AJ, Smith NR, Robinson PA, Markham AF. Vertebrate pseudogenes. FEBS Lett. 2000;468:109–114. doi: 10.1016/s0014-5793(00)01199-6. [DOI] [PubMed] [Google Scholar]
- 8.Harrison PM, Gerstein M. Studying genomes through the aeons: protein families, pseudogenes and proteome evolution. J. Mol. Biol. 2002;318:1155–1174. doi: 10.1016/s0022-2836(02)00109-2. [DOI] [PubMed] [Google Scholar]
- 9.Graur D, Shuali Y, Li WH. Deletions in processed pseudogenes accumulate faster in rodents than in humans. J. Mol. Evol. 1989;28:279–285. doi: 10.1007/BF02103423. [DOI] [PubMed] [Google Scholar]
- 10.Sasidharan R, Gerstein M. Genomics: protein fossils live on as RNA. Nature. 2008;453:729–731. doi: 10.1038/453729a. [DOI] [PubMed] [Google Scholar]
- 11.Zheng D. Asymmetric histone modifications between the original and derived loci of human segmental duplications. Genome Biol. 2008;9:R105. doi: 10.1186/gb-2008-9-7-r105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lam HY, Khurana E, Fang G, Cayting P, Carriero N, Cheung KH, Gerstein MB. Pseudofam: the pseudogene families database. Nucleic Acids Res. 2009;37:D738–D743. doi: 10.1093/nar/gkn758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Z, Carriero N, Zheng D, Karro J, Harrison PM, Gerstein M. PseudoPipe: an automated pseudogene identification pipeline. Bioinformatics. 2006;22:1437–1439. doi: 10.1093/bioinformatics/btl116. [DOI] [PubMed] [Google Scholar]
- 14.Lipman DJ, Altschul SF, Kececioglu JD. A tool for multiple sequence alignment. Proc. Natl Acad. Sci. USA. 1989;86:4412–4415. doi: 10.1073/pnas.86.12.4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gupta SK, Kececioglu JD, Schaffer AA. Improving the practical space and time efficiency of the shortest-paths approach to sum-of-pairs multiple sequence alignment. J. Comput. Biol. 1995;2:459–472. doi: 10.1089/cmb.1995.2.459. [DOI] [PubMed] [Google Scholar]
- 16.Li W, Graur D. Fundamentals of Molecular Evolution. Massachusetts, USA: Sinauer Associates Inc.; 1991. [Google Scholar]
- 17.Jiang Z, Tang H, Ventura M, Cardone MF, Marques-Bonet T, She X, Pevzner PA, Eichler EE. Ancestral reconstruction of segmental duplications reveals punctuated cores of human genome evolution. Nat. Genet. 2007;39:1361–1368. doi: 10.1038/ng.2007.9. [DOI] [PubMed] [Google Scholar]
- 18.Torrents D, Suyama M, Zdobnov E, Bork P. A genome-wide survey of human pseudogenes. Genome Res. 2003;13:2559–2567. doi: 10.1101/gr.1455503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ohshima K, Hattori M, Yada T, Gojobori T, Sakaki Y, Okada N. Whole-genome screening indicates a possible burst of formation of processed pseudogenes and Alu repeats by particular L1 subfamilies in ancestral primates. Genome Biol. 2003;4:R74. doi: 10.1186/gb-2003-4-11-r74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Z, Gerstein M. Patterns of nucleotide substitution, insertion and deletion in the human genome inferred from pseudogenes. Nucleic Acids Res. 2003;31:5338–5348. doi: 10.1093/nar/gkg745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang L, Lu HH, Chung WY, Yang J, Li WH. Patterns of segmental duplication in the human genome. Mol. Biol. Evol. 2005;22:135–141. doi: 10.1093/molbev/msh262. [DOI] [PubMed] [Google Scholar]
- 22.Innan H, Kondrashov F. The evolution of gene duplications: classifying and distinguishing between models. Nat. Rev. Genet. 2010;11:97–108. doi: 10.1038/nrg2689. [DOI] [PubMed] [Google Scholar]
- 23.Bischof JM, Chiang AP, Scheetz TE, Stone EM, Casavant TL, Sheffield VC, Braun TA. Genome-wide identification of pseudogenes capable of disease-causing gene conversion. Hum. Mutat. 2006;27:545–552. doi: 10.1002/humu.20335. [DOI] [PubMed] [Google Scholar]
- 24.Zhang ZD, Cayting P, Weinstock G, Gerstein M. Analysis of nuclear receptor pseudogenes in vertebrates: how the silent tell their stories. Mol. Biol. Evol. 2008;25:131–143. doi: 10.1093/molbev/msm251. [DOI] [PubMed] [Google Scholar]
- 25.Liu YJ, Zheng D, Balasubramanian S, Carriero N, Khurana E, Robilotto R, Gerstein MB. Comprehensive analysis of the pseudogenes of glycolytic enzymes in vertebrates: the anomalously high number of GAPDH pseudogenes highlights a recent burst of retrotrans-positional activity. BMC Genomics. 2009;10:480. doi: 10.1186/1471-2164-10-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pearson WR, Wood T, Zhang Z, Miller W. Comparison of DNA sequences with protein sequences. Genomics. 1997;46:24–36. doi: 10.1006/geno.1997.4995. [DOI] [PubMed] [Google Scholar]
- 27.Vilella AJ, Severin J, Ureta-Vidal A, Heng L, Durbin R, Birney E. EnsemblCompara GeneTrees: Complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 2009;19:327–335. doi: 10.1101/gr.073585.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Korbel JO, Urban AE, Affourtit JP, Godwin B, Grubert F, Simons JF, Kim PM, Palejev D, Carriero NJ, Du L, et al. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318:420–426. doi: 10.1126/science.1149504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Glusman G, Yanai I, Rubin I, Lancet D. The complete human olfactory subgenome. Genome Res. 2001;11:685–702. doi: 10.1101/gr.171001. [DOI] [PubMed] [Google Scholar]
- 30.Zhang ZD, Frankish A, Hunt T, Harrow J, Gerstein M. Identification and analysis of unitary pseudogenes: historic and contemporary gene losses in humans and other primates. Genome Biol. 2010;11:R26. doi: 10.1186/gb-2010-11-3-r26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuehn BM. 1000 Genomes project promises closer look at variation in human genome. JAMA. 2008;300:2715. doi: 10.1001/jama.2008.823. [DOI] [PubMed] [Google Scholar]
- 32.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.