

Abstract
The Baltimore Longitudinal Study of Aging (BLSA) was established in 1958 and is one the oldest prospective studies of aging in the USA and the world. The BLSA is supported by the National Institute of Aging (NIA) and its mission is to learn what happens to people as they get old and how to sort out changes due to aging and from those due to disease or other causes. In 1986, an autopsy program combined with comprehensive neurologic and cognitive evaluations was established in collaboration with the Johns Hopkins University Alzheimer’s Disease Research Center (ADRC). Since then, 211 subjects have undergone autopsy. Here we review the key clinical neuropathological correlations from this autopsy series. The focus is on the morphological and biochemical changes that occur in normal aging, and the early neuropathological changes of neurodegenerative diseases, especially Alzheimer’s disease (AD). We highlight the combined clinical, pathologic, morphometric, and biochemical evidence of asymptomatic AD, a state characterized by normal clinical evaluations in subjects with abundant AD pathology. We conclude that in some individuals, successful cognitive aging results from compensatory mechanisms that occur at the neuronal level (i.e., neuronal hypertrophy and synaptic plasticity) whereas a failure of compensation may culminate in disease.
Keywords: α-synuclein, Alzheimer’s disease, asymptomatic Alzheimer’s disease, amyloid-β, dementia, Parkinson’s disease, stereology, successful aging, tau
INTRODUCTION
The Baltimore Longitudinal Study of Aging (BLSA) was established in 1958 by Dr. Nathan Shock [1]. The overall mission of the BLSA is to learn what happens to people as they get old and how to sort out changes due to aging from those due to disease or other causes. Initial enrollment in the study was limited to men but women have been included since 1978. A total of 3006 subjects have been enrolled over the years. Current enrollment is 1362 subjects of which 35% are women. The racial composition of the study is predominantly non-Hispanic white and the cohort is highly educated (the mean education is approximately 17 years).
The autopsy program of the BLSA, along with enhanced neurologic and cognitive evaluations, were initiated in 1986 through a collaboration between the Johns Hopkins University Alzheimer’s Disease Research Center (ADRC) and the Laboratory of Personality and Cognition at the National Institute on Aging (NIA). Since the inception of the autopsy program, there have been 238 deaths and 211 subjects have undergone autopsy, most of them including complete body autopsies with neuropathological evaluation and banking of fixed and frozen brain tissues. The mean age at death is 86.9 ± 8.2 years (range 57–102), and the mean interval between last evaluation and death is 8.7± 6.7 months.
The clinical pathological investigations of the BLSA focus on the morphological and biochemical changes that occur in normal aging, independent of clinical disease, as well as the early neuropathological changes of neurodegenerative diseases especially Alzheimer’s disease (AD). A guiding principle of these investigations is the notion that understanding changes occurring during the early-stage of a disease process is more likely to shed light on the underlying pathogenesis than studying end-stage pathological changes, which are likely to represent a final common pathway across different forms of cellular damage. In addition, we seek to identify biological processes that discriminate participants with mild underlying AD pathology who develop dementia from those who remain non-demented. Although AD is the most common neurodegenerative pathology in our cohort, it is often associated with other neurological conditions characterized by specific pathologies such as coexisting vascular lesions or Parkinson’s disease (PD). Moreover, there is strong evidence that age-associated brain neuropathological changes often represent the participation of the brain in multisystemic disorders and, therefore, information from brain autopsies is substantially more valuable when it is conducted in individuals who were clinically well characterized prior to death. The large proportion of complete autopsies in the BLSA makes the examination of these interactions possible.
The BLSA autopsy cohort is not fully representative of the population at large because it is predominantly male, white, and most subjects have college or graduate education and good access to medical care. Therefore, we believe all of these factors raise caveats for the interpretation of clinical and pathological findings in this cohort. Currently, the BLSA is enrolling females and minorities in an effort to allay some of the limitations cited above.
CLINICAL AND NEUROPSYCHOLOGICAL EVALUATIONS OF THE BLSA
Baseline evaluations of BLSA participants enrolled in the autopsy study were performed by a neurologist who collected information on history of cerebrovascular disease, focal neurologic abnormalities, and impairment of cognitive or behavioral functions due to secondary causes or medical treatments. After the age of 75 years, physical and cognitive assessments were performed annually. The evaluation included a neuropsychological battery [2], neurologic exam, medication review, and informant- and subject-structured interview. The latter was based on the Clinical Dementia Rating scale [3,4] since 1998 and the Dementia Questionnaire [5] before 1998. All subjects were followed annually and were reviewed at a diagnostic consensus conference if their Blessed Information Memory Concentration score [6] was 3 or greater, if their informant or subject Clinical Dementia Rating score was 0.5 or greater, or if their Dementia Questionnaire was abnormal. All subjects, regardless of the screening tests, were evaluated at a diagnostic conference at the time of entry into the autopsy program and at withdrawal or death. The full panel of neuropsychological diagnostic tests and clinical data were available for review at the diagnostic conference. Diagnosis of dementia was based on Diagnostic and Statistical Manual of Mental Disorders, Revised Third Edition criteria [7]. A diagnosis of cognitive impairment not meeting criteria for dementia, defined as mild cognitive impairment (MCI), was based on the Mayo Clinic criteria [8]. Accordingly, the diagnosis of MCI was given to subjects who had deficits limited to 1 or 2 areas of cognition (usually memory), with preservation of normal activities of daily living compared with other people of similar age.
NEUROPATHOLOGY METHODS AND DIAGNOSTIC CRITERIA
All brains were examined in the Division of Neuropathology of the Johns Hopkins University. After weighing and external examination, the right hemi-brain is cut in 1-cm coronal slabs and a standard set of tissue blocks is removed for overnight fixation in 4% paraformaldehyde or snap freezing. The remaining coronal slabs are frozen on prechilled aluminum plates and then maintained at −80°C. The left hemi-brain is fixed in 10% buffered formaldehyde for at least 2 weeks and then cut coronally. For diagnostic purposes, tissue blocks are dissected from middle frontal gyrus, superior and middle temporal gyri, inferior parietal cortex, occipital cortex, cingulate gyrus, hippocampus, entorhinal cortex, amygdala, thalamus, basal ganglia, midbrain, pons, medulla, and cerebellum (Table 1). For diagnostic purposes, tissue blocks are processed and embedded in paraffin, cut at 10 μm, and stained with hematoxylin and eosin. Selected sections are silver-stained with the Hirano method [9] and immunostained for α-synuclein (BD Transduction Laboratories, Palo Alto, CA; dilution, 1:500) and phosphorylated tau (anti-phosphorylated tau, paired helical filament 1 clone; a gift of Dr. P. Davies, Albert Einstein College of Medicine, Bronx, NY; dilution, 1:100). In selected brains, we prepare fixed tissue sections for stereology. Presently, we are obtaining a set of large tissue blocks containing the entire temporal lobe including hippocampus and entorhinal cortex, and a set from the brainstem and lower diencephalon containing the entire substantia nigra and locus coeruleus.
Table 1.
List of brain regions blocked for histologic examination
| Anatomical region Brodmann area (BA) | Regions assessed for neuritic (CERAD) plaques (silver stains) | Regions assessed for neurofibrillary (Braak) tangles (silver stains) | Regions assessed for α-synuclein lesions | Regions assessed for vascular lesions and other neuropathologies (H&E stains) |
|---|---|---|---|---|
| Middle frontal gyrus BA 46 | √+ | √+ | √ | |
| Superior & middle temporal gyri BA 22 & 21 | √+ | √+ | √ | √ |
| Inferior parietal lobule BA 39 & 40 | √+ | √+ | √ | |
| Occipital cortex BA 17 & 18 | √+ | √+ | √ | |
| Cingulate gyrus BA 24 | √ | √ | ||
| Basal ganglia and basal forebrain | √ | |||
| Entorhinal cortex BA 28 | √+ | √ | √ | |
| Hippocampus | √+ | √ | √ | |
| Thalamus | √ | |||
| Amygdala | √ | √ | √ | |
| Midbrain | √ | √ | √ | |
| Pons | √ | √ | √ | |
| Medulla | √ | |||
| Cerebellum | √ |
The √ symbol indicates the staining of tissue sections from specific regions. Those sections marked with a + are the silver-stained sections [9] used for the assessment of the frequency of neuritic plaques according to CERAD guidelines [10] and the distribution of neurofibrillary tangles according to Braak [11].
The severity of neuritic plaques is assigned a semi-quantitative and age-adjusted score (0, A, B, or C) according to CERAD [10], and the distribution of neurofibrillary tangles is assigned a stage score (0–VI) according to Braak [11] (Table 1). Although immunostains for tau (PHF1) and the amyloid peptide Aβ (6E10, Signet Laboratories) are performed, all Braak and CER-AD staging is based on silver stains. The assessment of Lewy body diseases, specifically idiopathic PD and Dementia with Lewy bodies (DLB), is conducted on both H&E and α-synuclein stained tissue sections. For diagnosis of PD we follow the criteria of the London Brain Bank [12] and for DLB the criteria of the DLB consortium [13].
NEUROPATHOLOGY IN THE BRAIN OF CLINICALLY NORMAL OLDER SUBJECTS
We have taken advantage of the careful clinical characterization of the BLSA participants, as well as of the short interval between the last cognitive evaluation and death, to establish rigorous clinical-pathological correlations. Our first publication compared neuropathology in controls and demented subjects [14] and was based on observations on 22 subjects (69–97 years of age), of whom 15 had normal and stable cognitive performances and seven had dementia of variable severity. In the majority of normal subjects, few or no Aβ deposits or senile plaques were present in the neocortex, but neurofibrillary tangles were consistently found in CA1 of the hippocampus and layer II of the entorhinal cortex. In two normal individuals, the densities of senile plaques were consistent with the diagnosis of possible AD. We proposed that these subjects with normal cognitive states and abundant neocortical senile plaques may represent preclinical AD. We concluded that in the majority of cognitively intact individuals the neocortex remains free of Aβ deposits or senile plaques, even into the tenth decade of life.
NEUROPATHOLOGIC STUDIES OF PRECLINICAL/ASYMPTOMATIC ALZHEIMER’S DISEASE
The diagnostic category of cognitively normal individuals with pathology at autopsy has been termed pre-clinical AD by some investigators [15,16], high pathology controls [17], or high plaque non-demented subjects [18]. A substantial part of our efforts has been devoted to the understanding of the morphological substrate and underlying mechanisms by which some individuals remain cognitively intact in spite of considerable AD pathology. Initially, we defined this condition as preclinical AD, but subsequently we used the term asymptomatic AD (ASYMAD) [19,20], mostly because we do not know a priori whether with longer survival, these subjects would have remained clinically normal or would have eventually progressed to MCI. In the BLSA autopsy series, ASYMAD subjects represent approximately 50% of individuals with preserved cognition beyond 75 years of age. We believe that these participants are extremely important because they may represent a group of individuals resistant to the toxic effects of AD pathology. An important caveat to the interpretation of our findings is that participants in the BLSA cohort are highly educated (mean 17.4 ± 2.3 years), a factor that may contribute to the resistance to the clinical manifestations of the disease [21] or to “brain reserve” [22,23].
SYNAPTIC PROTEINS IN NORMAL AGING AND ALZHEIMER’S DISEASE
Normal cognition and memory as well as neuronal survival depend on synapses (Fig. 1). Regulated release of neurotransmitter-containing vesicles is necessary for normal synaptic function. In biochemical experiments, we found that synaptophysin levels were reduced in the hippocampus (Fig. 1) when comparing definite AD to controls, preclinical/ASYMAD to controls (25%, p < 0.005), and definite AD to preclinical/ASYMAD, but the levels in the entorhinal cortex, occipital cortex, and caudate nucleus were either unchanged or less significantly altered than in hippocampus. Synaptic abnormalities in the hippocampus correlated with the severity of neuropathology and memory deficit in individuals with AD [24]. Only some proteins that control synaptic operation are abnormal in preclinical/ASYMAD and in AD (Fig. 1). These proteins function in the presynaptic terminal by controlling the regulated exocytosis of neurotransmitter packets. In a follow-up experiment we found that synaptic proteins that control neurotransmitter vesicle translocation and priming at the release site of the presynaptic nerve terminal are more vulnerable than proteins involved in the vesicle exocytosis at the cell membrane [25,26]. Specific proteins, most notably synaptobrevin and synaptotagmin, are reduced in the hippocampus of individuals with preclinical/ASYMAD and in AD individuals who had moderate to severe abnormalities in memory (Fig. 1F). It appears that these proteins begin to be lost in individuals in the early stages of AD who do not yet have significant cognitive impairment, but as the synaptic defects progressively become more severe, individuals manifest severe memory impairment. Abnormalities in presynaptic proteins that regulate neurotransmitter release may be an early pathological process in the development of AD (Fig. 1).
Fig. 1.
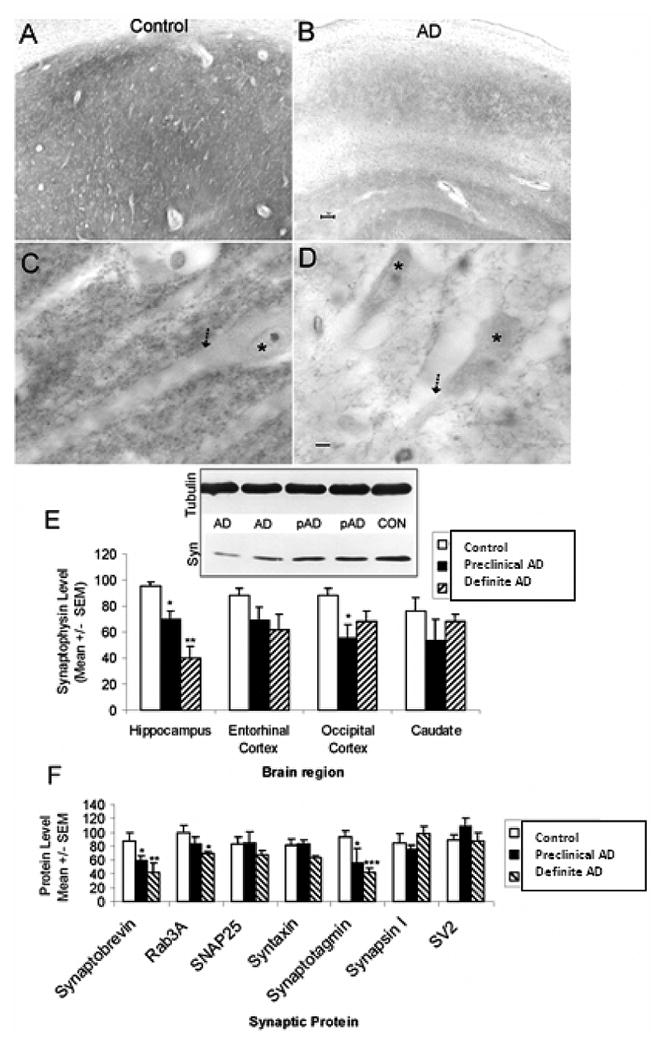
Abnormalities occur in selective synaptic proteins in preclinical/asymptomatic AD and AD brain. (A-D) Immunohistochemical localization (brown labeling) of the presynaptic vesicle protein synaptophysin in human control (A,C) and AD (B,D) CA1 region of hippocampus with cresyl violet counterstaining. In controls, the neuropil of CA1 is highly enriched in synaptophysin immunoreactivity (A) which can be seen as discrete small particle-like boutons that decorate the surfaces of dendrites (arrow) of CA1 pyramidal neurons (asterisk). In the AD brain, the CA1 region is severely depleted of synaptophysin immunoreactivity (B) and the labeling of synaptophysin-positive presynaptic terminals on dendrites (arrow) of CA1 neurons (asterisk) and in the surrounding neuropil is reduced markedly. Scale bars = 10 μm. (E) Graph showing the results of immunoblot experiments of synaptophysin protein levels in different brain regions of control, preclinical/asymptomatic AD (pAD), and definite AD cases. A representative blot for synaptophysin (Syn) is shown with β-tubulin immunoreactivity serving as a protein loading control. Significant differences in synaptophysin levels from control are indicated by single asterisk (p < 0.05) or double asterisk (p < 0.0001). For more about this study see Sze et al., 1997. (F) Graph showing the result of immunoblot experiments of different presynaptic vesicle exocytosis proteins (synaptobrevin, Rab3A, synaptotagmin, synapsin I, and SV2) and presynaptic plasma membrane proteins (SNAP25 and syntaxin) in CA1 hippocampus of control, preclinical/asymptomatic AD, and definite AD cases. Significant differences in protein levels from control are indicated by single asterisk (p < 0.05), double asterisk (p < 0.01) or triple asterisk (p < 0.005). For more about this study see Sze et al., 2000 [25]. (Colours are visible in the electronic version of the article at www.iospress.nl.)
MICROGLIA IN PRECLINICAL/ASYMPTOMATIC ALZHEIMER’S DISEASE
Since inflammatory changes are an important component of the pathological cascade of AD, we examined the association of immune reactive cells in senile plaques and cognitive decline [27]. To this end, we examined the associations of postmortem neocortical immunoreactivities for microglia, astrocytes, Aβ, and Tau with cognitive changes in clinically characterized subjects with pathological diagnoses of AD dementia [28], asymptomatic AD [29], and age-matched controls [30]. By measuring the fractional area (FA) of immunoreactivity, we found that Aβ deposits appear early in the pathogenesis of AD, but cannot account for cognitive decline. We found a significant increase in the FA of microglia in ASYMAD cases (clinically non-demented) compared to controls (p < 0.05) and in the FA of astrocytes in definite AD (demented) compared to ASYMAD (p < 0.01). Tau immunoreactivity was observed only in the neuropil of definite AD cases (p < 0.001). The significant increase in microglia in ASYM AD compared to controls suggests that activation of microglia occurs in the early pathogenesis of AD and might reflect an underlying synaptic pathology. The significant association between astrocytic reaction and dementia, however, suggests that these cells play a role in the late-stage of the disease, when dementia develops. Tau immunoreactivity appeared as the strongest morphological correlate of dementia [31].
TOTAL NUMBER OF NEURONS IN THE HIPPOCAMPUS IN NORMAL AGING, ALZHEIMER’S DISEASE, AND ASYMPTOMATIC ALZHEIMER’S DISEASE
In a previous study of hippocampal neurons in aging and AD [32], we had demonstrated that the loss of neurons in the CA1 region was disease-specific and not related to aging. In a follow-up study, we examined the loss of hippocampal neurons in preclinical or what we now call ASYMAD, a state in which individuals have abundant amyloid deposits in the brain but remain clinically normal. We examined the postmortem brains of 33 subjects from the Baltimore Longitudinal Study of Aging and the Johns Hopkins Alzheimer’s Disease Research Center. Using unbiased stereology, we estimated the total number of neurons in the granule cell layer, hilus, CA3-2, CA1, and subiculum of AD (n = 14), preclinical/ASYMAD (n = 8), and age-matched control subjects (n = 11). The results confirmed our previous finding of significant neuronal losses in the CA1 (48%), hilus (14%), and subiculum (24%) in AD [32]. However, we did not observe a significant loss of neurons in CA1 or any of the other subdivisions of the hippocampus in subjects with preclinical/ASYMAD.
NEURONAL HYPERTROPHY IN PRECLINICAL/ASYMPTOMATIC ALZHEIMER’S DISEASE
Since we did not observe loss of neurons in the hippocampus of preclinical/ASYMAD, we decided to examine whether the neurons had simply become atrophic. In an effort to understand what differentiates ASY-MAD subjects from other subjects with similar degrees of AD lesions, but who manifest cognitive deterioration, we performed stereological studies of the size of neurons, and their nuclei and nucleoli in CA1 region of hippocampus, anterior cingulate gyrus (ACG), posterior cingulate gyrus, and primary visual cortex (PVC). ASYMAD were defined as subjects assessed as clinically normal within the last year of life but with identifiable AD pathology at autopsy, i.e., CERAD neuritic plaque score B or higher [10] and a Braak score of III or higher [11]. Surprisingly, rather than atrophy, we found significant hypertrophy of the neuronal cell bodies (+48.7%, p < 0.0001), nuclei (+30.4%, p < 0.001), and nucleoli (+58.6%, p < 0.0001) in the CA1 region of the hippocampus in ASYMAD compared to clinically normal older adults without pathology. The neuronal cell bodies, nuclei, and nucleoli of CA1 in ASYMAD were also significantly larger than in MCI cases (Figs 2 and 3). Similar hypertrophy of neuronal cell bodies, nuclei, and nucleoli were present in the ACG of ASYMAD (Figs 2 and 3). In the posterior cingulate gyrus and primary visual cortex, the hypertrophy was limited to nuclei and nucleoli. We interpreted these findings as evidence that the hypertrophy of cortical neurons and their nuclei and nucleoli in ASY-MAD may represent an early reaction to the presence of neurotoxic species of Aβ or tau, or a compensatory mechanism that prevents the progression of the disease into dementia [19,20].
Fig. 2.
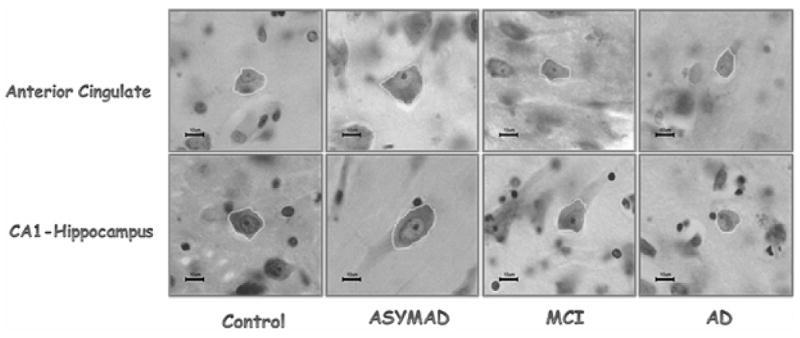
This panel illustrates representative neurons in the anterior cingulate (top row) and CA1-hippocampus (bottom row) in each study group. Note the neuronal hypertrophy in ASYMAD. Nissl stain sections. All magnification bars are 10 μm. (Colours are visible in the electronic version of the article at www.iospress.nl.)
Fig. 3.

A and D illustrate the anatomical sites from where tissue sections were taken: CA1-hippocampus (A) and anterior cingulate (D). The histograms (B, C, E, and F) show the mean volumes of neuronal cell bodies and nuclei. In CA1, comparing ASYMAD versus controls, the mean volume of neuronal cell bodies is 48.7% larger (p < 0.0001) in ASYMAD (B), and the nuclear volume 30.4% (p < 0.001) larger in ASYMAD (C). In anterior cingulate gyrus, the cell bodies are 43.5% (p < 0.0001) larger (E) and nuclei 55.5% (p < 0.0001) larger (F) in ASYMAD compared to controls. The bars correspond to the standard errors of the mean.
CLINICAL STUDIES OF PRECLINICAL/ASYMPTOMATIC ALZHEIMER’S DISEASE
Utilizing the rich database of longitudinal neurological and neuropsychological information that characterizes the BLSA, our first clinical study compared the cognitive trajectories of preclinical/asymptomatic AD with age-matched controls, MCI, and AD dementia. Clinically normal elderly individuals, with and without AD neuropathology, showed similar cognitive trajectories across different cognitive domains. In contrast, individuals with MCI or AD showed steeper rates of longitudinal decline, years before the diagnosis of dementia. The conclusion of this study, which was the first to use the term asymptomatic AD (ASYMAD), was that understanding the pathobiology of this subgroup of apparently ASYMAD individuals may shed some light on mechanisms that forestall the progression of the disease and maintain cognitive health [33].
A second clinical study focused on the rates of depressive symptoms in individuals with asymptomatic AD [34]. The prevalence of major depression is increased in AD, but the basis of this association remains unclear. In this study, we examined depressive symptoms in four groups of participants with postmortem examination from the BLSA: 1) cognitively normal controls with no Alzheimer pathology; 2) cognitively normal individuals with Alzheimer pathology (asymptomatic AD); 3) individuals with mild cognitive impairment with Alzheimer pathology; and 4) individuals with clinical diagnoses of dementia with Alzheimer pathology. Depressive symptoms were assessed using the Center for Epidemiologic Studies Depression Scale (CES-D). Individuals with Alzheimer pathology but no cognitive decline before death, i.e., ASYMAD, had significantly lower rates of depression than cognitively normal controls with no Alzheimer pathology and individuals with AD dementia. These findings suggested that subjects with ASYMAD are resilient not only to dementia, but also to symptoms of depression [35].
JOINT EFFECTS OF CEREBROVASCULAR DISEASE AND ALZHEIMER’S DISEASE NEUROPATHOLOGY IN BLSA AUTOPSIES
In advanced age, it is common to find both AD lesions and cerebrovascular disease. In this context, it becomes important to define the contribution of each pathologic change to the development of dementia and to ask whether AD and cerebrovascular disease are independent, additive, or synergistic. Several prospective autopsy studies [36,42] have reported on the role of vascular pathology in the cause of dementia. Although all agree on the importance of cerebral infarcts as a cause of dementia, the results conflict on the importance of vascular risk factors, asymptomatic infarcts, infarct size, and infarct location. Most studies agree that AD and vascular pathologies interact in an additive fashion, although the quantitative nature of this association has not been documented. The wealth of the longitudinal clinical information, the large number of autopsies, and the large number of subjects made the BLSA cohort unique for elucidating the contribution of cerebrovascular pathology to dementia. To this end, we examined the effects of brain infarcts and AD pathology on the risk for dementia in 179 subjects from the BLSA Autopsy Program. All subjects had longitudinal clinical and cognitive evaluations, and underwent a postmortem examination of the brain. Of the 179 subjects, 89 (50%) were demented and 79 (44%) had cerebral infarcts. Our observations indicate that brain infarcts were common in our cohort, and both symptomatic and asymptomatic infarcts conferred a significant increase in the odds of dementia. Risk factors for stroke in the absence of an infarct did not increase the odds of dementia, which was quantitatively related to the number but not the size of hemispheric infarcts; deep subcortical infarcts conferred no increased risk for dementia either. The contribution of microscopic infarcts to dementia was significant and equivalent to that of macroscopic infarcts (i.e., size did not matter). In subjects with intermediate AD pathology scores, a single macroscopic hemispheric infarct was sufficient to cause dementia. A logistic regression model of the effect of infarcts and AD pathology on dementia indicated that AD pathology alone accounts for 50% of the dementia seen in this cohort, and that hemispheric infarcts alone or in conjunction with AD pathology account for 35%. Based on these observations, we concluded that cerebrovascular disease is a significant and potentially preventable cause of dementia in the BLSA and that the burden and location of infarcts are significantly associated with cognitive decline. We also found that AD and cerebrovascular disease are independent pathologies and that their effects are additive, but not synergistic [43].
STUDIES RELEVANT TO PARKINSON’S DISEASE AND α-SYNUCLEONOPATHIES
Morphometry of the human substantia nigra in aging and PD
Idiopathic PD is a common age-associated neurodegenerative disorder clinically characterized primarily by motor manifestations: bradykinesia, rigidity, resting tremor, and abnormal postural reflexes. A sizeable percent of PD patients, however, eventually develop cognitive decline [44,45]. The neuropathology of PD is characterized by degeneration of the substantia nigra (SN) with loss of neurons and the accumulation of α-synuclein aggregates within neurons and neurites, known as Lewy bodies and Lewy neurites, respectively. Although the pathology of PD is not limited to the SN, as it also involves other brain stem nuclei, hippocampus, amygdala, neocortex, and olfactory bulb, the degeneration of the SN pars compacta and its dopaminergic projection to the striatum is the most salient morphological feature of PD and plays a preeminent role in the motor manifestations that characterize the disease.
Since PD is an age-associated disorder, it is important to assess how the loss of neurons associated with normal aging relates to the disease. Previous morphometric studies of the SN in PD agree that there is a loss of neuromelanin-containing neurons in normal aging, although there is no consensus on the rate of the loss. More important, there is no consensus regarding potential variability in size of SN neurons in normal aging. In this study, we measured the total number of neurons and the cell body volume of pigmented (neuromelanin) neurons in the substantia nigra (SN) of young (n = 7, mean age 19.9 years), middle-aged (n = 9, mean age 50.1 years), and older controls from the BLSA (n = 7, mean age 87.6 years). On random-systematically selected paraffin Nissl- and tyrosine hydroxylase (TH)-stained sections, we used the Optical fractionator to estimate the total number of neurons on one side of the SN. Then, using the Nucleator probe, we measured the volume of these neurons. We observed a significant loss of pigmented (−28.3%, p < 0.01) and tyrosine hydroxylase (TH) (+) (−36.2%, p < 0.001) neurons in older controls compared with younger subjects. Analysis of the size distribution of pigmented and TH (+) neurons showed a significant hypertrophy in older subjects (Nissl +39%, p < 0.01; TH (+) 23%, p < 0.01) compared to young controls. These data suggest that neuronal hypertrophy represents a compensatory mechanism within individual SN neurons that allows for normal motor function despite the loss of neurons in normal aging [46]. (Fig. 4A, B)
Fig. 4.
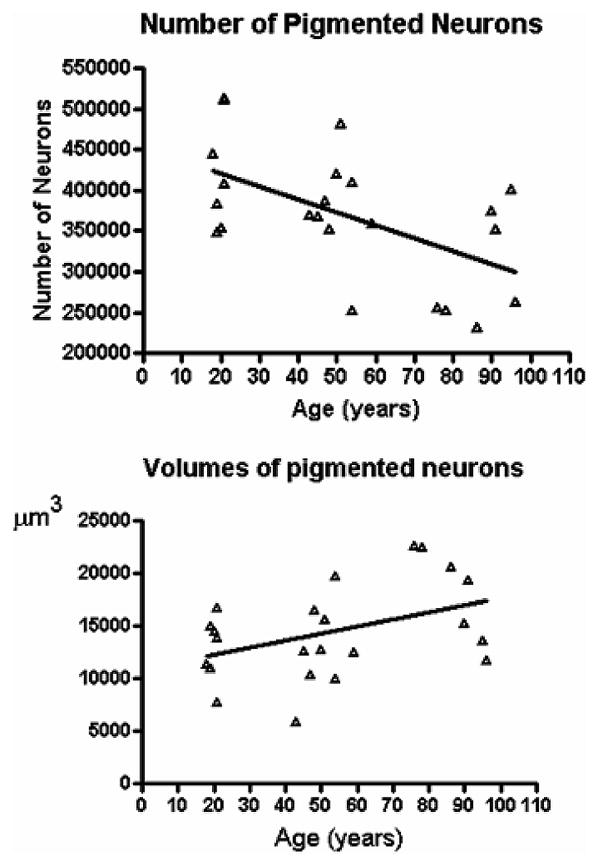
A) Linear regression of the total number of pigmented neurons in one side of the substantia nigra versus age is significant (p = 0.0060) with R2 = 0.337; CV = 0.20. B) Linear regression of the mean volume of the pigmented neurons in the substantia nigra versus age (p = 0.046) withR2 = 0.18; CV = 0.31.
α-SYNUCLEIN LESIONS IN NORMAL AGING, PARKINSON’S DISEASE, AND ALZHEIMER’S DISEASE
Since α-synuclein lesions are characteristic of idiopathic PD and other α-synucleopathies, it is important to know how frequent these lesions are in normal aging and how frequently they coexist with the lesions of AD. To explore these questions, we examined the autopsy brains from normal and demented subjects in the BLSA (n = 117). We found that the overall frequency of α-synuclein lesions was 25%, with 100% in 7 cases of Parkinson’s disease, 31.5% in 56 cases with AD lesions, and 8.3% among 36 older control brains. This latter group may constitute presymptomatic Lewy body disease. Among brains with AD lesions, the frequency of α-synuclein pathology was higher in those with higher scores for neuritic plaques, but not in those with higher scores for NFT. Our observations indicate that α-synuclein lesions are uncommon in aged control subjects. However, the coexistence of Aβ amyloid and α-synuclein lesions in AD brains suggests that common pathogenic mechanisms may be at play in the development of these two pathologies [47].
SUCCESSFUL AGING AND NEURONAL SIZE
An important conclusion from our series of neuropathologic studies is that many individuals can reach advanced age without clinical or neurological impairment while leading independent and fulfilling lives. This group of individuals who represent “successful aging” is not homogeneous. Whereas some individuals remain practically free of neuropathology, others bear abundant AD lesions (i.e., neuritic plaques score CERAD C and NFT Braak stage VI). This latter group, which we have called ASYMAD, shows marked hypertrophy of cerebral neurons, a change that may account for the functional compensation reflected in the different clinical outcome compared to MCI and AD subjects. But this possible “compensatory hypertrophy” is not limited to the cerebral cortex. We also observe neuronal hypertrophy in the substantia nigra, where the age-associated loss of neurons is accompanied by hypertrophy of a magnitude such that the total volume of SN remains similar to that of young controls. We believe that the hypertrophy of neurons, both in the cerebral cortex and the SN, is not limited to cell bodies and probably includes axonal terminals and dendritic spines that allow compensation for loss or injury of nerve cells. Overall, our studies suggest that in some individuals, successful cognitive aging results from compensatory mechanisms that occur at the neuronal level; whereas a failure of compensation (i.e., synaptic plasticity) may culminate in disease. Understanding and harnessing the underlying mechanism of neuronal hypertrophy in the cerebral cortex of ASYMAD and the SN of normal aging is an important endeavor bound to open new therapeutic approaches for age-associated neurodegenerative disease.
Acknowledgments
The authors thank Karen Wall for preparation of the article, the BLSA participants, their families, and National Institute of Aging and Johns Hopkins University staff, in particular to David Dolan and Hillary Dolan.
This work was supported by the Johns Hopkins University Alzheimer’s Disease Research Center (National Institutes of Health, AG05146), the Burroughs Wellcome Fund for Translational Research, and the Intramural Research Program of the National Institute of Aging and National Institutes of Health.
Footnotes
Authors’ disclosures available online (http://www.j-alz.com/disclosures/view.php?id=93).
References
- 1.Shock NW, Greulich RC, Costa PTJ, Andres R, Lakatta EG, Arenberg D, Tobin JD. Normal Human Aging: The Baltimore Longitudinal Study of Aging. U.S. Government Printing Office; Washington, D.C.: 1984. [Google Scholar]
- 2.Kawas C, Gray S, Brookmeyer R, Fozard J, Zonderman A. Age-specific incidence rates of Alzheimer’s disease: the Baltimore Longitudinal Study of Aging. Neurology. 2000;54:2072–2077. doi: 10.1212/wnl.54.11.2072. [DOI] [PubMed] [Google Scholar]
- 3.Hughes CP, Berg L, Danziger WL, Coben LA, Martin RL. A new clinical scale for the staging of dementia. Br J Psychiatry. 1982;140:566–572. doi: 10.1192/bjp.140.6.566. [DOI] [PubMed] [Google Scholar]
- 4.Morris JC, Ernesto C, Schafer K, Coats M, Leon S, Sano M, Thal LJ, Woodbury P. Clinical dementia rating training and reliability in multicenter studies: the Alzheimer’s Disease Cooperative Study experience. Neurology. 1997;48:1508–1510. doi: 10.1212/wnl.48.6.1508. [DOI] [PubMed] [Google Scholar]
- 5.Silverman JM, Breitner JC, Mohs RC, Davis KL. Reliability of the family history method in genetic studies of Alzheimer’s disease and related dementias. Am J Psychiatry. 1986;143:1279–1282. doi: 10.1176/ajp.143.10.1279. [DOI] [PubMed] [Google Scholar]
- 6.Blessed G, Tomlinson BE, Roth M. The association between quantitative measures of dementia and of senile change in the cerebral grey matter of elderly subjects. Br J Psychiatry. 1968;114:797–811. doi: 10.1192/bjp.114.512.797. [DOI] [PubMed] [Google Scholar]
- 7.Association AP. Diagnostic and Statistical Manual of Mental Disorders. 3. Washington D.C.: American Psychiatric Press, Inc; 1987. Revised (DSM-II-R) [Google Scholar]
- 8.Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med. 2004;256:183–194. doi: 10.1111/j.1365-2796.2004.01388.x. [DOI] [PubMed] [Google Scholar]
- 9.Yamamoto T, Hirano A. A comparative study of modified Bielschowsky, Bodian and thioflavin S stains on Alzheimer’s neurofibrillary tangles. Neuropathol Appl Neurobiol. 1986;12:3–9. doi: 10.1111/j.1365-2990.1986.tb00677.x. [DOI] [PubMed] [Google Scholar]
- 10.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 11.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 12.Hughes AJ, Daniel SE, Blankson S, Lees AJ. A clinicopathologic study of 100 cases of Parkinson’s disease. Arch Neurol. 1993;50:140–148. doi: 10.1001/archneur.1993.00540020018011. [DOI] [PubMed] [Google Scholar]
- 13.McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H, Cummings J, Duda JE, Lippa C, Perry EK, Aarsland D, Arai H, Ballard CG, Boeve B, Burn DJ, Costa D, Del Ser T, Dubois B, Galasko D, Gauthier S, Goetz CG, Gomez-Tortosa E, Halliday G, Hansen LA, Hardy J, Iwatsubo T, Kalaria RN, Kaufer D, Kenny RA, Korczyn A, Kosaka K, Lee VM, Lees A, Litvan I, Londos E, Lopez OL, Minoshima S, Mizuno Y, Molina JA, Mukaetova-Ladinska EB, Pasquier F, Perry RH, Schulz JB, Trojanowski JQ, Yamada M. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65:1863–1872. doi: 10.1212/01.wnl.0000187889.17253.b1. [DOI] [PubMed] [Google Scholar]
- 14.Troncoso JC, Martin LJ, Dal Forno G, Kawas CH. Neuropathology in controls and demented subjects from the Baltimore Longitudinal Study of Aging. Neurobiol Aging. 1996;17:365–371. doi: 10.1016/0197-4580(96)00028-0. [DOI] [PubMed] [Google Scholar]
- 15.Schmitt FA, Davis DG, Wekstein DR, Smith CD, Ashford JW, Markesbery WR. Preclinical AD revisited: neuropathology of cognitively normal older adults. Neurology. 2000;55:370–376. doi: 10.1212/wnl.55.3.370. [DOI] [PubMed] [Google Scholar]
- 16.Morris JC, Price AL. Pathologic correlates of non-demented aging, mild cognitive impairment, and early-stage Alzheimer’s disease. J Mol Neurosci. 2001;17:101–118. doi: 10.1385/jmn:17:2:101. [DOI] [PubMed] [Google Scholar]
- 17.Lue LF, Brachova L, Civin WH, Rogers J. Inflammation, A beta deposition, and neurofibrillary tangle formation as correlates of Alzheimer’s disease neurodegeneration. J Neuropathol Exp Neurol. 1996;55:1083–1088. [PubMed] [Google Scholar]
- 18.Benzing WC, Mufson EJ, Armstrong DM. Immunocytochemical distribution of peptidergic and cholinergic fibers in the human amygdala: their depletion in Alzheimer’s disease and morphologic alteration in non-demented elderly with numerous senile plaques. Brain Res. 1993;625:125–138. doi: 10.1016/0006-8993(93)90145-d. [DOI] [PubMed] [Google Scholar]
- 19.Riudavets MA, Iacono D, Resnick SM, O’Brien R, Zonderman AB, Martin LJ, Rudow G, Pletnikova O, Troncoso JC. Resistance to Alzheimer’s pathology is associated with nuclear hypertrophy in neurons. Neurobiol Aging. 2007;28:1484–1492. doi: 10.1016/j.neurobiolaging.2007.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iacono D, O’Brien R, Resnick SM, Zonderman AB, Pletnikova O, Rudow G, An Y, West MJ, Crain B, Troncoso JC. Neuronal hypertrophy in asymptomatic Alzheimer disease. J Neuropathol Exp Neurol. 2008;67:578–589. doi: 10.1097/NEN.0b013e3181772794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Katzman R. Education and the prevalence of dementia and Alzheimer’s disease. Neurology. 1993;43:13–20. doi: 10.1212/wnl.43.1_part_1.13. [DOI] [PubMed] [Google Scholar]
- 22.Katzman R, Terry R, DeTeresa R, Brown T, Davies P, Fuld P, Renbing X, Peck A. Clinical, pathological, and neurochemical changes in dementia: a subgroup with preserved mental status and numerous neocortical plaques. Ann Neurol. 1988;23:138–144. doi: 10.1002/ana.410230206. [DOI] [PubMed] [Google Scholar]
- 23.Stern Y. Cognitive reserve and Alzheimer disease. Alzheimer Dis Assoc Disord. 2006;20:S69–74. doi: 10.1097/00002093-200607001-00010. [DOI] [PubMed] [Google Scholar]
- 24.Sze CI, Troncoso JC, Kawas C, Mouton P, Price DL, Martin LJ. Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:933–944. doi: 10.1097/00005072-199708000-00011. [DOI] [PubMed] [Google Scholar]
- 25.Sze CI, Bi H, Kleinschmidt-DeMasters BK, Filley CM, Martin LJ. Selective regional loss of exocytotic presynaptic vesicle proteins in Alzheimer’s disease brains. J Neurol Sci. 2000;175:81–90. doi: 10.1016/s0022-510x(00)00285-9. [DOI] [PubMed] [Google Scholar]
- 26.Martin LJ. Neurodegenerative Disorders. In: Ramachandran VS, editor. Encyclopedia of the Human Brain. Vol. 3. Elsevier Science; 2002. pp. 441–463. [Google Scholar]
- 27.Vehmas AK, Kawas CH, Stewart WF, Troncoso JC. Immune reactive cells in senile plaques and cognitive decline in Alzheimer’s disease. Neurobiol Aging. 2003;24:321–331. doi: 10.1016/s0197-4580(02)00090-8. [DOI] [PubMed] [Google Scholar]
- 28.Crystal H, Dickson D, Fuld P, Masur D, Scott R, Mehler M, Masdeu J, Kawas C, Aronson M, Wolfson L. Clinicopathologic studies in dementia: nondemented subjects with pathologically confirmed Alzheimer’s disease. Neurology. 1988;38:1682–1687. doi: 10.1212/wnl.38.11.1682. [DOI] [PubMed] [Google Scholar]
- 29.Stern Y, Jacobs D, Goldman J, Gomez-Tortosa E, Hyman BT, Liu Y, Troncoso J, Marder K, Tang MX, Brandt J, Albert M. An investigation of clinical correlates of Lewy bodies in autopsy-proven Alzheimer disease. Arch Neurol. 2001;58:460–465. doi: 10.1001/archneur.58.3.460. [DOI] [PubMed] [Google Scholar]
- 30.Price JL, Morris JC. Tangles and plaques in nondemented aging and preclinical Alzheimer’s disease. Ann Neurol. 1999;45:358–368. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 31.West MJ, Kawas CH, Stewart WF, Rudow GL, Troncoso JC. Hippocampal neurons in pre-clinical Alzheimer’s disease. Neurobiol Aging. 2004;25:1205–1212. doi: 10.1016/j.neurobiolaging.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 32.West MJ, Coleman PD, Flood DG, Troncoso JC. Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer’s disease. Lancet. 1994;344:769–772. doi: 10.1016/s0140-6736(94)92338-8. [DOI] [PubMed] [Google Scholar]
- 33.Driscoll I, Resnick SM, Troncoso JC, An Y, O’Brien R, Zonderman AB. Impact of Alzheimer’s pathology on cognitive trajectories in nondemented elderly. Ann Neurol. 2006;60:688–695. doi: 10.1002/ana.21031. [DOI] [PubMed] [Google Scholar]
- 34.Morgan MD, Mielke MM, O’Brien R, Troncoso JC, Zonderman AB, Lyketsos CG. Rates of depression in individuals with pathologic but not clinical Alzheimer disease are lower than those in individuals without the disease: findings from the Baltimore Longitudinal Study on Aging (BLSA) Alzheimer Dis Assoc Disord. 2007;21:199–204. doi: 10.1097/WAD.0b013e3181461932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morgan D, Gitter BD. Evidence supporting a role for anti-Abeta antibodies in the treatment of Alzheimer’s disease. Neurobiol Aging. 2004;25:605–608. doi: 10.1016/j.neurobiolaging.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 36.Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. Jama. 1997;277:813–817. [PubMed] [Google Scholar]
- 37.Schneider JA, Wilson RS, Bienias JL, Evans DA, Bennett DA. Cerebral infarctions and the likelihood of dementia from Alzheimer disease pathology. Neurology. 2004;62:1148–1155. doi: 10.1212/01.wnl.0000118211.78503.f5. [DOI] [PubMed] [Google Scholar]
- 38.Esiri MM, Nagy Z, Smith MZ, Barnetson L, Smith AD. Cerebrovascular disease and threshold for dementia in the early stages of Alzheimer’s disease. Lancet. 1999;354:919–920. doi: 10.1016/S0140-6736(99)02355-7. [DOI] [PubMed] [Google Scholar]
- 39.Pathological correlates of late-onset dementia in a multicentre, community-based population in England and Wales. Neuropathology Group of the Medical Research Council Cognitive Function and Ageing Study (MRC CFAS) Lancet. 2001;357:169–175. doi: 10.1016/s0140-6736(00)03589-3. [DOI] [PubMed] [Google Scholar]
- 40.White L, Petrovitch H, Hardman J, Nelson J, Davis DG, Ross GW, Masaki K, Launer L, Markesbery WR. Cerebrovascular pathology and dementia in autopsied Honolulu-Asia Aging Study participants. Ann N Y Acad Sci. 2002;977:9–23. doi: 10.1111/j.1749-6632.2002.tb04794.x. [DOI] [PubMed] [Google Scholar]
- 41.Sonnen JA, Larson EB, Crane PK, Haneuse S, Li G, Schellenberg GD, Craft S, Leverenz JB, Montine TJ. Pathological correlates of dementia in a longitudinal, population-based sample of aging. Ann Neurol. 2007;62:406–413. doi: 10.1002/ana.21208. [DOI] [PubMed] [Google Scholar]
- 42.Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology. 2007;69:2197–2204. doi: 10.1212/01.wnl.0000271090.28148.24. [DOI] [PubMed] [Google Scholar]
- 43.Troncoso JC, Zonderman AB, Resnick SM, Crain B, Pletnikova O, O’Brien RJ. Effect of infarcts on dementia in the Baltimore longitudinal study of aging. Ann Neurol. 2008;64:168–176. doi: 10.1002/ana.21413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aarsland D, Andersen K, Larsen JP, Lolk A, Nielsen H, Kragh-Sorensen P. Risk of dementia in Parkinson’s disease: a community-based, prospective study. Neurology. 2001;56:730–736. doi: 10.1212/wnl.56.6.730. [DOI] [PubMed] [Google Scholar]
- 45.Jellinger KA. The morphological basis of mental dysfunction in Parkinson’s disease. J Neurol Sci. 2006;248:167–172. doi: 10.1016/j.jns.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 46.Rudow G, O’Brien R, Savonenko AV, Resnick SM, Zonderman AB, Pletnikova O, Marsh L, Dawson TM, Crain BJ, West MJ, Troncoso JC. Morphometry of the human substantia nigra in ageing and Parkinson’s disease. Acta Neuropathol. 2008;115:461–470. doi: 10.1007/s00401-008-0352-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mikolaenko I, Pletnikova O, Kawas CH, O’Brien R, Resnick SM, Crain B, Troncoso JC. Alpha-synuclein lesions in normal aging, Parkinson disease, and Alzheimer disease: evidence from the Baltimore Longitudinal Study of Aging (BLSA) J Neuropathol Exp Neurol. 2005;64:156–162. doi: 10.1093/jnen/64.2.156. [DOI] [PubMed] [Google Scholar]