

The protective anti-cancer effect of nonsteroidal anti-inflammatory drugs (NSAIDs) has attracted much attention, and clinical trials of several NSAIDs are under way for treatment or prevention of various cancers.[1] As NSAIDs are best known for their inhibition of cyclooxygenase 1 and 2 (COX-1/2), they are hypothesized to suppress tumor growth by blocking prostaglandin synthesis.[2] However, this hypothesis does not explain all of the available data.[3–5] Accumulated evidence suggests that some NSAIDs also target the Wnt/β-catenin signaling pathway in human cancer cells.[4,5a–c] For example, sulindac (Clinoril) has been shown to suppress canonical β-catenin–related Wnt signaling in breast cancer, lung cancer, and colon cancer cell lines.[4e] However, the molecular mechanism of this effect is not clear.
Wnt signaling plays crucial roles in embryonic development and in tissue maintenance in adults.[5] Abnormal activation of Wnt signaling is observed in several types of cancers.[5,6] Dishevelled (Dvl) is a key molecule in the Wnt pathways that, through its PDZ domain, relays Wnt signals from membrane-bound Wnt receptors to downstream components.[5,7] We and others have worked to develop small-molecule inhibitors of Dvl PDZ protein-protein interaction for use in elucidating biological processes and as potential cancer treatment and prevention agents.[8] Here we show that both sulindac and sulindac sulfone bind to the PDZ domain of Dvl and sulindac suppresses Wnt3A-induced β-catenin signaling at the level of Dvl. Our results suggest that the anticancer protective effect of sulindac (and its metabolite) reflect not only COX-1/2 inhibition but also the inhibition of abnormal canonical Wnt signaling via blockade of the Dvl PDZ domain.
To test the binding of sulindac and sulindac sulfone to the Dvl PDZ domain, we conducted chemical shift perturbation experiments with nuclear magnetic resonance (NMR) spectroscopy; this method is widely used to characterize protein-ligand interactions.[9] When added to a solution of 15N-labeled Dvl PDZ domain, both compounds generated chemical shift perturbations that indicated binding to the same region of the Dvl PDZ domain (Figures 1a and S1). The structure of the Dvl PDZ domain comprises six β-strands (βA–βF) and two α-helices (αA and αB). The chemical shift perturbations induced by binding indicated that both compounds bind to the Dvl PDZ domain between its αB and βB structures, as do the domain's native ligands.[7,10]
Figure 1.

Sulindac directly binds to the PDZ domain of Dishevelled. (a) the extended 15N-HSQC spectra of 15N-labeled Dvl PDZ domain during the titration of sulindac (blue,free state; red, bound state). (b) Ensemble of the 20 lowest-energy conformations of the PDZ-sulindac complex. Backbone of PDZ domain, magenta; sulindac, blue stick model.
Because there are many PDZ domains in the human proteome,[8c,11] we next examined whether sulindac and sulindac sulfone bind exclusively to the Dvl PDZ domain. We tested three other PDZ domains that are representative of most human PDZ domains: the first and second PDZ domains of PSD-95 protein (PDZ1 and PDZ2, respectively; class-1 PDZ domains) and the seventh PDZ domain of GRIP1 protein (PDZ7; a class 2 PDZ domain). The Dvl PDZ domain belongs to neither class 1 nor class 2.[7,11] Remarkably, in NMR titration experiments, little or no interaction was observed between sulindac or sulindac sulfone and these three PDZ domains (Figure S2 and Table S1), indicating that sulindac and sulindac sulfone bind specifically to the Dvl PDZ domain.
To further investigate the specificity with which sulindac and sulindac sulfone recognize the Dvl PDZ domain, we determined the structure of Dvl PDZ domain in complex with sulindac (Figure 1). Two-dimensional (2D) [15N, 13C]-double filtered nuclear Overhauser effect (NOE) spectroscopy experiments showed that sulindac bound to Dvl PDZ has a cis (Z) conformation (Figure S3 and Table S2).[12a] The complex structure was determined by using 45 intermolecular NOEs between bound sulindac and the PDZ domain, obtained from 3D 13C-F1-half-filtered, F2-edited NOE spectroscopy–heteronuclear single quantum coherence (HSQC) experiments, and 7 intramolecular NOEs of sulindac (Figure S4 and Table S3).[12] An ensemble of the 20 lowest-energy calculated structures of the complex is shown in Fig. 1b. In the complex, sulindac binds to the peptide pocket of the PDZ domain (Figure 2, and Table S4), in which the sulindac carboxylate group forms hydrogen bonds with the amide groups (L262, G263, and I264) in the PDZ-domain loop region (Fig. 2b). The sulindac methylene group (f in Figure 1a) contacts the side chains of PDZ-domain residues L262 and V325 and the sulindac aromatic rings (g, h, i in Fig. 1a) contact the hydrophobic pocket formed by PDZ-domain residues L262, I266, V325, L321, and V318. The sulindac benzyl group (b and c in Fig. 1a) and methyl group (a) contact PDZ-domain residues I266 and V318. Notably, the side chain of R322 on the PDZ-domain α-helix (αB-5′) forms a hydrogen bond with the sulfonyl oxygen atom of sulindac (S=O---H-Nη; dO---Nη ~ 2.9 Å) (Fig. 2b). Consistent with this finding, the R322A mutant PDZ domain interacted negligibly with sulindac (Figure S5). Therefore, residue R322 on the αB helix of Dvl PDZ appears to play a crucial role in determining the specificity of binding. The residue Arg in the αB-5′ position (R322) occurs in no PDZ domain except that of Dvl (Figure S6). Therefore, this interaction is likely to explain the specific binding of sulindac and sulindac sulfone to the Dvl PDZ domain. Indeed, sulindac sulfide, the most prominent metabolite of sulindac bearing the COX-1/2 inhibition and having a thioether instead of a sulfonyl group, binds to the PDZ domain with much weaker affinity (Figure S6).
Figure 2.
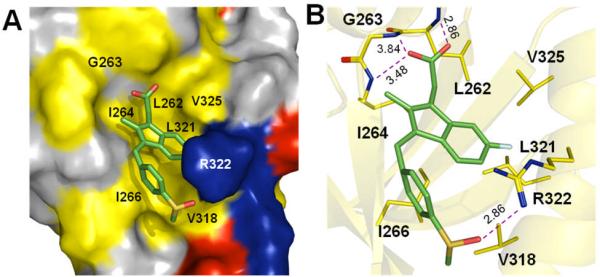
Three-dimensional structure of the Dvl PDZ domain in complex with sulindac. (a) Surface representation showing the binding interface between the Dvl PDZ domain and sulindac (stick model). Hydrophobic amino acid residues in Dvl PDZ are yellow, positively charged residues are blue, negatively charged residues are red, and uncharged polar residues are gray. (b) Sulindac (stick model) and Dvl PDZ (cartoon model). Dvl PDZ domain residues in contact with sulindac are yellow.
To obtain further insight into the binding mechanism of sulindac and sulindac sulfone to the Dvl PDZ domain, we performed a competitive binding assay using a peptide derived from the C-terminus of Dapper (Rox-SGSLKLMTTVCOOH, termed Rox-DprC). Dapper protein is known to antagonize Wnt signaling by binding to the PDZ domain of Dvl.[7,10] We first measured the binding affinity between this peptide and the PDZ domain of Dvl by monitoring fluorescence polarization during titration of unlabeled Dvl PDZ into a solution of fluorescence-labeled Rox-DprC (KD ~ 8.0 ± 1.0 μM).[7] We then added either sulindac or sulindac sulfone in the solution of the labeled peptide and repeated the same binding assay, and found that both compounds inhibited interaction of the peptide with Dvl PDZ (KI values, 10.7 ± 1.2 μM and 8.0 ± 0.2 μM, respectively) (Figure 3). As the binding affinities of Dapper, sulindac, and sulindac sulfone to the Dvl PDZ domain are similar, sulindac and sulindac sulfone, like Dapper, effectively block cellular Wnt signaling by disrupting the interactions between Dvl and the Wnt receptors Frizzled (Fz).[7]
Figure 3.

Competitive binding experiments. The KD value of the fluorescently labeled, Dapper-derived peptide Rox-DprC was obtained by plotting 1/ΔmP vs 1/ΔS, where ΔmP is the fluorescence polarization change (×1000) of Rox-DprC and ΔS is the concentration change of unlabeled PDZ domain.The KI values of sulindac and sulindac sulfone were obtained by using the equation KDapp = (KD/(1+[I]/KI).
To confirm that sulindac inhibits canonical Wnt signaling, we co-injected Wnt3A mRNA (1 pg) and the Siamois reporter construct[13] into the animal pole region of Xenopus embryos at the two-cell stage and cultured them with different concentrations of sulindac. A luciferase assay was performed on ectodermal explants dissected at the blastula stage. As shown in Figure 4a, sulindac inhibited Siamois promoter-driven luciferase activity induced by Wnt3A, consistent with our previous results.[8a] To establish whether inhibition of Wnt signaling by sulindac occurs upstream or downstream of Dvl, we also co-injected the animal pole region of Xenopus embryos with the Siamois reporter construct and Dvl mRNA (100 pg) or β-catenin mRNA (500 pg), treated the embryos with sulindac (3.5 μg/ml, 9.8 μM), and assayed luciferase activity (Fig. 4b). As expected, sulindac attenuated Dvl-mediated Wnt signaling but not β-catenin-mediated signaling. Therefore, sulindac blocks canonical Wnt signaling at the level of Dvl.
Figure 4.
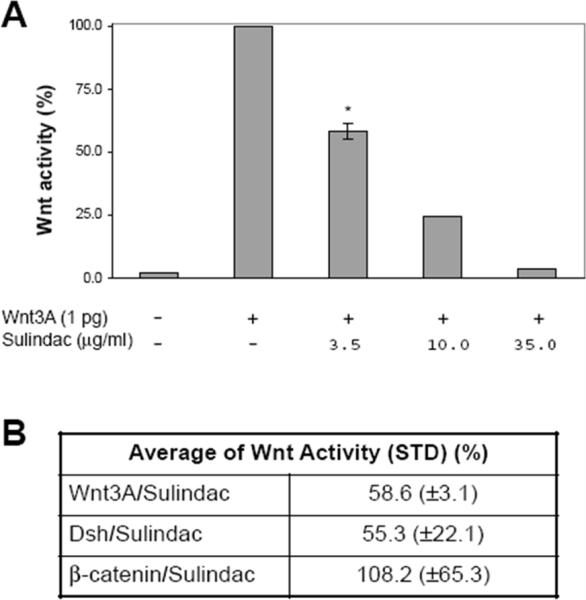
Effect of sulindac on canonical Wnt signaling in Xenopus embryos. (a) Sulindac inhibits canonical Wnt signaling induced by Wnt3A in a concentration-dependent manner. Luciferase activity was measured in ectodermal explants dissected at the late blastula stage. Activity in the untreated control was assigned a value of 100%. Four embryos were treated with 3.5 μg/ml of sulindac; the mean ± SD is shown. The remaining data represent single embryos. (b) Sulindac attenuates canonical Wnt signaling at the level of Dvl. Values are mean luciferase activity ± SD from four independent experiments.
Dvl is a critical regulator of Wnt signaling pathways.[5] In the canonical Wnt signaling pathway, Wnt signal is passed from the membrane Wnt receptor Fz to Dvl, which then relays the signal to downstream components.[5,7] The direct recognition between Dvl PDZ domain and a conserved sequence (KTXXXW) in Fz, localized two residues after the seventh transmembrane domain, is the key interaction in the pathway.[7] Indeed, Dvl PDZ-binding peptides as well as small molecules targeting the Dvl PDZ domain can effectively block Wnt signaling.[8] Here we show that sulindac and sulindac sulfone bind the peptide-binding site of the Dvl PDZ domain with an affinity comparable to that of the native binding partners[7] and that, like other Dvl PDZ inhibitors, sulindac blocks Wnt signaling at the Dvl level in Xenopus. Therefore, we propose that sulindac exerts its chemoprotective anticancer effect not only by inhibiting COX enzymes[2] but also by inhibiting Wnt signaling via Dvl.
Abnormal activation of Dvl has been linked to several types of cancer, including mesothelioma, breast cancer, prostate cancer, cervical cancer, and non-small-cell lung cancer.[6] In many cases, over-expression of Dvl is implicated in the activation of Wnt signaling. Therefore, inhibition of Dvl PDZ-domain interactions by sulindac or sulindac sulfone should work counter to this cancer-promoting effect. However, Wnt signaling may be activated in various ways in cancer,[14a] and tumor development and progression often result from cumulative events abnormally activating Wnt signaling.[15,16] For example, most cases of colorectal cancer are initiated by activation of the Wnt pathway in the presence of a mutant adenomatous polyposis coli (APC) tumour suppressor, but in many cases, the level of Wnt signaling increases during tumor progression.[14b] Epigenetic inactivation of Wnt antagonist genes, such as DICKKOPF and SFRP, often occurs early in colorectal cancer progression and could induce the constitutive Wnt signaling that is a necessary complement to APC mutations in the evolution of colorectal cancer.[15] Therefore, even though APC is downstream of Dvl in the canonical Wnt signaling pathway, inhibition of Wnt signaling through sulindac blockade of the Dvl PDZ domain should help to suppress the progression and evolution of colorectal cancer. For example, in a recent study, restoration of expression of DACT3 (Dapper/Frodo), a protein antagonist of Dvl PDZ domain that is epigenetically repressed in colorectal cancer, resulted in attenuation of Wnt signaling and apoptosis of colorectal cancer cells.[16]
The testing of NSAIDs as anticancer chemopreventive agents is an excellent example of current efforts to “repurpose” old drugs to new applications.[4c, 17] However, unresolved questions such as mechanism of action of NSAIDs limit their clinical application to the cancer prevention or treatment.[1a] Our findings suggest that at least one of the chemopreventive mechanisms of sulindac is the blockade of canonical Wnt signaling downstream of the Dvl PDZ domain. This conclusion is consistent with the fact that sulindac and sulindac sulfone exert similar chemopreventive effects although they have very different effects on the COX enzymes (sulindac is a non-specific COX inhibitor, whereas sulindac sulfone does not inhibit COX).[4f] It also provides a rationale to explore other NSAIDs and their analogs and metabolites as potential anticancer therapeutic and preventive agents. Moreover, since sulindac interacts with Dvl PDZ domain at low micromolar affinity, further development of more potent inhibitors is clearly needed.
Experimental Section
Structure Determination of the Dvl PDZ–sulindac Complex
Distance restraints were obtained by conducting several types of NOE spectroscopy experiments, including 2D-[13C,15N]-double filtered 1H-NOE spectroscopy, 2D 13C F1-half-filtered 1H-1H NOE spectroscopy, and 3D 13C-F1-half-filtered and F2-edited HSQC-NOE spectroscopy, on a sample of 13C/15N-double-labeled Dvl PDZ domain bound to unlabeled sulindac in a ratio of 1 (PDZ) to 10 (sulindac).[13b] NOE restraints were grouped into distance ranges according to their relative intensity (Tables S2 and S3). Topology and parameter files for sulindac were generated by using xplo2d,[18a] and the charge parameters of sulindac were calculated by using the antechamber module in the AMBER8 software package.[18b] To calculate the structure of the complex, we used the simulated annealing protocol in the program CNS[18c] with the modified X-ray structure of Xenopus PDZ domain (1L6O)[10] as the starting point; all calculations were done in the program HADDOCK.[18d] In the final calculation, two different restraints were used to elucidate the complex structure: (1) NOE-based unambiguous distance restraints; and (2) the hydrogen bond restraints, which were set at 1.6 Å < dNH-O < 2.4 Å and 2.4 Å < dN-O < 3.5 Å by using the donor-acceptor pairs identified in initial rounds of structural calculation. Two thousand initial structures of the Dvl1 PDZ–sulindac complex were generated. The 20 lowest-energy conformations among the final 100 water-refinement structures of the complex were selected (Table S4). The refined structures were analyzed by the programs PROCHECK[18e] and MOLMOL[18f] and have been deposited into the Protein Data Bank (PDB code 2KAW).
Fluorescence Binding Studies
A Fluorolog-3 spectrofluorometer (Jobin-Yvon Inc.) and a 10 × 4 mm quartz cell (Hellma Inc.) with magnetic stirring were used to conduct the competition binding experiments. The details are described in supporting information.
Luciferase Assay
Xenopus eggs were obtained from females that had received injections of 500 IU of human chorionic gonadotropin (Sigma)and had been artificially fertilized. Synthesis and microinjection of capped mRNAs were carried out as previously described.[19] The mRNA and the Siamois reporter DNA[13] were co-injected into the animal pole region at the two-cell stage. Injected embryos were cultured in the presence of sulindac at different concentrations. Animal cap explants from untreated and treated embryos were dissected at the late blastula stage and a luciferase assay (Promega) was performed by preparing cell lysates from 10 explants and measuring the luciferase activity in a Lumat LB 9507 luminometer.[13]
Supplementary Material
Acknowledgments
The financial support of this work by NIH grants CA21765 (Cancer Center Support Grant) and GM081492, by the American Lebanese Syrian Associated Charities (ALSAC) (JJZ), and by grants from the Centre National de la Recherche Scientifique (D-LS). The authors thank Sharon Naron for editing the manuscript; the Hartwell Center for Bioinformatics and Biotechnology at St. Jude for computational time; S. Malone, M. Zhou and Dr. C. Ross for computer-related technical support; Dr. W. Zhang for assistance with the NMR experiments; and Y. Shao for producing proteins.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org.
References
- [1].a) Thun MJ, Henley SJ, Patrono C. J. Natl. Cancer Inst. 2002;94(4):252. doi: 10.1093/jnci/94.4.252. [DOI] [PubMed] [Google Scholar]; b) Brown JR, DuBois RN. J Clin. Oncol. 2005;23:2840. doi: 10.1200/JCO.2005.09.051. [DOI] [PubMed] [Google Scholar]; c) Webster WS, Leibovich BC. Expert Rev. Anticancer Ther. 2005;5:957. doi: 10.1586/14737140.5.6.957. [DOI] [PubMed] [Google Scholar]; d) Lim JT, Joe AK, Suzui M, Shimizu M, Masuda M, Weinstein IB. Clin. Cancer Res. 2006;12:3478. doi: 10.1158/1078-0432.CCR-05-2051. [DOI] [PubMed] [Google Scholar]
- [2].Ulrich CM, Bigler J, Potter JD. Nat. Rev. Cancer. 2006;6:130. doi: 10.1038/nrc1801. and references therein. [DOI] [PubMed] [Google Scholar]
- [3].a) Zhang X, Morham SG, Langenbach R, Young DA. J. Exp. Med. 1999;190:451. doi: 10.1084/jem.190.4.451. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Babbar N, Ignatenko NA, Casero RA, Jr., Gerner EW. J. Biol. Chem. 2003;278:47762. doi: 10.1074/jbc.M307265200. [DOI] [PubMed] [Google Scholar]; c) Smith ML, Hawcroft G, Hull MA. Eur. J. Cancer. 2000;36:664. doi: 10.1016/s0959-8049(99)00333-0. [DOI] [PubMed] [Google Scholar]
- [4].a) Boon EM, Keller JJ, Wormhoudt TA, Giardiello FM, Offerhaus GJ, van der Neut R, Pals ST. Br. J. Cancer. 2004;90:224. doi: 10.1038/sj.bjc.6601505. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gardner SH, Hawcroft G, Hull MA. Br. J. Cancer. 2004;91:153. doi: 10.1038/sj.bjc.6601901. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Barker N, Clevers H. Nat..Rev. Drug Discov. 2006;5:997. doi: 10.1038/nrd2154. and references therein. [DOI] [PubMed] [Google Scholar]; d) Sauter A, Matharu R, Braun T, Schultz J, Sadick H, Hormann K, Naim R. Arch. Med. Res. 2007;38:367. doi: 10.1016/j.arcmed.2006.11.005. [DOI] [PubMed] [Google Scholar]; e) Han A, Song Z, Tong C, Hu D, Bi X, Augenlicht LH, Yang W. Eur. J. Pharmacol. 2008;583:26. doi: 10.1016/j.ejphar.2007.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Piazza GA, Alberts DS, Hixson LJ, Paranka NS, Li H, Finn T, Bogert C, Guillen JM, Brendel K, Gross PH, Sperl G, Ritchie J, Burt RW, Ellsworth L, Ahnen DJ, Pamukcu R. Cancer Res. 1997;57:2909. [PubMed] [Google Scholar]
- [5].a) Moon RT, Kohn AD, De Ferrari GV, Kaykas A. Nat. Rev. Genet. 2004;5:691. doi: 10.1038/nrg1427. and references therein. [DOI] [PubMed] [Google Scholar]; b) Logan CY, Nusse R. Annu. Rev. Cell Dev. Biol. 2004;20:781. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]; c) Klaus A, Birchmeier W. Nat. Rev. Cancer. 2008;8:387. doi: 10.1038/nrc2389. [DOI] [PubMed] [Google Scholar]; d) Wallingford JB, Habas R. Development. 2005;132:4421. doi: 10.1242/dev.02068. and references therein. [DOI] [PubMed] [Google Scholar]; e) Pan W, Choi SC, Wang H, Qin Y, Volpicelli-Daley L, Swan L, Lucast L, Khoo C, Zhang X, Li L, Abrams CS, Sokol SY, Wu D. Science. 2008;321:1350. doi: 10.1126/science.1160741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Uematsu K, He B, You L, Xu Z, McCormick F, Jablons DM. Oncogene. 2003;22:7218. doi: 10.1038/sj.onc.1206817. [DOI] [PubMed] [Google Scholar]; b) Uematsu K, Kanazawa S, You L, He B, Xu Z, Li K, Peterlin BM, McCormick F, Jablons DM. Cancer Res. 2003;63:454. [PubMed] [Google Scholar]; c) Mizutani K, Miyamoto S, Nagahata T, Konishi N, Emi M, Onda M. Tumori. 2005;91:546. doi: 10.1177/030089160509100616. [DOI] [PubMed] [Google Scholar]; d) Okino K, Nagai H, Hatta M, Nagahata T, Yoneyama K, Ohta Y, Jin E, Kawanami O, Araki T, Emi M. Oncol. Rep. 2003;10:1219. doi: 10.3892/or.10.5.1219. [DOI] [PubMed] [Google Scholar]
- [7].Wong HC, Bourdelas A, Krauss A, Lee HJ, Shao YM, Wu D, Mlodzik M, Shi DL, Zheng J. Mol. Cell. 2003;12:1251. doi: 10.1016/s1097-2765(03)00427-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Shan J, Shi DL, Wang J, Zheng J. Biochemistry. 2005;44:15495. doi: 10.1021/bi0512602. [DOI] [PubMed] [Google Scholar]; b) Fujii N, You L, Xu Z, Uematsu K, Shan J, He B, Mikami I, Edmondson LR, Neale G, Zheng J, Guy RK, Jablons DM. Cancer Res. 2007;67:573. doi: 10.1158/0008-5472.CAN-06-2726. [DOI] [PubMed] [Google Scholar]; c) Wang NX, Lee H-J, Zheng JJ. Drug News Perspect. 2008;21:137. [PMC free article] [PubMed] [Google Scholar]; d) Shan J, Zheng JJ. J Comput. Aided. Mol. Des. 2009;23:37. doi: 10.1007/s10822-008-9236-1. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Lee H-J, Wang NX, Shao Y, Zheng JJ. Bioorg. Med. Chem. 2009;17:1701. doi: 10.1016/j.bmc.2008.12.060. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Grandy D, Shan J, Zhang X, Rao S, Akunuru S, Li H, Zhang Y, Alpatov I, Zhang XA, Lang RA, Shi DL, Zheng JJ. J. Biol. Chem. 2009;12:16256. doi: 10.1074/jbc.M109.009647. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Zhang Y, Appleton BA, Wiesmann C, Lau T, Costa M, Hannoush RN, Sidhu SS. Nat. Chem. Biol. 2009;5:217. doi: 10.1038/nchembio.152. [DOI] [PubMed] [Google Scholar]
- [9].Pellecchia M, Sem DS, Wuthrich K. Nat. Rev. Drug Discov. 2002;1:211. doi: 10.1038/nrd748. [DOI] [PubMed] [Google Scholar]
- [10].Cheyette BN, Waxman JS, Miller JR, Takemaru K, Sheldahl LC, Khlebtsova N, Fox EP, Earnest T, Moon RT. Dev.Cell. 2002;2:449. doi: 10.1016/s1534-5807(02)00140-5. [DOI] [PubMed] [Google Scholar]
- [11].a) Bhattacharyya RP, Remenyi A, Yeh BJ, Lim WA. Annu. Rev. Biochem. 2006;75:655. doi: 10.1146/annurev.biochem.75.103004.142710. [DOI] [PubMed] [Google Scholar]; b) Feng W, Fan JS, Jiang M, Shi YW, Zhang M. J.Biol.Chem. 2002;277:41140. doi: 10.1074/jbc.M207206200. [DOI] [PubMed] [Google Scholar]; c) Long JF, Tochio H, Wang P, Fan JS, Sala C, Niethammer M, Sheng M, Zhang M. J.Mol.Biol. 2003;327:203. doi: 10.1016/s0022-2836(03)00113-x. [DOI] [PubMed] [Google Scholar]
- [12].a) Iwahara J, Wojciak JM, Clubb RT. J. Biomol. NMR. 2001;19:231. doi: 10.1023/a:1011296112710. [DOI] [PubMed] [Google Scholar]; b) Zwahlen C, Legault P, Vincent S, Greenblatt J, Konrat R, Kay LE. J.Am.Chem.Soc. 1997;119:6711. [Google Scholar]
- [13].Brannon M, Gomperts M, Sumoy L, Moon RT, Kimelman D. Genes Dev. 1997;11:2359. doi: 10.1101/gad.11.18.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) Polakis P. Curr. Opin. Genet. Dev. 2007;17:45. doi: 10.1016/j.gde.2006.12.007. and references therein. [DOI] [PubMed] [Google Scholar]; b) Segditsas S, Rowan AJ, Howarth K, Jones A, Leedham S, Wright NA, Gorman P, Chambers W, Domingo E, Roylance RR, Sawyer EJ, Sieber OM, Tomlinson IP. Oncogene. 2009;28:146. doi: 10.1038/onc.2008.361. [DOI] [PubMed] [Google Scholar]
- [15].a) Suzuki H, Watkins DN, Jair KW, Schuebel KE, Markowitz SD, Chen WD, Pretlow TP, Yang B, Akiyama Y, Van Engeland M, Toyota M, Tokino T, Hinoda Y, Imai K, Herman JG, Baylin SB. Nat. Genet. 2004;36:417. doi: 10.1038/ng1330. [DOI] [PubMed] [Google Scholar]; b) Sato H, Suzuki H, Toyota M, Nojima M, Maruyama R, Sasaki S, Takagi H, Sogabe Y, Sasaki Y, Idogawa M, Sonoda T, Mori M, Imai K, Tokino T, Shinomura Y. Carcinogenesis. 2007;28:2459. doi: 10.1093/carcin/bgm178. [DOI] [PubMed] [Google Scholar]; c) Caldwell GM, Jones CE, Soon Y, Warrack R, Morton DG, Matthews GM. Br. J. Cancer. 2008;98:1437. doi: 10.1038/sj.bjc.6604327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Jiang X, Tan J, Li J, Kivimae S, Yang X, Zhuang L, Lee PL, Chan MT, Stanton LW, Liu ET, Cheyette BN, Yu Q. Cancer Cell. 2008;13:529. doi: 10.1016/j.ccr.2008.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chong CR, Sullivan DJ., Jr. Nature. 2007;448:645. doi: 10.1038/448645a. [DOI] [PubMed] [Google Scholar]
- [18].a) Schuttelkopf AW, van Aalten DM. Acta Crystallogr D Biol Crystallogr. 2004;60:1355. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]; b) Case DA, Cheatham TE, III, Darden T, Gohlke H, Luo R, Merz KM, Jr., Onufriev A, Simmerling C, Wang B, Woods RJ. J. Comput. Chem. 2005;26:1668. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Crystallogr D Biol Crystallogr. 1998;54:905. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]; d) Dominguez C, Boelens R, Bonvin AM. J. Am. Chem. Soc. 2003;125:1731. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]; e) Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. J. Biomol. NMR. 1996;8:477. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]; f) Koradi R, Billeter M, Wuthrich K. J. Mol. Graph. 1996;14:51. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- [19].Umbhauer M, Djiane A, Goisset C, Penzo-Mendez A, Riou JF, Boucaut JC, Shi D-L. EMBO J. 2000;19:4944. doi: 10.1093/emboj/19.18.4944. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.