

The conjugate addition of enolate or enol nucleophiles to activated double bonds (the Michael reaction) is an efficient method for the synthesis of 1,5-dicarbonyl compounds.[1] The potential for the direct formation of a new carbon–carbon bond with control of up to three new stereogenic centers has driven continued and significant development of this reaction. Two central strategies are the use of metalloenolates and the addition of latent nucleophiles such as enol silanes in combination with a Lewis acid.[2] Studies by the research groups of Yamaguchi,[3] MacMillan,[4] Jørgensen,[5] and List[6] have demonstrated that secondary amines catalyze Michael reactions by the generation of activated unsaturated electrophiles through iminium ions. The corresponding strategy to the iminium approach is the catalytic generation of an enolate or enol nucleophile.[7,8] Herein we report that N-heterocyclic carbenes (NHCs) are highly selective catalysts for the intramolecular Michael reaction of substrates 1 to afford dicarbonyl compounds 2 after the addition of an exogenous nucleophile [Eq. (1)].
 |
(1) |
The Michael reaction traditionally relies on the stoichiometric generation of an enolate or enol. In our recent studies that involved NHCs and carbonyl compounds,[9] the opportunity to catalyze the formation of enol intermediates became apparent.[10] We anticipated that intramolecular Michael reactions catalyzed by NHCs could be achieved by using a conjugate acceptor with the general structure of 1. The proposed pathway for this process involves the addition of the NHC to an α,β-unsaturated aldehyde to afford the extended diene intermediate I (Scheme 1).[11] The key enol nucleophile is revealed by β-protonation of I followed by a Michael addition to generate the enol III. An intramolecular acylation event releases the NHC catalyst to afford the bicyclic acylated enol IV that readily opens to give products such as 2 on exposure to mild nucleophiles.[12]
Scheme 1.

Proposed catalytic pathway.
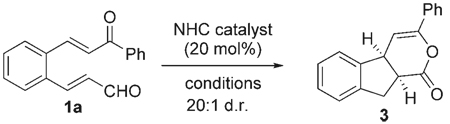
The search for optimal conditions for this process started with the parent enal 1a, imidazolium salt A, and typical reaction conditions for NHC catalysis (Table 1, entry 1, Mes = 2,4,6-trimethylphenyl). Unfortunately, only a small amount of the desired product 3 was observed. When the base was changed from DBU to iPr2EtN, we were pleased to observe an increased yield of 3 with greater than 20:1 selectivity favoring the cis diastereomer (entry 3).[13] The use of the achiral triazolium salt B in toluene with THF as a cosolvent to induce homogeneity of the reaction provided good yields (entry 4). Solvents that are not Lewis basic, such as toluene or CH2Cl2, were crucial to promote the β-protonation process with all the azolium precatalysts. This protonation step in turn generates the desired enol which participates in the reaction. When the concentration of 1a in the reaction was decreased, the intramolecular manifold was favored with a resulting increase in yield (entry 9).
Table 1.
Optimization of the Michael reaction.
 | ||||
|---|---|---|---|---|
| Entry | Catalyst | Conditions[a] | Yield[b] | ee [%][c] |
| 1 | A | DBU, THF | 6 | – |
| 2 | A | KN(SiMe3)2, THF[d] | 11 | – |
| 3 | A | iPr2EtN, THF, 45°C | 50 | – |
| 4 | B | iPr2EtN, toluene/THF[e] | 61 | – |
| 5 | C | iPr2EtN, toluene/THF[e,f] | 61 | 93 |
| 6 | C | Et3N (0.1 m), toluene/THF[e] | 62 | 93 |
| 7 | C | iPr2EtN (0.1 m), CH2Cl2, −20°C | 49 | 93 |
| 8 | D | iPr2EtN (0.1 m), toluene/THF[e] | 53 | 99 |
| 9 | D | iPr2EtN (0.05 m), toluene/THF[e] | 66 | 99 |
| 10 | D | iPr2EtN (0.05 m), CH2Cl2 | 68 | 99 |
| 11 | D[g] | iPr2EtN (0.05 m), CH2Cl2 | 68 | 99 |
Base (20 mol %), 1a (0.2 m) at 23°C unless otherwise noted. DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene.
Yield of isolated product.
Determined by HPLC (Chiracel AD-H). Absolute and relative configuration of 3 assigned by X-ray crystallography.[16] See the Supporting Information for details.
Carbene generated prior to addition of substrate.
10:1 toluene/THF.
Base (1.2 equiv).
D (10 mol %). ![[g]](https://cdn.ncbi.nlm.nih.gov/pmc/blobs/f179/2978499/db6e8c90974d/nihms248707t2.jpg)
Importantly, NHCs derived from the triazolium salts C and D were found to be the most efficient catalysts and provided a platform to control the stereochemical outcome for this process. Accordingly, the use of the phenylalanine-derived salt C afforded good yields of 3 with 93% ee (entries 5–7). When the structure of the catalyst was tuned to that from the salt D, which was derived from amino indanol and was first disclosed by Bode and co-workers,[14] we observed excellent levels of enantioselectivity with catalyst loadings of 10 mol% (99% ee, entry 11). It is important to note that the inclusion of N-mesityl substitution on the azolium salts was required for any reasonable conversion into the desired product.
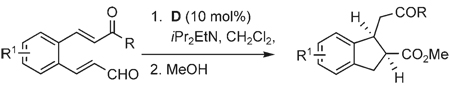
Once the triazolium salt D had been identified as the most selective precatalyst, we surveyed potential substrates for this intramolecular process (Table 2). The use of methanol to quench the reaction avoids the propensity for several of the bicyclic products to undergo hydrolysis when purified on silica gel. The optimized reaction conditions allowed both electron-withdrawing and -donating groups on the enone (entries 1–3), and additionally, electron-withdrawing and -donating substituents could be placed on the aromatic tether (entries 6 and 7). This intramolecular reaction was not restricted to the use of aromatic substituents. The α,β-unsaturated methyl ketone 1d provided a moderate yield of the cyclopentane product with excellent enantioselectivity (entry 4). The bisaldehyde 1e underwent an interesting desymmetrization reaction in which one aldehyde became the nucleophile when exposed to an NHC while the other unsaturated moiety became the conjugate acceptor (entry 5). The cyclization of the aliphatic substrate 13 (entry 8) proceeded in good yield after ten hours with a catalyst loading of 20 mol%. When the tether length was increased to access six-membered rings, cyclohexene products were afforded but with reduced enantioselectivity and yield (62% ee, 52%; entry 9).[15] Interestingly, product 16 did not open after the addition of methanol unlike the cyclopentane compound.
Table 2.
Substrate Scope.
 | ||||
|---|---|---|---|---|
| Entry | Substrate | Product | Yield [%][a] | ee [%][b] |
| 1 | 1a R = Ph | 4 | 69 | 99 |
| 2 | 1b R = 4-BrC6H4 | 5 | 62 | 99 |
| 3 | 1c R = 4- = MeC6H4 | 6 | 80 | 99 |
| 4 | 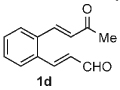 |
7 | 59 | 99 |
| 5 | 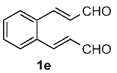 |
8 | 68 | 99 |
| 6 | 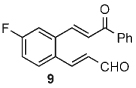 |
10 | 68 | 99 |
| 7 | 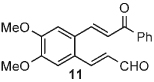 |
12 | 73 | 99 |
| 8[c] | 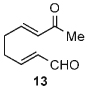 |
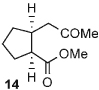 |
66 | 99 |
| 9[d] | 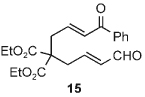 |
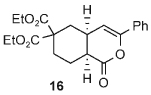 |
52 | 62 |
Yield of isolated product after purification.
Determined by HPLC on Chiracel AD-H or OD-H columns.
D (20 mol %) with the enantioselectivity determined by GC (BetaDex column).
D (20 mol %).
The efficient formation of methyl esters by the simple addition of methanol to the reaction in Table 2 indicated that bicyclic intermediates such as 3 are good acylating agents. Accordingly, substituted cyclopentyl amides, such as primary amide 17 and secondary amide 18, could be accessed in good yield directly by the addition of the corresponding primary and secondary amines to the reaction mixture after the starting material 1a had been consumed (Scheme 2).
Scheme 2.

Amide formation from the acylated enol products.
In summary, a highly diastereo- and enantioselective intramolecular Michael reaction catalyzed by N-heterocyclic carbenes has been developed. The addition of the carbene catalyst to an α,β-unsaturated aldehyde, followed by subsequent β-protonation generated a reactive enol intermediate that underwent addition to a pendant conjugate acceptor. Aryl and alkyl substituents are suitable for the reaction, and high enantioselectivity is achieved when chiral enantiopure triazolium salts are used. The turnover of the catalyst is facilitated by the generation of a cyclic O-acylated enol which is intercepted by alcohols and amines to provide different esters and amides. Further studies to generate nucleophiles by using N-heterocyclic carbenes are ongoing and will be reported in due course.
Experimental Section
The azolium salt D (4.2 mg, 0.01 mmol) and the corresponding enal (0.1 mmol) were added to a flame-dried round-bottom flask (10 mL) containing a magnetic stirring bar. The flask was sealed with a rubber septum and placed under a positive pressure of N2. The heterogeneous mixture was then diluted with CH2Cl2 (2 mL, 0.05 m). Once the material had dissolved, diisopropylethylamine (2 µL, 0.01 mmol) was added through a syringe. The reaction mixture was stirred at 23 °C under N2 until the enal had been completely consumed (as observed by TLC). Methanol (5 mL) was then added and the reaction mixture stirred at 23°C under N2 for 5 h. The reaction mixture was partially concentrated under reduced pressure and the remaining residue was purified by chromatography (silica gel, 5% EtOAc/Hexanes) to afford the pure methyl ester. Analytical data for 4: IR (film): ν̃ = 3024, 2947, 1730, 1685, 1442, 1365 cm−1; 1H NMR (500 MHz, CDCl3): δ= 7.95 (d, J = 7.3 Hz, 2H), 7.57 (t, J = 7.3 Hz, 1H), 7.46 (t, J = 7.6 Hz, 2H), 7.27 (d, J = 6.4 Hz, 1H), 7.19 (m, 3H), 4.28 (q, J = 7.3 Hz, 1H), 3.59 (m, 4H), 3.42 (m, 2H), 3.15 ppm (m, 2H); 13C NMR (125 MHz, CDCl3): δ =198.72, 174.72, 144.87, 137.26, 133.32, 128.84, 128.26, 127.53, 127.08, 124.81, 124.35, 51.90, 47.69, 42.75, 40.81, 34.73 ppm; LRMS (ES): calcd for C18H16O3 [M]+, 294.32; found [M+H]+, 295.5; [α]D = −16.6 (CH2Cl2, c = 1.0, 99.5:0.5 er). The enantiomeric ratio was determined by HPLC (Chiralcel AD-H, 15% 2-propanol/hexanes, 1 mL min−1, tr1 = 8.84, tr2 = 13.88).
Supplementary Material
Footnotes
Research support was generously provided by the NIH/NIGMS (RO1 GM73072), Abbott Laboratories, Amgen, 3M, and Boerhinger-Ingelheim. A.C. is the recipient of a Dow Chemical Company Fellowship. We thank T. Reynolds and C. Stern (NU) for assistance with X-ray crystallography. Funding for the NU Analytical Services Laboratory has been furnished in part by the NSF (CHE-9871268).
Supporting information for this article (detailed experimental procedures and full characterization of new compounds) is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.a) Michael A. J Prakt. Chem. 1887;36:349–356. [Google Scholar]; b) Bergmann ED, Ginsburg D, Pappo R. Org. React. 1959;10:179–555. [Google Scholar]; c) Oare DA, Heathcock CA. In: Topics in Stereochemistry. Eliel EL, Wilen SH, editors. Vol. 20. New York: Wiley; 1991. pp. 124–170. [Google Scholar]; d) Little RD, Masjedizadeh MR. Org. React. 1995;47:315–552. [Google Scholar]; e) Krause N, Hoffmann-Röder A. Synthesis. 2001:171–196. [Google Scholar]
- 2.a) Narasaka K, Soai K, Mukaiyama T. Chem. Lett. 1974:1223–1224. [Google Scholar]; b) Mukaiyama T, Kobayashi S. Org. React. 1994;46:1–103. [Google Scholar]; c) Evans DA, Scheidt KA, Johnston JN, Willis MC. J. Am. Chem. Soc. 2001;123:4480–4491. doi: 10.1021/ja010302g. and references therein. [DOI] [PubMed] [Google Scholar]; d) Jacobsen EN, Pfaltz A, Yamamoto H, editors. Comprehensive Asymmetric Catalysis. New York: Springer; 1999. chap. 31. [Google Scholar]
- 3.a) Yamaguchi M, Shiraishi T, Hirama M. Angew. Chem. 1993;105:1243–1245. Angew. Chem. Int. Ed. Engl.1993, 32, 1176 – 1178. [Google Scholar]; b) Yamaguchi M, Shiraishi T, Hirama M. J. Org. Chem. 1996;61:3520–3530. [Google Scholar]
- 4.Brown SP, Goodwin NC, MacMillan DWC. J. Am. Chem. Soc. 2003;125:1192–1194. doi: 10.1021/ja029095q. [DOI] [PubMed] [Google Scholar]
- 5.a) Melchiorre P, Jørgensen KA. J. Org. Chem. 2003;68:4151–4157. doi: 10.1021/jo026837p. [DOI] [PubMed] [Google Scholar]; b) Halland N, Aburel PS, Jørgensen KA. Angew. Chem. 2003;115:685–689. Angew. Chem. Int. Ed.2003, 42, 661 – 665. [Google Scholar]; c) Halland N, Aburel PS, Jørgensen KA. Angew. Chem. 2004;116:1292–1297. Angew. Chem. Int. Ed.2004, 43, 1272 – 1277. [Google Scholar]
- 6. List B. Tetrahedron. 2002;58:5573–5590. Hechavarria Fonseca MT, List B. Angew. Chem. 2004;116:4048–4050. doi: 10.1002/anie.200460578. Angew. Chem. Int. Ed.2004, 43, 3958 – 3960. c) For an excellent review of enamine catalysis, see: List B. Acc. Chem. Res. 2004;37:548–557. doi: 10.1021/ar0300571.
- 7.For examples of metal-catalyzed enolate generation, see: Yoshikawa N, Yamada YMA, Das J, Sasai H, Shibasaki M. J. Am. Chem. Soc. 1999;121:4168–4178. Trost BM, Ito H. J. Am. Chem. Soc. 2000;122:12003–12004. Evans DA, Tedrow JS, Shaw JT, Downey CW. J. Am. Chem. Soc. 2002;124:392–393. doi: 10.1021/ja0119548. Evans DA, Thomson RJ. J. Am. Chem. Soc. 2005;127:10506–10507. doi: 10.1021/ja053386s.
- 8.For examples of amine-catalyzed enamine generation, see: List B, Lerner RA, Barbas CF., III J. Am. Chem. Soc. 2000;122:2395–2396. Betancort JM, Sakthivel K, Thayumanavan R, Barbas CF., III Tetrahedron Lett. 2001;42:4441–4444. List B. Synlett. 2001:1675–1686. Dalko PI, Moisan L. Angew. Chem. 2001;113:3840–3864. Angew. Chem. Int. Ed.2001, 40, 3726 – 3748. List B. Tetrahedron. 2002;58:5573–5590. Hayashi Y, Gotoh H, Tamura T, Yamaguchi H, Masui R, Shoji M. J. Am. Chem. Soc. 2005;127:16028–16029. doi: 10.1021/ja055740s.
- 9.a) Mattson AE, Bharadwaj AR, Scheidt KA. J. Am. Chem. Soc. 2004;126:2314. doi: 10.1021/ja0318380. [DOI] [PubMed] [Google Scholar]; b) Bharadwaj AR, Scheidt KA. Org. Lett. 2004;6:2465. doi: 10.1021/ol049044t. [DOI] [PubMed] [Google Scholar]; c) Mattson AE, Scheidt KA. Org. Lett. 2004;6:4363. doi: 10.1021/ol0481129. [DOI] [PubMed] [Google Scholar]; d) Myers MC, Bharadwaj AR, Milgram BC, Scheidt KA. J. Am. Chem. Soc. 2005;127:14675. doi: 10.1021/ja0520161. [DOI] [PubMed] [Google Scholar]; e) Chan A, Scheidt KA. Org. Lett. 2005;7:905. doi: 10.1021/ol050100f. [DOI] [PubMed] [Google Scholar]; f) Chan A, Scheidt KA. J. Am. Chem. Soc. 2006;128:4558. doi: 10.1021/ja060833a. [DOI] [PubMed] [Google Scholar]
- 10.Enols have also been implemented as intermediates in reactions that combine NHCs and aldehydes; see: Reynolds NT, Rovis T. J. Am. Chem. Soc. 2005;127:16406–16407. doi: 10.1021/ja055918a. He M, Struble JR, Bode JW. J. Am. Chem. Soc. 2006;128:8418–8420. doi: 10.1021/ja062707c.
- 11.The potential hetero-Diels–Alder pathway is unlikely given the constrained nature of the intramolecular substrates and the high levels of cis diastereoselectivity. For a related intermolecular process using triazolium salt D as the precatalyst which invokes a hetero-Diels–Alder pathway, see: He M, Uc GJ, Bode JW. J. Am. Chem. Soc. 2006;128:15088–15089. doi: 10.1021/ja066380r.
- 12.The high diastereoselectivity in this reaction may arise because of a reversible Michael reaction followed by an irreversible lactonization occurring through the cis isomer. Investigations of these mechanistic pathways are ongoing.
- 13.For an approach to the trans diastereomers of compounds such as 4 using the Hantzsch ester and a chiral secondary amine, see Yang JW, Fonseca MTH, List B. J. Am. Chem. Soc. 2005;127:15036–15037. doi: 10.1021/ja055735o.
- 14.See Ref [10b]. For relevant examples of chiral triazolium salts used in asymmetric catalysis, see: Enders D, Breuer K, Runsink J, Teles JH. Helv. Chim. Acta. 1996;79:1899–1902. Kerr MS, de Alaniz JR, Rovis T. J. Am. Chem. Soc. 2002;124:10298–10299. doi: 10.1021/ja027411v. Enders D, Kallfass U. Angew. Chem. 2002;114:1822–1824. doi: 10.1002/1521-3773(20020517)41:10<1743::aid-anie1743>3.0.co;2-q. Angew. Chem. Int. Ed.2002, 41, 1743. Reynolds NT, de Alaniz JR, Rovis T. J. Am. Chem. Soc. 2004;126:9518. doi: 10.1021/ja046991o. Enders D, Balensiefer T. Acc. Chem. Res. 2004;37:534. doi: 10.1021/ar030050j. de Alaniz JR, Rovis T. J. Am. Chem. Soc. 2005;127:6284. doi: 10.1021/ja0425132. Enders D, Niemeier O, Balensiefer T. Angew. Chem. 2006;118:1491. doi: 10.1002/anie.200503885. Angew. Chem. Int. Ed.2006, 45, 1463.
- 15.The remaining mass balance is primarily the five-membered ring which resulted from the conjugate addition of the homoenolate formed in situ.
- 16.CCDC-63 1931 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.