

Abstract
Multiple sclerosis (MS) is a CD4+ T cell-mediated autoimmune disease affecting the CNS. MS and its animal model, experimental autoimmune encephalomyelitis (EAE), have been thought to be Th1-mediated diseases. However, recent studies provide strong evidence that the major pathogenic T cell subsets in EAE are Th17 cells. IL-9, a hematopoietic growth factor, is considered to be a mediator of Th17 cells, but the precise mechanisms of its action are largely unknown. The present study was designed to investigate the role of IL-9 in autoimmune demyelination. IL-9 blockade with anti-IL-9 monoclonal antibody (mAb) inhibited the development of EAE, reduced the serum levels of IL-17, the CNS mRNA expression of IL-17, IL-6, IFN-γ and TNF-α, and the MOG-induced IL-17, IFN-γ secretion of lymphocytes. Further, anti-IL-9 mAb in culture suppressed IL-17 production of MOG-reactive T cells and their potency in adoptive transfer EAE. These findings indicate that the protective effect of IL-9 blockade in EAE was likely mediated via inhibition of the development of MOG-peptide-specific T cells, which in turn led to reduced infiltration of T cells into the CNS. Thus, anti-IL-9 mAb treatment may provide an effective therapeutic strategy against autoimmune diseases.
Keywords: IL-9, T helper cells, Autoimmunity, EAE, Multiple sclerosis
Introduction
Experimental autoimmune encephalomyelitis (EAE), a CD4+ T cell-mediated inflammatory demyelinating disease of the central nervous system (CNS), serves as a model of the human disease multiple sclerosis (MS) (1). EAE can be induced by immunization with myelin antigens or by adoptive transfer of myelin-reactive CD4+ T cells into naive recipients.
Although there is evidence linking CD4+ T cells such as Th17 (2, 3), Th1 (4, 5), and Th9 cells (6) with the pathogenesis of EAE, the precise contribution of these T cell subtypes or their associated cytokines is still unclear. There is growing evidence that clinically similar forms of autoimmune demyelinating disease can be driven by myelin-specific T cells of distinct lineages, with different degrees of dependence on IL-17A production by Th17 cells, to achieve their pathological effects (2, 7-9). Moreover, IL-17-deficient mice have been reported to develop EAE with delayed onset and reduced severity (10, 11). In humans, Th17 cells have been identified in the CNS of patients with MS (12, 13). The pathogenic role of IL-17 in MS has been suggested in numerous studies (14-16). Because IL-17 signaling is important for the production of various chemokines from fibroblasts and epithelial cells (14-16), which attract APCs to the CNS, resulting in demyelination, the suppression of the development or the proliferation of Th17 cells may represent a promising therapy for MS. However, recently, Axtell RC et al. showed regular susceptibility to EAE induction in the absence of IL-17A and IL-17F, as well as suppression of EAE in the presence of unaltered Th17 responses, and they suggested that both Th1 and IL-17 producing CD4+ T cells are important for the development of EAE.
It has been demonstrated that IL-9 together with TGF-β can induce the differentiation of naïve CD4+ T cells into Th17 cells in vitro (17, 18), and that IL-9 produced by Th17 cells themselves amplifies Th17 development in a positive autocrine loop (17). Thus, IL-9 represents a potential target for the inhibition of Th17 development in vivo. Temporal blockade of IL-9 using neutralizing antibody may produce results inconsistent with those obtained in IL-9 receptor-deficient mice, given that IL-9 acts as a hematopoietic growth factor and its complete absence may influence hematopoietic development (19, 20). Treatment with anti-IL-9 mAb has been reported in one study to be protective against EAE (17). However, in another study, IL-9 receptor knockout mice were more susceptible to EAE than WT mice (21), and they were partially resistant in another study (17).
Additionally, IL-9 inhibited lymphokine production by IFN-γ-producing CD4+ T cells (22), exogenous IL-9 reduced IFN-γ mRNA expression in PBMC from latent tuberculosis infection by 30%, and neutralization of IL-9 restored IFN-γ mRNA expression (23). Activation of naïve T cells in the presence of TGF-β and anti-IFN-γ significantly enhanced IL-9 production (24). These results suggest pleiotropic effects on effecter CD4+ T cells. The role of IL-9 in the pathogenesis of MS/EAE thus remains unclear.
In the present study, we investigated the in vivo role of IL-9 in the development of T cells, particularly Th17 cells, in EAE, using an anti-IL-9 mAb, which has shown significant protective effect in other inflammatory diseases (25, 26). Our results show that early anti-IL-9 mAb treatment reduces encephalitogenic Th1 and Th17 cells and inflammatory myeloid cell invasion into the CNS.
Methods
Mice
C57BL/6 mice were obtained from The Jackson Laboratory (Bar Harbor, ME). For all experiments 8- to 10-wk-old female mice were used. All animals were housed under specific pathogen-free conditions and animal protocols were approved by the Thomas Jefferson University Animal Care and Use Committee. Paralyzed mice were afforded easy access to food and water.
Induction of EAE
Mice were immunized s.c. with 200 μg of MOG35-55 peptide emulsified in CFA and injected twice with pertussis toxin (27). The severity of EAE was monitored and graded on a scale of 0-5: 0 = no disease; 1 = limp tail; 2 = hind limb weakness; 3 = hind limb paralysis; 4 = hind and fore limb paralysis; 5 = moribund and death.
Anti-IL-9 mAb treatment
Anti-IL-9 mAb (clone D9302C12, BD Pharmingen, San Diego, CA) was confirmed in vitro to have inhibitory effects upon IL-9 bioactivity (28). In a pilot study we sought to ascertain the optimal dose of anti-IL-9 mAb in vivo. In brief, anti-IL-9 mAb at doses of 0.3 mg, 1.2 mg, 4.8 mg (n = 3 in each group) or isotype-matched hamster IgG at a dose of 4.8 mg (n = 3) was injected i.p. once starting with day -1 of EAE induction. Blood from the orbital vein was obtained via a capillary tube on day 19. Anti-IL-9 mAb was measured by ELISA (eBioscience). We found that a dose of 0.3 mg is optimal (data not shown).
For IL-9 blockade, mice were i.p. treated with 0.3 mg of hamster no-immune isotype control I (clone Ha4/8, hamster Gig, BD Pharmingen) or anti-IL-9 mAb (clone D9302C12, hamster IgG, BD Pharmingen) every other day starting on day -1 post immunization (p.i.). Other groups of mice received 10 ng rIL-9 (Akron Biotech, Philadelphia, PA) dissolved in 0.3 ml of PBS daily beginning on the day of EAE induction.
CNS histology
Spinal cords were carefully dissected and immersion fixed in 4% formaldehyde in PBS (27). In brief, fixed tissues were embedded in paraffin wax and sections were cut from the mid lumbar spinal cord (L3). Sections were stained with H&E or Luxol fast blue (myelin stain) for analyses of inflammation and demyelization, respectively. Slides were assessed in a blind fashion for inflammation and demyelination as described previously (29, 30). Briefly, inflammation: 0, none; 1, a few inflammatory cells; 2, organization of perivascular infiltrates; and 3, increasing severity of perivascular cuffing with extension into the adjacent tissue. For demyelination: 0, none; 1, rare foci; 2, a few areas of demyelination; 3, large (confluent) areas of demyelination.
Isolation of CNS-infiltrating cells
Mice were perfused via the left atrium with glucose containing PBS, and spinal cords were removed. Mononuclear cells were isolated by digestion of the spinal cord homogenate with collagenase and DNAse, followed by Percoll gradient centrifugation. CNS homogenates were pooled for each treatment group prior to analysis. For flow cytometry analyses, these cells were incubated with Abs to murine CD11b, CD11c, CD4, CD45, CD8a, and B220 (all from BD Pharmingen). The absolute numbers for each population were calculated by multiplying the frequency of each population by the total number of cells isolated per treatment group.
Gene expression analysis by real-time PCR
Mice were sacrificed at the peak of actively induced EAE disease: IgG-treated group (n=8, mean score 3.25±0.14, range 3-3.5). Anti-IL-9 mAb-treated group (n = 4). Additionally, six healthy mice were sacrificed to serve as controls. Animals were perfused transcardially with PBS, and spinal cords were removed and stored in RNAlater (Ambion, Austin, TX). Tissues were homogenized with a TissueLyser (Qiagen, Valencia, CA) and RNA was extracted using RNeasy Lipid Tissue Midi kits (Qiagen). cDNA was synthesized using Superscript II first-strand synthesis kits (Invitrogen, Carlsbad, CA) and gene expression was analyzed by Taqman real-time PCR (Applied Biosystems, Foster City, CA). β-actin was used as an endogenous control in all samples and levels of gene expression were compared with healthy controls. Relative expression was calculated following the previously described protocol (27).
Adoptive EAE
Female 8-10 week-old wild type mice were immunized s.c. with 100 μg of MOG35-55 containing 200 μg of Mycobacterium tuberculosis H37Ra (Disco, Detroit, MI) on day 0 and day 7. The immunized mice were treated with anti-IL-9 mAb (300 μg) once on days -1, 0, 2, 4, 6, 9, 10 or with 20 ng of IL-9 every day beginning on the day after injection, Control mice were treated with PBS. Lymph node cells were harvested on day 12 p.i. and cultured for 3 d in RPMI 1640 supplemented with 10% FCS, penicillin/streptomycin, L-glutamine, HEPES, sodium private and 2-ME. All cells were cultured in the presence of MOG35-55 (50μg/ml), and IL-23 (10 ng/ml). After 72 hr, cells were harvested, washed in PBS and transferred to naïve recipient mice (2×107 cells/mouse) via the tail vein. The mice were given two doses (250 ng/mouse) of pertussis toxin i.p. on days 0 and 2 post cell transfer. EAE disease induction was assessed as described above.
Intracellular cytokine staining
Draining lymph node cells were stimulated with 15 μg/ml MOG35-55 peptide for 72 h and restimulated with 50 ng/ml phorbol 12-myristate 13-acetate (PMA) and 750 ng/ml ionomycin for the last 4 h in the presence of 10 μg/ml Brefeldin A. To analyze lymphocytes infiltrated into the CNS, spinal cords were removed as described above, followed by digestion with collagenase D (5.0 mg/ml; Roche Diagnostics). Cells were isolated by Percoll centrifugation as previously described (27), and surface-stained with antibodies against CD4 (RM4-5;BD Biosciences, San Jose, CA). Intracellular cytokine staining and FoxP3 staining were performed as described in (27, 31). Cells were analyzed by FACSAria flow cytometer (BD Biosciences), and data obtained were analyzed by FlowJo software (Tree Star, Ashland, OR).
Recall response and T cell cytokine production
Spleens were taken from mice sacrificed 8 d after MOG35-55 immunization. Cells were gently dispersed through nylon mesh into a single cell suspension, washed, and cultured at 106 cells per milliliter in complete RPMI (RPMI 1640 containing 10% heat-inactivated FCS [CSL, Parkville, Victoria, Australia], 2 mM l-glutamine, 100 U/ml of penicillin, and 100 μg/ml of streptomycin). Cells were either stimulated with MOG35-55 (15 μg/ml) or anti-CD3 (1 μg/ml) + CD28 (1 μg/ml) or left without any exogenous stimuli. For proliferation, [3H] thymidine (1 μCi/well) was added at 60 hr and incorporation of thymidine in DNA was measured after 12 hr by 1450 Microbeta Wallac Trilux liquid scintillation counter (Perkin Elmer Life Sciences, Foster City, CA). Supernatants were collected at 72 h and quantitative ELISA was performed for cytokines using paired mAbs as recommended by the manufacturer (R&D Systems, Minneapolis, MN). For serum cytokine ELISA, blood was taken during the course of EAE via the tail vein, and serum samples were prepared using Z-Gel microtubes (Sarstedt, Newton, NC). Mouse IL-17 was detected with a Quantikine immunoassay kit (R&D Systems).
In vitro Th17 cell differentiation
Spleens were removed from naïve mice and passed through a 100- μm cell strainer. Erythrocytes were lysed. CD4+ T cells, purified using magnetic bead separation kits (Miltenyi Biotec, Auburn, CA), were cultured with anti-CD3 (1 μg/ml) and anti-CD28 (1 μg/ml) Abs (BD Pharmingen), TGF-β (3 ng/ml), IL-6 (30 ng/ml) and anti-IL-4 (10 μg/ml) (all from Invitrogen) in the presence or absence of recombinant mouse IL-9 (10 ng/ml) (Akron Biotech), or anti-IL-9 mAb (100 ng/ml).
Statistical analysis
The two-tailed Student t test and the χ2 test were used to analyze the significance of results. Additionally, the Mann-Whitney U test was performed for nonparametric analyses. Differences were considered significant at p ≤ 0.05.
Results
Anti-IL-9 mAb treatment suppressed acute EAE
To determine the preventive efficacy of IL-9 blockade in autoimmune inflammatory disease, we tested the effect of anti-IL-9 mAb on EAE. Anti-IL-9 mAb treatment delayed the onset of clinical disease, ameliorated the severity of EAE (Fig. 1A), and reduced its incidence from 100% to 25% (Fig. 1B). This result is consistent with studies by Nowak et al. (17) demonstrating that targeted genetic disruption of IL-9 receptor results in resistance to EAE.
Figure 1. Anti-IL-9 mAb suppressed actively induced EAE.

C57BL/6 mice immunized for EAE induction were treated with anti-IL-9 mAb, or control IgG every other day starting on day -1 p.i. (A). Filled circles indicate the IgG-treated group (n = 10), and open circles indicate the anti-IL-9 mAb-treated group (n = 10). Clinical scores (averages ± SEM) combining three independent experiments are shown. Incidence of EAE in these mice is shown in (B). Statistics were calculated with the Mann-Whitney U test (A) and the χ2 test (B). ***, p<0.001
Treatment with anti-IL-9 mAb reduces CNS inflammation and demyelination
At the peak of EAE control IgG-treated mice had extensive infiltration of mononuclear cells into the white matter of spinal cords (Fig. 2A-C). In contrast, cellular infiltration and demyelination were markedly reduced in spinal cords of anti-IL-9 mAb-treated mice, which was consistent with their decreased clinical scores (Fig. 2D-F). The difference between the pathological scores of control IgG-treated mice and anti-IL-9 mAb treatment groups of mice was highly significant (p<0.01) (Fig. 2G).
Figure 2. Anti-IL-9 mAb-treated mice have reduced pathology in the CNS.
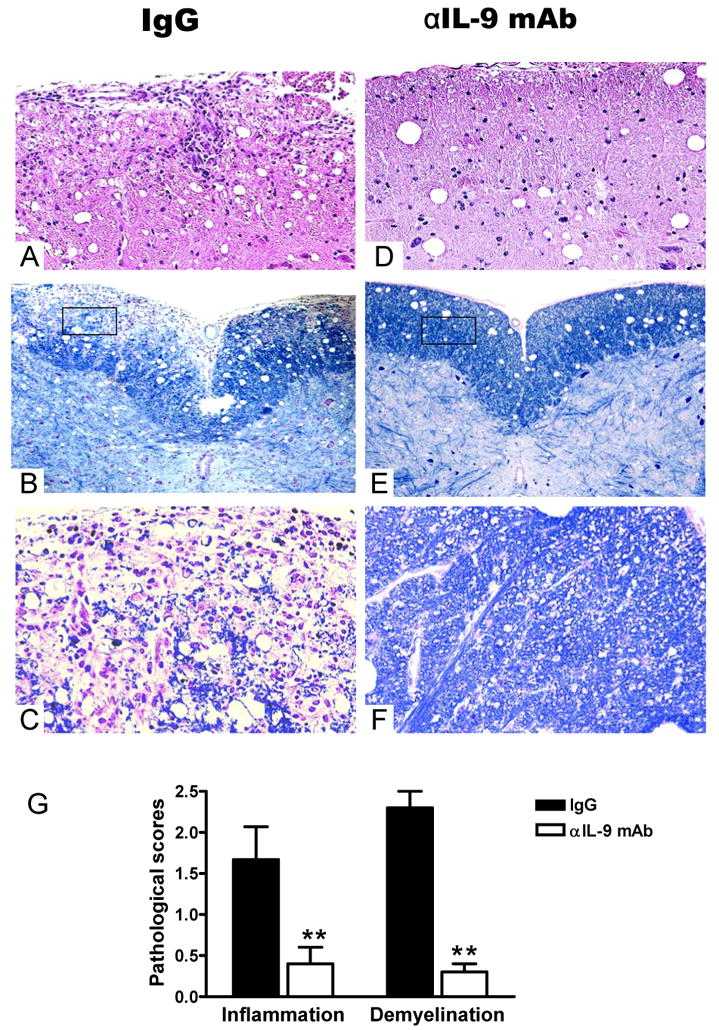
At disease peak, spinal cords were harvested after extensive perfusion, and 5 μm sections were stained with H&E and Luxol fast blue. Examples of mice receiving control IgG i.p. infiltration (A), demyelination (B and C), anti-IL-9 mAb i.p. infiltration (D), demyelination (E and F) are shown. The differences between control IgG -i.p. and anti-IL-9 mAb i. p groups were significant (all p <0.01). (G) Mean scores of inflammation and demyelination ± SD combining three independent experiments are shown (n =15 each group). **, p <0.01.
Anti-IL-9 mAb treated mice are devoid of CNS-infiltrated cells
To investigate the population of mononuclear cells that infiltrated into the CNS, we recovered mononuclear cells from spinal cords and stained them with CD4, CD8, B220, and CD11b antibodies. At the peak of EAE, CD4+ T cells, CD8+ T cells, B cells, and macrophages were detected in control IgG-treated mice (Fig. 3A). In anti-IL-9 mAb-treated mice, the numbers of these cells were significantly reduced (Fig. 3A). Compared to control IgG-treated mice, the CNS of anti-IL-9 mAb-treated mice expressed significantly lower levels of IL-17, IL-6, IFN-γ and TNF-α, but higher levels of FoxP3 mRNA (all p<0.05∼0.001, Fig. 3B).
Figure 3. Anti-IL-9 mAb-treated mice are devoid of CNS-infiltrated cells.

(A) Analysis of mononuclear cells infiltrating into the spinal cords. Mononuclear cells were recovered from spinal cord at day 19 p.i. Isolated cells were stained with antibodies against CD45, CD4, CD8, B220, and CD11b. CD45 high populations were gated. (B) RNA was made from spinal cord samples; the mean values of mRNA expression relative to β-actin are shown. Filled bars indicate the control IgG-treated group (n = 8), and open bars indicate the anti-IL-9 mAb-treated group (n = 4). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Induction of MOG-specific T cells was suppressed in anti-IL-9 mAb treated mice
To investigate whether the deficiency of CNS-infiltrating CD4+ T cells in anti-IL-9 mAb treated mice was due to a T cell priming defect, we analyzed MOG35-55-peptide-specific CD4+ T cells from draining lymph nodes. In the lymphocytes prepared from inguinal lymph nodes at priming stage (8 days after with immunization), the development of Th17 cells as well as Th1 cells was significantly suppressed in anti-IL-9 mAb-treated mice compared with IgG-treated mice (Fig. 4A). The population of FoxP3-positive Treg cells was not significantly changed in anti-IL-9-mAb-treated mice (Fig. 4B). To investigate the effect of IL-9 blockade on MOG35-55-peptide-specific cytokine production, we quantified the concentrations of cytokine levels secreted into the culture supernatant of lymphocytes from inguinal lymph nodes. We found that the production of IL-17 and IFN-γ was significantly suppressed in anti-IL-9 mAb-treated mice compared with control mice (Fig. 4C). Serum IL-17 level was also lower in anti-IL-9-mAb treated mice (Fig. 4D). Additionally, anti-IL-9 mAb-treated T cells showed a significantly lower proliferative response to anti-CD3 stimulation and MOG35-55-peptide than did wild type T cells (Fig. 4E). These results suggest that suppression of the differentiation of MOG35-55-reactive Th17 and Th1 cells by IL-9 blockade contributes to the protective effect against EAE.
Figure 4. IL-9 blockade suppressed the induction of MOG35-55 peptide-specific T cells in peripheral lymphoid tissue.
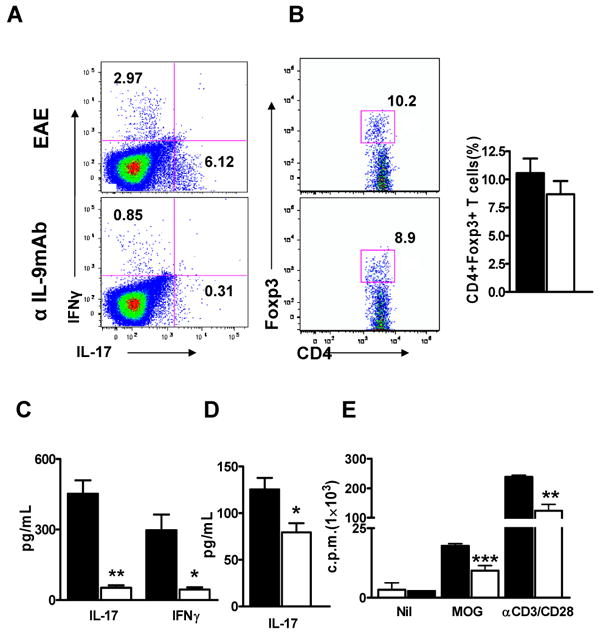
Intracellular staining of lymphocytes stimulated with MOG35-55 peptide. Inguinal lymph node cells were recovered at day 8 p.i. All plots were gated on CD4+ T cells (A and B). (C) IL-9 blockade suppressed the antigen-specific cytokine production of IL-17 and IFN-γ from lymphocytes. Splenocytes were recovered at the peak of disease after immunization and restimulated with 50 μg/ml MOG35-55 peptide for 72 h. IL-17 and IFN-γ concentrations in the supernatants were determined. (D) IL-9 blockade suppressed the serum level of IL-17. Mice were treated with anti-IL-9 mAb or control IgG, and serum samples were prepared at the peak of EAE after antigen immunization. IL-17 cytokine concentrations in the serum were analyzed. (E) Spleen cells from MOG35-55-immunized mice obtained 8 days p.i. were re-stimulated ex vivo with MOG35-55 peptide (10 μg/ml) or anti-CD3 (1 μg/ml) and anti-CD28 (1 μg/ml). Proliferation was determined by [3H] thymidine incorporation as described in the Materials and Methods section. n=5 in each group. All p values were determined by using the Student's t test. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Effect of anti-IL-9 mAb on Th17 differentiation in vitro
Given the role of IL-9 as a mediator of Th17-driven inflammatory disease (17), we next examined whether anti-IL-9 mAb had a direct effect on Th17 cell differentiation. Indeed, CD4+ T cells cultured with anti-IL-9 mAb showed significantly lower levels (3-fold decrease) of IL-17 production compared with wild type CD4+ T cells when cultured with IL-6 plus TGF-β, suggesting that neutralizing IL-9 with anti-IL-9 mAb decreased the development of Th17 cells (Fig. 5A). IL-17 production was enhanced in the presence of recombinant IL-9 (Fig. 5A), suggesting an amplification loop in which IL-9 produced by Th17 cells participates in enhancing further differentiation of Th17 cells.
Figure 5. Effect of anti-IL-9 mAb on the encephalitogenicity of MOG-specific Th17 cells.

(A) IL-17 production by naïve CD4+ T cells cultures under optimal Th17 conditions and in the presence of either IL-9 (10 ng/ml), or anti-IL-9 mAb (100 ng/ml). (B) Donor mice were immunized with MOG, IFA, and M. tuberculosis. Immunized mice were treated with either rIL-9 (20 ng/mouse), anti-IL-9 mAb (300 μg/mouse), or PBS. On day 12 post immunization, mice were sacrificed, and total lymph node cells were further primed with MOG35-55 (50 μg/ml), IL-23 (10 ng/ml) for 3 days. A total of 2 × 107 viable MOG-specific Th17 cells were adoptively transferred to naive recipient mice. n=5 in each group. Mice were examined for clinical signs every day until 20 dpt. *, comparison between rIL-9 -treated mice and wildtype mice. *, p < 0.05; **, p < 0.01; ***, p < 0.001. #, comparison between rIL-9 -treated mice and wildtype mice. #, p < 0.05.
Anti-IL-9 mAb inhibits the generation of MOG-specific T cells in vivo
Because in vitro treatment of T cells with IL-9 mAb inhibited the differentiation of Th17 cells, we next investigated whether IL-9 mAb treatment is capable of inhibiting the generation of encephalitogenic T cells in vivo. Donor mice were treated with anti-IL-9 mAb or rIL-9; T cells from these donor mice were stimulated with MOG and IL-23 for 3 d in vitro, and then transferred to recipient mice. In the control group, EAE was induced by adoptive transfer of enriched Th17 cells from donor mice that received only PBS. Mice that received MOG-specific T cells from IL-9 mAb-treated donor mice exhibited significantly less severe clinical signs and disease severity compared with the control EAE group (Fig. 5B). On the other hand, mice receiving enriched Th17 cells from recombinant IL-9-treated donor mice displayed a greater degree of disease severity compared with the control EAE group (Fig. 5B). These results suggest that IL-9 plays an important role in the generation of encephalitogenic T cells and that neutralization of IL-9 by mAb suppresses the production of encephalitogenic T cells in donor mice.
Discussion
CD4+ T-cell subsets are important for understanding adaptive immunity in humans and in animal models of autoimmune disease. Our knowledge of CD4+ T cell differentiation has changed significantly, and new subsets continue to be recognized. Importantly, the recent discovery of the CD4+ T cell subset that produces IL-9 has expanded significantly (32). IL-9 has largely been regarded as a Th2 cytokine that plays a significant role in inflammatory disease (33, 34). Under certain conditions, IL-9 can cause pleiotropic effects on Th1 (23, 35), Th17 (21, 36), regulatory T cells (21, 37) and the Th9 subset (21, 23, 36), playing different roles depending on microenvironments. Biological actions of IL-9 in autoimmune disease are continuing to be refined. Focusing on how anti-IL-9 mAb affects MOG-specific CD4+ T cells in EAE will provide a better mechanistic understanding of the pathogenesis of various autoimmune diseases.
In this study, we demonstrated that administration of anti-IL-9 mAb treatment effectively suppressed disease incidence and severity of EAE. This was associated with significantly decreased levels of both MOG-specific Th1 and Th17 cells in the periphery and the CNS. Our results, together with the finding that humanized anti-IL-9 mAb can be treated repeatedly without antigenicity (39), suggest that anti-IL-9 mAb treatment may provide an effective therapeutic strategy against autoimmune diseases.
Infiltration of MOG35–55-peptide-specific CD4+ T cells into the CNS is an important step in EAE disease onset (39), and IL-9 has been reported to increase levels of expression of eotaxin, MIP-1α, and MCP- 1, -3, and -5 in vitro and in vivo (40). These proteins have been suggested to be involved in the infiltration of lymphocytes through the blood-brain barrier (39). We therefore investigated the effect of anti-IL-9 mAb treatment against induction of EAE to elucidate the effect of IL-9 blockade against infiltration of activated lymphocytes into the spinal cord. We observed significantly reduced levels of inflammation and demyelination in the CNS of anti-IL-9 mAb-treated mice compared with control mice. Furthermore, anti-IL-9 mAb-treated mice exhibited a significant decrease in the absolute number of CD4+ T cells, CD8+ T cells, B cells, and macrophages infiltrating into the CNS. The absence of activated lymphocytes in the spinal cord of anti-IL-9-mAb-treated mice is likely to be mediated via the inhibition of the induction of MOG35–55-peptide-specific CD4+ T cells, including Th17, Th1 cells, and CD8+ T cells in the peripheral lymphoid tissue and also via the inhibition of infiltration of activated lymphocytes into the spinal cord.
Th17 cells directed against self-Ags cause severe autoimmune disease in mice, including EAE, non-obese diabetes and collagen-induced arthritis (7, 41-43). These studies highlight the importance of understanding the regulation of Th17 cell development in autoimmune disease. In mice, autoantigen-specific Th17 cells have been shown to be the dominant pathogenic T cell subset in EAE (7, 44, 45). Previous studies have suggested a pathogenic role of IL-9 as a Th17-derived cytokine that can contribute to inflammatory disease (17). We provide evidence that functional blocking of IL-9 inhibited the differentiation of Th17 cells in vitro. We investigated the in vivo role of IL-9 in T cell development in EAE, using an anti-IL-9 mAb and adoptive transfer EAE model. We showed that treatment with anti-IL-9 mAb effectively suppressed the incidence and severity of EAE. These results are consistent with previous studies using anti-IL-9 mAb treatment and IL-9 receptor deficient mice (17). Anti-IL-9 mAb-treated mice were devoid of mononuclear cells in the spinal cord. Importantly, treatment of donor mice with anti-IL-9 mAb also inhibited the generation of encephalitogenic T cells in vivo. In contrast, treatment of donor mice with recombinant IL-9 stimulated the generation of encephalitogenic T cells. As previously reported, IL-9 synergizes with TGF-β differentiated naïve CD4+ T cells into Th17 cells in vitro (21). Our data support the importance of IL-9 in the differentiation of Th17 cells in vivo.
Furthermore, we found that anti-IL-9 mAb potently suppressed IL-17 production in culture. IL-23-plus anti-IL-9 mAb treated cells transferred significantly less severe EAE to naive animals. Importantly, anti-IL-9 mAb negated this effect, as demonstrated by a low level of IL-17 production in anti-IL-9 mAb-treated cultures. Consistent with these observations, rIL-9 in culture enhanced IL-17 production and the potency of MOG-reactive T cells to transfer EAE. These findings provide further evidence that suppression of adoptive transfer EAE by anti-IL-9 mAb is mediated by Th17 suppression.
In this study, we observed that anti-IL-9 mAb treatment decreased the induction of Th1 cells during the priming stage of lymph nodes, suggesting that IL-9 may regulate the differentiation of Th1 cells. In agreement with this, it has been reported that IL-9 can induce IFN-γ production and suppress Th2 immune response in Mycobacterium infections (35). The effects of IL-9 were not limited to antagonizing suppressive factors in Mycobacterium infections; it also amplified the stimulatory activities of IL-2 (35, 46) and IL-6 (35). This general stimulation of cytolytic activities was accompanied by and dependent on IFN-γ, as the IFN-γ message was up-regulated by IL-9 (35). Moreover, antigen- and IL-18-stimulated Th1 cells can strongly produce IFN-γ and IL-9 (47). Culturing of sorted IFN-γ+ Th1 cells with OVA, IL-2, and IL-18 in the presence of APC led to an increase in IL-9, IL-13, IFN-γ and GM-CSF, but not IL-4 production (48). These results indicate a positive correlation between IL-9 and Th1 cells. Thus, decreased Th1 response after anti-IL-9 treatment, together with suppressed Th17 cells, represents an important mechanism underlying anti-IL-9 induced EAE suppression.
When comparing the developmental suppression of Th17 and? Th1 in peripheral lymphoid tissue of anti-IL-9 mAb-treated mice, the frequency of Foxp3+ cells was also slightly diminished at priming stage. At the same time, the effect of IL-9 blockade against the proliferation of MOG35-55-peptide-specific periphery T cells suggests that the deficiency of CNS-infiltrating CD4+ T cells in anti-IL-9 mAb treated mice was perhaps due to a T cell priming defect. However, at the peak stage, Foxp3mRNA increased in the CNS. The reason for the preferential accumulation of Treg cells in the CNS under anti-IL-9 mAb treatment should be further investigated.
In conclusion, our studies suggest that the protective effect of anti-IL-9 mAb treatment in EAE is mediated not only via suppression of IL-9-induced inflammatory reactions, but also via inhibition of the induction of MOG35-55-peptide-specific Th17 and Th1 cells, which in turn leads to reduced infiltration of T cells into the CNS. These findings not only provide evidence for an important pathogenic role of IL-9 in EAE, but also indicate that anti-IL-9 mAb treatment might represent a promising therapy for human MS and other Th17-mediated chronic autoimmune diseases.
Acknowledgments
We would like to thank Katherine Regan for editorial assistance.
This work was supported by grants from the National Institutes of Health, the National Multiple Sclerosis Society (NMSS), and the Commonwealth of Pennsylvania Department of Health (to A.R.).
Abbreviations used in this paper
- EAE
experimental autoimmune encephalomyelitis
- MOG
myelin oligodendrocyte glycoprotein
- MS
multiple sclerosis
- pi
postimmunization
Footnotes
Disclosures: The authors have no financial conflict of interest.
References
- 1.Steinman L. Assessment of animal models for MS and demyelinating disease in the design of rational therapy. Neuron. 1999;24:511–514. doi: 10.1016/s0896-6273(00)81107-1. [DOI] [PubMed] [Google Scholar]
- 2.Segal BM. Th17 cells in autoimmune demyelinating disease. Semin Immunopathol. 32:71–77. doi: 10.1007/s00281-009-0186-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carlson T, Kroenke M, Rao P, Lane TE, Segal B. The Th17-ELR+ CXC chemokine pathway is essential for the development of central nervous system autoimmune disease. J Exp Med. 2008;205:811–823. doi: 10.1084/jem.20072404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Axtell RC, de Jong BA, Boniface K, van der Voort LF, Bhat R, De Sarno P, Naves R, Han M, Zhong F, Castellanos JG, Mair R, Christakos A, Kolkowitz I, Katz L, Killestein J, Polman CH, de Waal Malefyt R, Steinman L, Raman C. T helper type 1 and 17 cells determine efficacy of interferon-beta in multiple sclerosis and experimental encephalomyelitis. Nat Med. 16:406–412. doi: 10.1038/nm.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang J. Yin and yang interplay of IFN-gamma in inflammation and autoimmune disease. J Clin Invest. 2007;117:871–873. doi: 10.1172/JCI31860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jager A, Dardalhon V, Sobel RA, Bettelli E, Kuchroo VK. Th1, Th17, and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. J Immunol. 2009;183:7169–7177. doi: 10.4049/jimmunol.0901906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stockinger B, Veldhoen M, Martin B. Th17 T cells: linking innate and adaptive immunity. Semin Immunol. 2007;19:353–361. doi: 10.1016/j.smim.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 10.Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- 11.Nakae S, Iwakura Y, Suto H, Galli SJ. Phenotypic differences between Th1 and Th17 cells and negative regulation of Th1 cell differentiation by IL-17. J Leukoc Biol. 2007;81:1258–1268. doi: 10.1189/jlb.1006610. [DOI] [PubMed] [Google Scholar]
- 12.Hofstetter H, Gold R, Hartung HP. Th17 Cells in MS and Experimental Autoimmune Encephalomyelitis. Int MS J. 2009;16:12–18. [PubMed] [Google Scholar]
- 13.Montes M, Zhang X, Berthelot L, Laplaud DA, Brouard S, Jin J, Rogan S, Armao D, Jewells V, Soulillou JP, Markovic-Plese S. Oligoclonal myelin-reactive T-cell infiltrates derived from multiple sclerosis lesions are enriched in Th17 cells. Clin Immunol. 2009;130:133–144. doi: 10.1016/j.clim.2008.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pilette C, Ouadrhiri Y, Van Snick J, Renauld JC, Staquet P, Vaerman JP, Sibille Y. Oxidative burst in lipopolysaccharide-activated human alveolar macrophages is inhibited by interleukin-9. Eur Respir J. 2002;20:1198–1205. doi: 10.1183/09031936.02.00005402. [DOI] [PubMed] [Google Scholar]
- 15.Matusevicius D, Kivisakk P, He B, Kostulas N, Ozenci V, Fredrikson S, Link H. Interleukin-17 mRNA expression in blood and CSF mononuclear cells is augmented in multiple sclerosis. Mult Scler. 1999;5:101–104. doi: 10.1177/135245859900500206. [DOI] [PubMed] [Google Scholar]
- 16.Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, Fugger L. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol. 2008;172:146–155. doi: 10.2353/ajpath.2008.070690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nowak EC, Weaver CT, Turner H, Begum-Haque S, Becher B, Schreiner B, Coyle AJ, Kasper LH, Noelle RJ. IL-9 as a mediator of Th17-driven inflammatory disease. J Exp Med. 2009;206:1653–1660. doi: 10.1084/jem.20090246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Locksley RM. Nine lives: plasticity among T helper cell subsets. J Exp Med. 2009;206:1643–1646. doi: 10.1084/jem.20091442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Donahue RE, Yang YC, Clark SC. Human P40 T-cell growth factor (interleukin-9) supports erythroid colony formation. Blood. 1990;75:2271–2275. [PubMed] [Google Scholar]
- 20.Williams DE, Morrissey PJ, Mochizuki DY, de Vries P, Anderson D, Cosman D, Boswell HS, Cooper S, Grabstein KH, Broxmeyer HE. T-cell growth factor P40 promotes the proliferation of myeloid cell lines and enhances erythroid burst formation by normal murine bone marrow cells in vitro. Blood. 1990;76:906–911. [PubMed] [Google Scholar]
- 21.Elyaman W, Bradshaw EM, Uyttenhove C, Dardalhon V, Awasthi A, Imitola J, Bettelli E, Oukka M, van Snick J, Renauld JC, Kuchroo VK, Khoury SJ. IL-9 induces differentiation of TH17 cells and enhances function of FoxP3+ natural regulatory T cells. Proc Natl Acad Sci U S A. 2009;106:12885–12890. doi: 10.1073/pnas.0812530106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gessner A, Blum H, Rollinghoff M. Differential regulation of IL-9-expression after infection with Leishmania major in susceptible and resistant mice. Immunobiology. 1993;189:419–435. doi: 10.1016/S0171-2985(11)80414-6. [DOI] [PubMed] [Google Scholar]
- 23.Wu B, Huang C, Kato-Maeda M, Hopewell PC, Daley CL, Krensky AM, Clayberger C. IL-9 is associated with an impaired Th1 immune response in patients with tuberculosis. Clin Immunol. 2008;126:202–210. doi: 10.1016/j.clim.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 24.Schmitt E, Germann T, Goedert S, Hoehn P, Huels C, Koelsch S, Kuhn R, Muller W, Palm N, Rude E. IL-9 production of naive CD4+ T cells depends on IL-2, is synergistically enhanced by a combination of TGF-beta and IL-4, and is inhibited by IFN-gamma. J Immunol. 1994;153:3989–3996. [PubMed] [Google Scholar]
- 25.Cheng G, Arima M, Honda K, Hirata H, Eda F, Yoshida N, Fukushima F, Ishii Y, Fukuda T. Anti-interleukin-9 antibody treatment inhibits airway inflammation and hyperreactivity in mouse asthma model. Am J Respir Crit Care Med. 2002;166:409–416. doi: 10.1164/rccm.2105079. [DOI] [PubMed] [Google Scholar]
- 26.Richard M, Grencis RK, Humphreys NE, Renauld JC, Van Snick J. Anti-IL-9 vaccination prevents worm expulsion and blood eosinophilia in Trichuris muris-infected mice. Proc Natl Acad Sci U S A. 2000;97:767–772. doi: 10.1073/pnas.97.2.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H, Zhang GX, Chen Y, Xu H, Fitzgerald DC, Zhao Z, Rostami A. CD11c+CD11b+ dendritic cells play an important role in intravenous tolerance and the suppression of experimental autoimmune encephalomyelitis. J Immunol. 2008;181:2483–2493. doi: 10.4049/jimmunol.181.4.2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nabel G, Galli SJ, Dvorak AM, Dvorak HF, Cantor H. Inducer T lymphocytes synthesize a factor that stimulates proliferation of cloned mast cells. Nature. 1981;291:332–334. doi: 10.1038/291332a0. [DOI] [PubMed] [Google Scholar]
- 29.Gran B, Zhang GX, Yu S, Li J, Chen XH, Ventura ES, Kamoun M, Rostami A. IL-12p35-deficient mice are susceptible to experimental autoimmune encephalomyelitis: evidence for redundancy in the IL-12 system in the induction of central nervous system autoimmune demyelination. J Immunol. 2002;169:7104–7110. doi: 10.4049/jimmunol.169.12.7104. [DOI] [PubMed] [Google Scholar]
- 30.Zhang GX, Yu S, Gran B, Li J, Siglienti I, Chen X, Calida D, Ventura E, Kamoun M, Rostami A. Role of IL-12 receptor beta 1 in regulation of T cell response by APC in experimental autoimmune encephalomyelitis. J Immunol. 2003;171:4485–4492. doi: 10.4049/jimmunol.171.9.4485. [DOI] [PubMed] [Google Scholar]
- 31.Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, Shen Y, Du J, Rubtsov YP, Rudensky AY, Ziegler SF, Littman DR. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. 2008;453:236–240. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li H, Rostami A. IL-9: Basic Biology, Signaling Pathways in CD4+ T Cells and Implications for Autoimmunity. J Neuroimmune Pharmacol. 2009 doi: 10.1007/s11481-009-9186-y. [DOI] [PubMed] [Google Scholar]
- 33.Uyttenhove C, Simpson RJ, Van Snick J. Functional and structural characterization of P40, a mouse glycoprotein with T-cell growth factor activity. Proc Natl Acad Sci U S A. 1988;85:6934–6938. doi: 10.1073/pnas.85.18.6934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hultner L, Kolsch S, Stassen M, Kaspers U, Kremer JP, Mailhammer R, Moeller J, Broszeit H, Schmitt E. In activated mast cells, IL-1 up-regulates the production of several Th2-related cytokines including IL-9. J Immunol. 2000;164:5556–5563. doi: 10.4049/jimmunol.164.11.5556. [DOI] [PubMed] [Google Scholar]
- 35.Finiasz MR, Franco MC, de la Barrera S, Rutitzky L, Pizzariello G, del Carmen Sasiain M, Renauld JC, Van Snick J, Fink S. IL-9 promotes anti-Mycobacterium leprae cytotoxicity: involvement of IFNgamma. Clin Exp Immunol. 2007;147:139–147. doi: 10.1111/j.1365-2249.2006.03241.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Veldhoen M, Uyttenhove C, van Snick J, Helmby H, Westendorf A, Buer J, Martin B, Wilhelm C, Stockinger B. Transforming growth factor-beta ‘reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat Immunol. 2008;9:1341–1346. doi: 10.1038/ni.1659. [DOI] [PubMed] [Google Scholar]
- 37.Elyaman W, Bradshaw EM, Uyttenhove C, Dardalhon V, Awasthi A, Imitola J, Bettelli E, Oukka M, van Snick J, Renauld JC, Kuchroo VK, Khoury SJ. IL-9 induces differentiation of TH17 cells and enhances function of FoxP3+ natural regulatory T cells. Proc Natl Acad Sci U S A. 2009 doi: 10.1073/pnas.0812530106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu Y, Teige I, Birnir B, Issazadeh-Navikas S. Neuron-mediated generation of regulatory T cells from encephalitogenic T cells suppresses EAE. Nat Med. 2006;12:518–525. doi: 10.1038/nm1402. [DOI] [PubMed] [Google Scholar]
- 39.White B, Leon F, White W, Robbie G. Two first-in-human, open-label, phase I dose-escalation safety trials of MEDI-528, a monoclonal antibody against interleukin-9, in healthy adult volunteers. Clin Ther. 2009;31:728–740. doi: 10.1016/j.clinthera.2009.04.019. [DOI] [PubMed] [Google Scholar]
- 40.Engelhardt B, Ransohoff RM. The ins and outs of T-lymphocyte trafficking to the CNS: anatomical sites and molecular mechanisms. Trends Immunol. 2005;26:485–495. doi: 10.1016/j.it.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 41.Dong Q, Louahed J, Vink A, Sullivan CD, Messler CJ, Zhou Y, Haczku A, Huaux F, Arras M, Holroyd KJ, Renauld JC, Levitt RC, Nicolaides NC. IL-9 induces chemokine expression in lung epithelial cells and baseline airway eosinophilia in transgenic mice. Eur J Immunol. 1999;29:2130–2139. doi: 10.1002/(SICI)1521-4141(199907)29:07<2130::AID-IMMU2130>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 42.Emamaullee JA, Davis J, Merani S, Toso C, Elliott JF, Thiesen A, Shapiro AM. Inhibition of Th17 cells regulates autoimmune diabetes in NOD mice. Diabetes. 2009;58:1302–1311. doi: 10.2337/db08-1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Furuzawa-Carballeda J, Vargas-Rojas MI, Cabral AR. Autoimmune inflammation from the Th17 perspective. Autoimmun Rev. 2007;6:169–175. doi: 10.1016/j.autrev.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 44.Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, Sedgwick JD, Cua DJ. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. 2003;198:1951–1957. doi: 10.1084/jem.20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aranami T, Yamamura T. Th17 Cells and autoimmune encephalomyelitis (EAE/MS) Allergol Int. 2008;57:115–120. doi: 10.2332/allergolint.R-07-159. [DOI] [PubMed] [Google Scholar]
- 46.Uyttenhove C, Sommereyns C, Theate I, Michiels T, Van Snick J. Anti-IL-17A autovaccination prevents clinical and histological manifestations of experimental autoimmune encephalomyelitis. Ann N Y Acad Sci. 2007;1110:330–336. doi: 10.1196/annals.1423.035. [DOI] [PubMed] [Google Scholar]
- 47.Hultner L, Druez C, Moeller J, Uyttenhove C, Schmitt E, Rude E, Dormer P, Van Snick J. Mast cell growth-enhancing activity (MEA) is structurally related and functionally identical to the novel mouse T cell growth factor P40/TCGFIII (interleukin 9) Eur J Immunol. 1990;20:1413–1416. doi: 10.1002/eji.1830200632. [DOI] [PubMed] [Google Scholar]
- 48.Sugimoto T, Ishikawa Y, Yoshimoto T, Hayashi N, Fujimoto J, Nakanishi K. Interleukin 18 acts on memory T helper cells type 1 to induce airway inflammation and hyperresponsiveness in a naive host mouse. J Exp Med. 2004;199:535–545. doi: 10.1084/jem.20031368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yoshimoto T, Takeda K, Tanaka T, Ohkusu K, Kashiwamura S, Okamura H, Akira S, Nakanishi K. IL-12 up-regulates IL-18 receptor expression on T cells, Th1 cells, and B cells: synergism with IL-18 for IFN-gamma production. J Immunol. 1998;161:3400–3407. [PubMed] [Google Scholar]