

Abstract
Δ8-Tetrahydrocannabinol (26), 3-(1′,1′-dimethylbutyl)-(12), 3-(1′,1′-dimethylpentyl)-(13), 3-(1′,1′-dimethylhexyl)-(14) and 3-(1′,1′-dimethylheptyl)-Δ8-tetrahydrocannabinol (15) have been converted into the corresponding 1-bromo-1-deoxy-Δ8-tetrahydrocannabinols (25, 8–11). This was accomplished using a protocol developed in our laboratory in which the trifluoromethanesulfonate of a phenol undergoes palladium mediated coupling with pinacolborane. Reaction of this dioxaborolane with aqueous-methanolic copper (II) bromide provides the aryl bromide. The affinities of these bromo cannabinoids for the cannabinoid CB1 and CB2 receptors were determined. All of these compounds showed selectivity for the CB2 receptor and one of them, 1-bromo-1-deoxy-3-(1′,1′-dimethylhexyl)-Δ8-tetrahydrocannabinol (10), exhibits 52-fold selectivity for this receptor with good (28 nM) affinity.
Keywords: CB1 receptor, CB2 receptor, nontraditional cannabinoids, 1-deoxy cannabinoids
1. Introduction
The modern era of the study of cannabinoids began in 1964 with the elucidation of the structure of Δ9-tetrahydrocannabinol (Δ9-THC, 1) by Gaoni and Mechoulam.1 Subsequently, a comprehensive set of structure-activity relationships (SAR) was developed based upon the dibenzopyran skeleton of THC.2–4 These SAR include inter alia the principles that a C-3 side chain of five to seven carbon atoms and a C-1 phenolic hydroxyl group are necessary for cannabinoid activity. Shortly after these SAR were formulated, a cannabinoid receptor in rat brain was identified.5 This G-protein coupled receptor, which is expressed primarily in the central nervous system, is now known as the CB1 receptor and has been cloned.6 A nearly identical (97% homology) human receptor has also been identified.7 In 1993 a second cannabinoid (CB2) receptor was identified and cloned; however, this receptor shows only 44% homology (68% in the transmembrane helical regions) with the initially described receptor.8 The CB2 receptor is expressed primarily in the periphery, particularly in the immune system.10–14
CB1 receptor agonists, including endogenous compounds, are generally considered to be responsible for the overt centrally mediated effects of cannabinoids, such as their psychotropic, appetite stimulant and anti-nausea effects. It has been suggested that the CB2 receptor is responsible for the immunomodulatory effects of cannabinoids,10 a conclusion that is supported by the fact that these effects are absent in CB2 receptor knockout mice.15 Both CB1 and CB2 receptors are expressed in a variety of cancer cells and CB1 and CB2 receptor agonists have been found to inhibit tumor growth.16,17 There is also evidence that the CB2 receptor is involved in inflammatory pain.18–25 Recent reviews have noted that the endocannabinoid system represents a potential therapeutic target and have suggested that development of selective ligands for the CB2 receptor may result in new useful drugs for the treatment of diseases.26,27
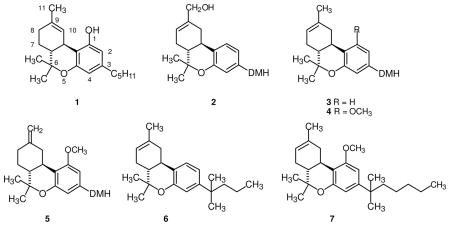
In contrast to the SAR data cited above, several years ago we found that 11-hydroxy-3-(1′,1′-dimethylheptyl)-1-deoxy-Δ8-THC (2, DMH = dimethylheptyl), a THC analog lacking the 1-hydroxyl, has very high affinity for the CB1 receptor (Ki = 1.2 ± 0.1 nM), and is a potent cannabinoid in vivo in the mouse. Cannabinoid 2 also has exceptionally high affinity for the CB2 receptor (Ki = 0.032 ± 0.019 nM).28 A second 1-deoxycannabinoid, 3-(1′,1′-dimethylheptyl)-1-deoxy-Δ8-THC (3), is also potent in vivo, has good affinity for the CB1 receptor (Ki = 23 ± 7 nM), and has nearly ten-fold selectivity for the CB2 receptor (Ki = 2.9 ± 1.6 nM). A group at Merck Frosst also described 1-deoxy-Δ8-THC-DMH (3) at approximately the same time. In addition, they reported that 1-methoxy-Δ8-THC-DMH (4), and 1-methoxy-Δ9(11)-THC-DMH (5) have affinity for the CB2 receptor in the 20 nM range and negligible affinity for the CB1 receptor.29
Based upon a combination of our results and those of the Merck Frosst group, we synthesized a series of 1-deoxy-Δ8-THC analogs.30 Several of these compounds have high affinity for the CB2 receptor with little affinity for the CB1 receptor and one in particular, JWH-133, 3-(1′,1′-dimethylbutyl)-1-deoxy-Δ8-THC (6), has very high affinity for the CB2 receptor (Ki = 3.4 ± 1 nM) and exhibits 200-fold selectivity relative to the CB1 receptor. Subsequently, series of 1-methoxy-11-hydroxy-1-deoxy- and 11-hydroxy-1-methoxy-Δ8-THC analogs were synthesized and their pharmacology was evaluated.31 Of this group of compounds, 3-(1′,1′-dimethylhexyl)-1-methoxyΔ8-THC (JWH-229, 7) showed the greatest selectivity with Ki = 18 ± 2 nM at the CB2 receptor and Ki = 3134 ± 110 nM at the CB1 receptor.
We have recently developed a concise and efficient procedure for converting a phenol to the corresponding aryl bromide.32 This sequence proceeds from the phenol to the corresponding trifluoromethanesulfonate, followed by palladium mediated reaction with pinacolborane to afford a boronate. Reaction of this boronate with copper (II) bromide provides the aryl bromide. Simple aryl halides and those containing electron releasing groups proceed in 60% to 88% overall yield. The presence of electron withdrawing groups in the phenol considerably attenuates the yield. To extend the scope of this synthetic protocol and to investigate the effect upon affinity for the cannabinoid receptors of 1-bromo-Δ8-THC analogs, we have applied this reaction sequence to Δ8-THC and four 3-(1′,1′-dimethylalkyl)-Δ8-THCs. Also, based upon the pharmacology of the 1-deoxy- and 1-methoxy-Δ8-THCs, it was considered possible that one or more of these bromo cannabinoids would exhibit selectivity for the CB2 receptor.
2. Results
Based upon the selectivity for the CB2 receptor shown by JWH-133 and JWH-229 and related compounds,29–31 the decision was made to synthesize 1-bromo-3-(1′,1′-dimethylalkyl)-1-deoxy-Δ8-THC analogs with a side chain of four to seven carbon atoms (8–11, Scheme 1). The 3-(1′,1′-dimethylalkyl)-Δ8-THC analogs (12–15) were prepared as previously described by acid catalyzed reaction of the appropriate substituted resorcinol (16–19) with menthadienol (20).33 Conversion into the trifluoromethanesulfonate, followed by palladium mediated coupling with pinacolborane provided boronates 21 to 24 in 62% to 78% yield. Although initially the conversion of boronates 21, 23 and 24 to the corresponding aryl bromides was accomplished using our published conditions,32 considerable difficulty was encountered in successfully repeating the reaction. In the original report of the conversion of boronic acids to aryl halides, Nesmejanow et al. carried out the reaction of the boronic acid with copper (II) bromide in aqueous media.34 When our earlier procedure was modified to employ an approximately 1:3 mixture of water and methanol, the reaction of boronates 21 to 24 proceeded smoothly and reproducibly to provide 1-bromo-1-deoxy-Δ8-THC analogs 8 (JWH-382), 9 (JWH-458), 10 (JWH-393) and 11 (JWH-393) in 66 % to 85 % yield. In addition to the four 1-bromo-3-(1′,1′-dimethylalkyl)-1-deoxy-Δ8-THC analogs (8–11), the parent compound, 1-bromo-1-deoxy-Δ8-THC (25, JWH-460) was prepared by the sequence shown in Scheme 1, but using Δ8-THC (26) in place of 12 to 15.
Scheme 1.

(a) HOTs, benzene, reflux 12h; (b) Tf2O, CH2Cl2, pyridine, 0 °C to 25 °C, 3 h (c) Pinacolborane, Pd(dppf)Cl2, CH3CN, Et3N, reflux 18 h (d) CuBr2, MeOH, H2O (3:1), reflux 10 h.
The affinities of 1-bromo-Δ8-THC analogs, 8 to 11 and 25, for the CB1 receptor were determined by measuring their ability to inhibit binding of the potent synthetic cannabinoid [3H]CP-55,940 to a membrane preparation from rat brain as described by Compton et al.35 Affinities for the CB2 receptor were determined by measuring the ability of the compounds to inhibit [3H]CP-55,940 binding to a cloned human receptor preparation using the procedure described by Showalter et al.36 The results of these determinations are summarized in Table 1. Also included in Table 1 are the receptor affinities for Δ8-THC (1), JWH-133 (6) and JWH-229 (7).
Table 1.
Receptor Affinities (mean ± SEM) of 1-bromo-Δ8-THC analogs 8 to 11 and 26, Δ9-THC (1) and JWH-133 (6) and JWH-229 (7).
| Compound | Ki (nM) |
||
|---|---|---|---|
| CB1 | CB2 | CB1/CB2 | |
| Δ9-THC (1) | 41 ± 2a | 36 ± 10b | 1.1 |
| JWH-133 (6) | 677 ± 132c | 3.4 ± 1c | 294 |
| JWH-229 (7) | 3134 ± 110d | 18 ± 2d | 174 |
| JWH-460 (25) | > 10,000 | 555 ± 72 | 18 |
| JWH-382 (8) | > 10,000 | 265 ± 17 | 38 |
| JWH-458 (9) | 1145 ± 137 | 71 ± 17 | 16 |
| JWH-393 (10) | 1444 ± 20 | 28 ± 2 | 52 |
| JWH-383 (11) | 562 ± 21 | 34 ± 2 | 27 |
As presented in Table 1, none of these 1-bromo-1-deoxy-Δ8-THC analogs have significant affinity for the CB1 receptor and two compounds, JWH-460 and JWH-382, respectively, 1-bromo-1-deoxy-Δ8-THC (25) and the 3-dimethylbutyl analog (8), have virtually no affinity for this receptor with Ki > 10,000 nM. In contrast to their lack of affinity for the CB1 receptor, all five of these 1-bromo-Δ8-THC analogs bind to the CB2 receptor and three of them, JWH-458 (9), JWH-383 (10) and JWH-383 (11), have moderate to good affinity for this receptor with Ki = 28 – 71 nM. Selectivity for the CB2 (vs. CB1) receptor ranges from 16- to 52-fold. JWH-393 (10) has the greatest (52-fold) selectivity and highest affinity for this receptor, with Ki = 28 ± 2 nM.
The receptor binding data are somewhat similar to those we reported several years ago for a series of 3-(1′,1′-dimethylalkyl)-1-methoxy-Δ8-THC analogs.31 In the methoxy series, the dimethylbutyl compound (JWH-214) has no affinity for the CB1 receptor with Ki > 10,000 nM, which is the same as that of the corresponding 1-bromo-1-deoxy-Δ8-THC (JWH-382, 8). The 1-methoxy-dimethylpentyl-(JWH-226), hexyl-(JWH-229, 7) and heptyl-(JWH-143) Δ8-THCs have Ki values for the CB1 receptor that range from 713 – 4001 nM. While CB1 affinities are slightly better for the corresponding analogs of the 1-bromo-1-deoxy-Δ8-THC series, the Ki values for 1′,1′-dimethylpentyl (JWH-458, 9), dimethylhexyl (JWH-393, 10) and dimethylheptyl (JWH-383, 11) analogs are still poor, ranging from 562 – 1444 nM.
The CB2 receptor affinities of the corresponding analogs of the 1-methoxy-Δ8-THC and 1-bromoΔ8-THC series are also very similar. The 1-bromo-3-(1′,1′-dimethylbutyl) compound (JWH-382, 8) and the 1-methoxy THC analog (JWH-214) have the least affinity, with Ki = 265 ± 17 nM and 325 ± 70 nM,31 respectively, whereas the dimethylhexyl Δ8-THC analogs, 1-bromo (JWH-393, 10) and 1-methoxy (JWH-229, 7), have the best affinities, with Ki = 28 ± 2 and 18 ± 2, respectively. Notably, all analogs in both the 1-bromo and 1-methoxy series show some degree of selectivity for the CB2 receptor, with the highest selectivity seen in the dimethylhexyl Δ8-THC analogs. The 1-methoxy compound, (JWH-229, 7) is 174-fold selective for this receptor, while the 1-bromo analog (JWH-393, 10) is 52-fold selective. While still maintaining selectivity for the CB2 receptor, 1-bromo-3-(1′,1′-dimethylheptyl)-1-deoxy-Δ8-THC (JWH-383, 11) and the 1-methoxy analog (JWH-143) exhibit only 27- and 12-fold31 selectivity, respectively.
Two of these bromocannabinoids, JWH-393 (10) and JWH-383 (11), have significant affinity for the CB2 receptor (Ki = 28 ± 2 and 34 ± 2 nM, respectively) and are from moderately to highly selective for the CB2 receptor. In order to evaluate the efficacy of these compounds, their ability to stimulate GTPγS binding was determined. This is a functional assay which measures G-protein coupled receptor activation using [34S]GTPγS binding.37 Chinese Hamster Ovary (CHO) cells stably expressing the human CB2 receptor were employed in this determination (see Experimental). The results of these determinations are summarized in Table 2. The stimulation is normalized to that produced by 3 μM CP-55,940, a maximally effective concentration of this very efficacious standard cannabinoid agonist. In addition to JWH-393 (10) and JWH-383 (11), the [34S]GTPγS binding for two highly CB2 selective cannabinoids, JWH-133 (6) and JWH-229 is included in Table 2.38
Table 2.
EC50 and Emax Values (mean ± SEM) for GTPγS Binding of CB2 for Selective Ligands.a
| Compound | EC50 (nM) | Emax (% CP-55940) |
|---|---|---|
| 1-Deoxy-3-(1′,1′-dimethylbutyl)-Δ8-THC (JWH-133, 6) | 4.0 ± 1.0 | 111.5 ± 13.6 |
| 1-Methoxy-3-(1′,1′-dimethylhexyl)-Δ8-THC (JWH-229, 7) | 4.6 ± 2.0 | 75.7 ± 8.3 |
| 1-Bromo-1-deoxy-3-(1′,1′-dimethylhexyl)-Δ8-THC (JWH-393, 10) | 28.6 ± 5.3 | 92.3 ± 6.8 |
| 1-Bromo-1-deoxy-3-(1′,1′-dimethylheptyl)-Δ8-THC (JWH-383, 11) | 15.0 ± 2.3 | 91.0 ± 6.7 |
Assays were carried out in Human CB2 Receptor-Expressing CHO Cells. Emax Values are Reported as Percent Relative to 3 μM CP-55,940.
As indicated in Table 2 both JWH-393 (10) and JWH-383 (11) are moderately potent in the [34S]GTPγS assay with EC50 values 28.6 ± 6.3 nM for JWH-393 (10) and 15.0 ± 2.3 nM for JWH-383 (11). Both of these CB2 receptor ligands are full agonists at the CB2 receptor with Emax values of 98.3 ± 6.8% (JWH-393) and 91.0 ± 6.7% (JWH-383), relative to CP-55,940.
3. Discussion and Conclusions
In both the 1-bromo and 1-methoxy series, the 3-(1′,1′-dimethylhexyl) compounds have useful selectivity for the CB2 receptor and both have similar affinity for this receptor, however the bromo analog is more efficacious. The similarity between the 1-methoxy and 1-bromo cannabinoids may be due to electronic rather than steric effects. As aromatic substituents, both bromine and methoxyl are inductively electron withdrawing and electron releasing by resonance. Based upon the rate of racemization of chiral biaryls, the effective size of a bromine substituent is larger than that of a methoxyl.39 If steric effects contributed significantly to the receptor affinities of 1-methoxy and 1-bromo THC derivatives, greater differences in the affinities between the two series of compounds would be expected.
In summary, we have developed a new class of selective ligands for the cannabinoid CB2 receptor and have extended the scope of our procedure for converting phenols to alkyl bromides to the moderately hindered 1-position of the traditional cannabinoid molecule.32
4. Experimental
4.1 General
1H and 13C NMR spectra were recorded on Bruker 300AC and JEOL 500 spectrometers. Mass spectral analyses were performed on a Shimadzu QP2010 capillary gas chromatograph/mass spectrometer equipped with a mass sensitive detector at 1.01 kV. HRMS data were obtained in the Mass Spectrometry Laboratory, School of Chemical Sciences, University of Illinois. Ether and THF were distilled from Na-benzophenone ketyl immediately before use, and other solvents were purified using standard procedures. Column chromatography was carried out on Sorbent Technologies silica gel (32 – 63 μ) using the indicated solvents as eluents. All new compounds were homogeneous to GLC and 13C NMR. All target compounds were at least 95% pure by GLC.
4.2 Δ8-Tetrahydrocannabinyl trifluoromethanesulfonate
To a solution of 0.71 g (2.26 mmol) of Δ8-tetrahydrocannabinol33 (26) and 0.89 g (3.17 mmol) of trifluoromethanesulfonic anhydride in 12 mL of dichloromethane at 0 °C, was added 0.38 g (4.80 mmol) of pyridine. The solution was allowed to warm to ambient temperature, stirred for 3 h, diluted with ether and quenched with 10 mL of 1M HCl. The organic layer was washed with successive portions of aqueous NaHCO3, brine, dried (MgSO4) and concentrated in vacuo. The crude product was purified by flash chromatography (petroleum ether/ether, 7:3) to afford 0.73 g (73%) of Δ8-tetrahydrocannabinyl trifluoromethanesulfonate: 1H NMR (500 MHz, CDCl3) δ 0.91 (t, J = 7.0 Hz, 3H), 1.27 (s, 3H), 1.29–1.34 (m, 4H), 1.41 (s, 3H), 1.56–1.62 (m, 2H), 1.73 (s, 3H), 1.78–1.93 (m, 3H), 2.12–2.21 (m, 1H), 2.54 (t, J = 7.0 Hz, 2H), 2.79–2.99 (m, 2H), 5.48 (d, J = 3.0 Hz, 1H), 6.62 (d, J = 2.0 Hz, 1H), 6.69 (d, J = 2.0 Hz, 1H); 13C NMR (125.8 MHz, CDCl3) δ 13.9, 18.3, 22.5, 23.3, 27.3, 27.7, 30.4, 31.3, 31.8, 35.3, 35.7, 44.6, 113.5, 116.9, 117.6, 119.4, 133.9, 143.6, 148.5, 155.1; MS (EI) m/z (rel intensity) 363 (97), 403 (90), 446(100).
4.3 3-(1′,1′-Dimethylbutyl)-Δ8-tetrahydrocannabinyl trifluoromethanesulfonate
This triflate was prepared by the procedure employed for the preparation of Δ8-tetrahydrocannabinyl trifluoromethanesulfonate. From 0.33 g (1.0 mmol) of 3-(1′,1′-dimethylbutyl)-Δ8-tetrahydrocannabinol33 (12), there was obtained after chromatography (petroleum ether), 0.38 g (82%) of 3-(1′,1′-dimethylbutyl)-Δ8-tetrahydrocannabinyl trifluoromethanesulfonate as a clear liquid: 1H NMR (500 MHz, CDCl3) δ 0.82 (t, J = 7.3 Hz, 3H), 1.02–1.12 (m, 2H), 1.13 (s, 3H), 1.23 (s, 3H), 1.24 (s, 3H), 1.40 (s, 3H), 1.48–1.54 (m, 2H), 1.71 (s, 3H), 1.78 (td, J = 4.3, 11.4 Hz, 1H), 1.81–1.96 (m, 2H), 2.12–2.20 (m, 1H), 2.84 (td, J = 4.6, 11.0 Hz, 1H), 2.93 (dd, J = 4.4, 16.2 Hz, 1H), 5.44 (d, J = 3.2 Hz, 1H), 6.72 (d, J = 1.8 Hz, 1H), 6.79 (d, J = 1.8 Hz, 1H); 13C NMR (125.8 MHz, CDCl3) δ 14.6, 17.8, 18.3, 23.3, 27.3, 27.6, 28.3, 28.7, 31.6, 35.6, 37.7, 44.6, 46.7, 77.5, 111.4, 115.4, 116.5, 118.6 (q, JC,F = 321 Hz), 119.9, 133.9, 148.6, 150.8, 154.8; GC/MS (EI) m/z (rel intensity) 460 (61), 445 (6), 419 (11), 418 (37), 417 (100), 404 (6), 377 (46), 349 (24), 285 (12), 242 (4), 201 (6), 85 (31).
4.4 3-(1′,1′-Dimethylpentyl)-Δ8-tetrahydrocannabinyl trifluoromethanesulfonate
This triflate was prepared by the procedure described for the synthesis of Δ8-tetrahydrocannabinyl trifluoromethanesulfonate. From 0.55 g (1.61 mmol) of 3-(1′,1′-dimethylpentyl)-Δ8-tetrahydrocannabinol33 (13) there was obtained 0.53 g (70%) of pure 3-(1′,1′-dimethylpentyl)-Δ8-tetrahydrocannabinyl trifluoromethanesulfonate after flash chromatography (petroleum ether/ether, 7:3): 1H NMR (500 MHz, CDCl3) δ 0.84 (t, J = 7.0 Hz, 3H), 1.03–1.11 (m, 2H), 1.13 (s, 3H), 1.15–1.21 (m, 6H),1.26 (s, 3H), 1.41 (s, 3H), 1.51–1.54 (m, 3H), 1.72, (s, 3H), 2.10–2.14 (m, 2H), 2.81 (td, J = 4.9, 10.7 Hz, 1H), 2.94 (dd, J = 4.0, 17.0 Hz, 1H), 5.45 (d, J = 3.0 Hz, 1H), 6.72 (d, J = 2.0 Hz, 1H), 6.79 (d, J = 2.0 Hz, 1H); 13C NMR (125.8 MHz, CDCl3) δ 14.1, 18.5, 22.5, 23.2, 27.1, 27.7, 28.4, 28.6, 31.5, 31.7, 35.5, 37.4., 44.2, 44.7, 77.2, 111.3, 115.4, 116.4.6, 116.2, 118.4, 119.3, 134.2, 148.5, 150.1, 154.7; MS (EI) m/z (rel intensity) 391 (50), 417 (100), 431 (30), 474 (90).
4.5 3-(1′,1′-Dimethylhexyl)-Δ8-tetrahydrocannabinyl trifluoromethanesulfonate
This triflate was prepared using by the procedure employed for the synthesis of Δ8-tetrahydrocannabinyl trifluoromethanesulfonate. From 0.29 g (0.81 mmol) of 3-(1′,1′-dimethylhexyl)-Δ8-tetrahydrocannabinol33 (14), there was obtained, after chromatography (petroleum ether), 0.37 g (93%) of 3-(1′,1′-dimethylhexyl)-Δ8-tetrahydrocannabinyl trifluoromethanesulfonate as a viscous yellow oil: 1H NMR (500 MHz, CDCl3) δ 0.82 (t, J = 7.4 Hz, 3H), 1.01–1.08 (m, 2H), 1.13 (s, 3H), 1.14–1.22 (m, 4H), 1.23 (s, 3H), 1.24 (s, 3H), 1.41 (s, 3H), 1.49–1.54 (m, 2H), 1.71 (s, 3H), 1.78 (td, J = 4.0, 11.3 Hz, 1H), 1.81–1.95 (m, 2H), 2.13–2.20 (m, 1H), 2.84 (td, J = 4.8, 10.9 Hz, 1H), 2.93 (dd, J = 3.9, 16.2 Hz, 1H), 5.44 (d, J = 4.2 Hz, 1H), 6.71 (d, J = 1.8 Hz, 1H), 6.79 (d, J = 1.8 Hz, 1H); 13C NMR (125.8 MHz) δ 14.0, 18.3, 22.5, 23.3, 24.2, 27.3, 27.6, 28.4, 28.6, 31.6, 32.4, 35.6, 37.7, 44.2, 44.5, 77.5, 111.4, 115.4, 116.5, 118.7 (q, JC,F = 308), 119.4, 134.0, 148.6, 150.9, 154.8; GC/MS (EI) m/z (rel intensity) 488 (36), 473 (3), 445 (14), 432 (6), 419 (12), 418 (41), 417 (100), 405 (43), 349 (37), 299 (6), 285 (33), 269 (8), 257 (9), 241 (16), 227 (9), 216 (17), 201 (34), 187 (15), 121 (22), 107 (15), 91 (16), 71 (48), 57 (45).
4.6 3-(1′,1′-Dimethylheptyl)-Δ8-tetrahydrocannabinyl trifluoromethanesulfonate
This triflate was prepared by the procedure employed for the preparation of Δ8-tetrahydrocannabinyl trifluoromethanesulfonate. From 0.31 g (0.84 mmol) of 3-(1′,1′-dimethylheptyl)-Δ8-tetrahydrocannabinol33 (15), there was obtained, after chromatography (petroleum ether), 0.30 g (71%) of 3-(1′,1′-dimethylheptyl)-Δ8-tetrahydrocannabinyl trifluoromethanesulfonate as a viscous yellow oil: 1H NMR (500 MHz, CDCl3) δ 0.84 (t, J = 7.1 Hz, 3H), 0.99–1.08 (m, 2H), 1.13 (s, 3H), 1.15–1.22 (m, 6H), 1.23 (s, 3H), 1.24 (s, 3H), 1.40 (s, 3H), 1.49–1.55 (m, 2H), 1.71 (s, 3H), 1.78 (td, J = 4.1, 11.3 Hz, 1H), 1.81–1.95 (m, 2H), 2.13–2.20 (m, 1H), 2.84 (td, J = 4.8, 10.9 Hz, 1H), 2.93 (dd, J = 4.1, 16.5 Hz, 1H), 5.44 (d, J = 4.2 Hz, 1H), 6.71 (d, J = 1.8 Hz, 1H), 6.79 (d, J = 1.8 Hz, 1H); 13C NMR (125.8 MHz, CDCl3) δ 14.0, 18.3, 22.6, 23.3, 24.5, 27.3, 27.6, 28.4, 28.6, 29.8, 31.6, 31.7, 35.6, 37.7, 44.2, 44.6, 77.5, 111.4, 115.4, 116.5, 118.6 (q, JC,F = 320 Hz), 119.4, 133.9, 148.6, 150.9, 154.8; GC/MS (EI) m/z (rel intensity) 502 (69), 487 (6), 459 (17), 446 (7), 434 (5), 419 (50), 418 (41), 417 (100), 349 (20), 285 (16), 201 (9), 121 (11).
4.7 1-Deoxy-Δ8-tetrahydrocannabinyl-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (27)
To a suspension of 0.20 g (0.448 mmol) of Δ8-tetrahydrocannabinyl trifluoromethanesulfonate and 0.010 g (0.0135 mmol) of 1′,1″-bis(diphenylphosphino)ferrocene-palladium(II)dichloride dichloromethane complex in 15 mL of acetonitrile under argon was slowly added 1.41 g (13.5 mmol) of triethylamine and the reaction was stirred at reflux overnight. The mixture was cooled to 0 °C, quenched by the addition of 10 mL of 1M HCl, diluted with diethyl ether, washed with brine, dried (MgSO4), and concentrated in vacuo. The resultant red oil was purified by flash chromatography (petroleum ether/ether, 8:2) to give 0.12 g (65%) of 1-deoxy-Δ8-tetrahydrocannabinyl-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (27): 1H NMR (500 MHz, CDCl3) δ 0.91 (t, J = 7.0 Hz, 3H), 1.13 (s, 3H), 1.28–1.33 (m, 4H), 1.34 (s, 3H), 1.36 (s, 6H), 1.38 (s, 6H), 1.41 (s, 3H), 1.58–1.64 (m, 2H), 1.69 (s, 3H), 1.73–1.93 (m, 2H), 2.11–2.22 (m, 1H), 2.53 (t, J = 7.0 Hz, 1H), 2.98 (dt, J = 4.0, 17.0 Hz, 1H), 5.41 (d, J = 3.0 Hz, 1H), 6.72 (d, J = 2.0 Hz, 1H), 7.11 (d, J = 2.0 Hz, 1H); 13C NMR (125.8 MHz, CDCl3) δ 14.0, 18.5, 22.6, 23.2, 24.1, 24.7, 25.1, 27.6, 28.2, 29.7, 30.9, 31.7, 33.5,35.4, 40.5, 45.5, 83.5, 119.9, 120.0, 128.1, 128.6, 134.5, 141.4, 152.9; MS (EI) m/z (rel intensity) 281 (65), 341 (100), 424 (80).
4.8 1-Deoxy-3-(1′,1′-dimethylbutyl)-Δ8-tetrahydrocannabinyl-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (21)
Dioxaborolane 21 was prepared using the procedure described for dioxaborolane 27. From 0.37 g (0.80 mmol) of 3-(1′,1′-dimethylbutyl)-Δ8-tetrahydrocannabinyl trifluoromethanesulfonate there was obtained, after 4 h at reflux 0.22 g (62%) of 21 as a yellow oil: 1H NMR (500 MHz, CDCl3) δ 0.81 (t, J = 7.3 Hz, 3H), 1.05–1.14 (m, 2H), 1.15 (s, 3H), 1.25 (s, 6H), 1.35 (s, 6H), 1.36 (s, 6H), 1.39 (s, 3H), 1.50–1.57 (m, 2H), 1.68 (s, 3H), 1.74 (td, J = 4.4, 11.4 Hz, 1H), 1.82–1.93 (m, 2H), 2.10–2.19 (m, 1H), 2.60 (dd, J = 3.9, 14.9 Hz, 1H), 2.98 (td, J = 4.7, 11.0 Hz, 1H), 5.44 (d, J = 3.2 Hz, 1H), 6.83 (d, J = 2.3 Hz, 1H), 7.22 (d, J = 1.8 Hz, 1H); 13C NMR (125.8 MHz, CDCl3) δ 14.8, 18.0, 18.5, 23.2, 24.5, 25.2, 27.6, 28.1, 28.6, 28.9, 33.4, 37.3, 40.4, 45.4, 46.9, 76.1, 83.4, 117.8, 120.0, 125.3, 128.1, 134.5, 148.4, 152.6; GC/MS (EI) m/z (rel intensity) 438 (90), 397 (8), 396 (47), 395 (100), 394 (20), 355 (68), 327 (23), 311 (15), 295 (31), 281 (16), 207 (89), 191 (12), 133 (10), 119 (11), 101 (22), 85 (34).
4.9 1-Deoxy-3-(1′,1′-dimethylpenty)-Δ8-tetrahydrocannabinyl-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (22)
Dioxaborolane 22 was prepared by the procedure employed for the synthesis of dioxaborolane 27. From 0.21 g (0.421 mmol) of 3-(1′,1′-dimethylpentyl)-Δ8-tetrahydrocannabinyl trifluoromethanesulfonate there was obtained 0.151 g (78%) of pure dioxaborolane 22 after flash chromatography (petroleum ether/ether, 9:1): 1H NMR (500 MHz, CDCl3) δ 0.82 (t, J = 7.0 Hz, 3H), 1.03–1.13 (m, 2H), 1.15 (s, 3H), 1.16–1.22 (m, 6H), 1.24 (s, 3H), 1.33 (s, 6H), 1.35 (s, 6H), 1.41 (s, 3H), 1.51–1.53 (m, 3H), 1.69, (s, 3H), 2.10–2.18 (m, 2H), 2.62 (td, J = 4.9, 10.7 Hz, 1H), 2.97 (dd, J = 4.0, 17.0 Hz, 1H), 5.44 (d, J = 3.0 Hz, 1H), 6.82 (d, J = 2.0 Hz, 1H), 7.21 (d, J = 2.0 Hz, 1H); 13C NMR (125.8 MHz) δ 14.2, 18.5, 22.5, 23.1, 27.4, 28.0, 28.6, 28.9, 31.8, 32.4, 37.6, 40.1., 44.6, 45.1, 76.1, 83.2, 117.6, 119.8, 125.1,127.9, 134.4, 148.5, 152.4; MS (EI) m/z (rel intensity) 368 (20), 369 (75), 395 (100), 452 (80).
4.10 1-Deoxy-3-(1′,1′-dimethylhexyl)-Δ8-tetrahydrocannabinyl-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (23)
Dioxaborolane 23 was prepared using the procedure employed for the synthesis of Δ8-tetrahydrocannabinyl-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (27). From 0.36 g (0.74 mmol) of 3-(1′,1′-dimethylhexyl)-Δ8-tetrahydrocannabinyl trifluoromethanesulfonate there was obtained, after 4 h at reflux, 0.26 g (76%) of 24 as a yellow oil: 1H NMR (500 MHz, CDCl3) δ 0.82 (t, J = 7.3 Hz, 3H), 1.09 (sextet, J = 7.3 Hz, 2H), 1.15 (s, 3H), 1.16–1.23 (m, 4H), 1.24 (s, 3H), 1.25 (s, 3H), 1.35 (s, 6H), 1.36 (s, 6H), 1.39 (s, 3H), 1.51–1.58 (m, 2H), 1.68 (s, 3H), 1.74 (td, J = 4.3, 11.6 Hz, 1H), 1.82–1.93 (m, 2H), 2.10–2.19 (m, 1H), 2.60 (dd, J = 4.3, 14.9 Hz, 1H), 2.99 (td, J = 4.9, 11.1 Hz, 1H), 5.44 (d, J = 3.7 Hz, 1H), 6.83 (d, J = 2.3 Hz, 1H), 7.22 (d, J = 1.8 Hz, 1H); 13C NMR (125.8 MHz, CDCl3) δ 14.1, 18.5, 22.5, 23.2, 24.3, 24.5, 25.2, 27.6, 28.1, 28.6, 29.0, 32.6, 33.4, 37.2, 40.4, 44.3, 45.4, 76.1, 83.4, 117.8, 120.0, 125.3, 128.1, 134.5, 148.5, 152.6; GC/MS (EI) m/z (rel intensity) 468 (6), 467 (33), 466 (100), 465 (25), 395 (71), 383 (32), 323 (9), 311 (6), 101 (8), 83 (13), 71 (8), 57 (14).
4.11 1-Deoxy-3-(1′,1′-dimethylheptyl)-Δ8-tetrahydrocannabinyl-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (24)
Dioxaborolane 24 was prepared using the procedure employed for the synthesis of dioxaborolane 27. From and 0.29 g (0.58 mmol) of 3-(1′,1′-dimethylheptyl)-Δ8-tetrahydrocannabinyl trifluoromethanesulfonate there was obtained, after 4 h at reflux 0.21 g (76%) of 24 as a yellow oil: 1H NMR (500 MHz, CDCl3) δ 0.84 (t, J = 6.9 Hz, 3H), 1.02–1.12 (m, 2H), 1.15 (s, 3H), 1.16–1.23 (m, 6H), 1.24 (s, 3H), 1.25 (s, 3H), 1.35 (s, 6H), 1.36 (s, 6H), 1.39 (s, 3H), 1.52–1.58 (m, 2H), 1.68 (s, 3H), 1.74 (td, J = 4.4, 11.6 Hz, 1H), 1.82–1.93 (m, 2H), 2.10–2.19 (m, 1H), 2.60 (dd, J = 4.4, 14.9 Hz, 1H), 2.99 (td, J = 4.8, 11.1 Hz, 1H), 5.44 (d, J = 3.7 Hz, 1H, 1H), 6.82 (d, J = 1.8 Hz, 1H), 7.22 (d, J = 2.2 Hz, 1H); 13C NMR (125.8 MHz, CDCl3) δ 14.1, 18.5, 22.7, 23.2, 24.5, 24.6, 25.2, 27.6, 28.1, 28.6, 29.0, 30.0, 31.8, 33.4, 37.2, 40.4, 44.4, 45.4, 76.1, 83.4, 117.8, 120.0, 125.3, 128.1, 134.5, 148.5, 152.6; GC/MS (EI) m/z (rel intensity) 482 (6), 481 (35), 480 (100), 479 (23), 395 (93), 337 (16), 327 (10), 311 (10), 211 (4), 101 (10), 83 (16), 71 (10).
4.12 1-Bromo-1-deoxy-Δ8-tetrahydrocannabinol (25, JWH-460)
To a solution of 0.20 g (0.472 mmol) of dioxaborolane 27 suspended in 20 mL of H2O was added 73 mL of MeOH, followed by 0.34 g (1.52 mmol) of copper(II) bromide in 5 mL of H2O. The solution was heated at reflux for 10 h and concentrated in vacuo. The solution was cooled to ambient temperature, diluted with ether, washed with brine, dried (MgSO4), and concentrated in vacuo. The resultant yellow oil was purified by flash chromatography (petroleum ether/ether, 8:2) to give 0.13 g (75%) of 1-bromo-1-deoxy-Δ8-tetrahydrocannabinol (25) as a colorless oil: 1H NMR (500 MHz, CDCl3) δ 0.91 (t, J = 7.0 Hz, 3H), 1.12 (s, 3H), 1.28–1.34 (m, 4H), 1.35 (s, 3H), 1.64 (s, 3H), 1.81–1.92 (m, 3H), 2.09–2.21 (m, 1H), 2.53 (t, J = 7.0 Hz, 2H), 2.54–2.59 (m, 2H), 2.71 (m, 1H), 3.41 (dt, J = 4.0, 17.0 Hz, 1H), 5.42 (d, J = 3.0 Hz, 1H), 6.59 (d, J = 2.0 Hz, 1H), 6.95 (d, J = 2.0 Hz, 1H); 13C NMR (125.8 MHz, CDCl3) δ 14.1, 18.1, 22.5, 23.4, 27.4, 28.3, 30.6, 31.5, 35.3, 36.4, 46.5, 117.1, 119.5, 123.5, 125.8, 134.8, 143.4, 154.8; MS (EI) m/z (rel intensity) 254 (45), 293 (100), 335 (30), 376 (75); HRMS calcd for C21H29OBr 376.1402, found 376.1402; [α]D 20 −221° (c 0.094, CHCl3).
4.13 1-Bromo-1-deoxy-3-(1′,1′-dimethylbutyl)-Δ8-tetrahydrocannabinol (8, JWH-382)
JWH-382 was prepared by the method employed for the synthesis of JWH-460, From 0.21 g (0.48 mmol) of 3-(1′,1′-dimethylbutyl)-Δ8-tetrahydrocannabinyl-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (21) and 0.33 g (1.5 mmol) of copper (II) bromide there was obtained, after 4 h at reflux and column chromatography (petroleum ether), 0.16 g (85%) of JWH-382 as a viscous light yellow oil: 1H NMR (500 MHz, CDCl3) δ 0.82 (t, J = 7.3 Hz, 3H), 1.02–1.13 (m, 5H), 1.22 (s, 6H), 1.39 (s, 3H), 1.46–1.53 (m, 2H), 1.72 (s, 3H), 1.73–1.83 (m, 1H), 1.83–1.91 (m, 2H), 2.11–2.22 (m, 1H), 2.68 (td, J = 4.4, 10.6 Hz, 1H), 3.46 (dd, J = 3.9, 16.7 Hz, 1H), 5.44 (s, 1H), 6.72 (d, J = 1.8 Hz, 1H), 7.06 (d, J = 1.8 Hz, 1H); 13C NMR (125.8 MHz, CDCl3) δ 14.7, 17.9, 18.1, 23.4, 27.4, 28.3, 28.5, 28.7, 35.2, 36.3, 37.5, 46.4, 46.8, 76.9, 115.0, 119.5, 122.6, 123.1, 123.5, 134.8, 150.6, 154.5; GC/MS (EI) m/z (rel intensity) 392 (65), 390 (65), 349 (70), 347 (64), 309 (38), 307 (37), 279 (20), 185 (17), 121 (26), 91 (32), 85 (100), 77 (20); HRMS: m/z calcd for C22H31BrO: 390.1558; found: 390.1554, [α]D 20 −177° (c 0.214, CHCl3).
4.14 1-Bromo-1-deoxy-3-(1′,1′-dimethylpentyl)-Δ8-tetrahydrocannabinol (9, JWH-458)
Dioxaborolane 22 was converted to 1-bromo-1-deoxy-3-(1′,1′-dimethylpentyl)-Δ8-tetrahydrocannabinol by the procedure described for the synthesis of JHW-460. From 0.050 g (1.11 mmol) of 22 and 0.079 g (0.35 mmol) of copper(II) bromide there was obtained 0.36 g (80%) of pure JWH-458 as a colorless oil after flash chromatography (petroleum ether/ether, 8:2): 1H NMR (500 MHz, CDCl3) δ 0.91 (t, J = 7.0 Hz, 3H), 1.12–1.21 (s, 5H), 1.22–1.35 (m, 9H), 1.41 (s, 3H), 1.51–1.65 (m, 2H), 1.86 (s, 3H), 1.85–1.95 (m, 2H), 2.13–2.28 (m, 1H), 2.69 (t, J = 7.0 Hz, 1H), 3.51 (dt, J = 4.0, 17.0 Hz, 1H), 5.47 (d, J = 3.0 Hz, 1H), 6.76 (d, J = 2.0 Hz, 1H), 7.08 (d, J = 2.0 Hz, 1H); 13C NMR (125.8 MHz, CDCl3) δ 14.1, 18.2, 23.3, 23.5, 26.9, 27.4, 28.3, 28.6, 28.7, 35.3, 36.4, 44.1, 46.5, 115.0, 119.5, 120.9, 122.9, 123.6, 150.7, 154.0; MS (EI) m/z (rel intensity) 321 (80), 349 (90), 364 (70), 404(100); HRMS calcd for C23H33OBr 404.1715, found 404.1714; [α]D 20 −263° (c 0.25, CHCl3).
4.15 1-Bromo-1-deoxy-3-(1′,1′-dimethylhexyl)-Δ8-tetrahydrocannabinol (10, JWH-393)
JWH-393 was prepared by the procedure employed for the synthesis of JWH-460. From 0.25 g (0.54 mmol) of dioxaborolane (23) and 0.38 g (1.7 mmol) of copper (II) bromide there was obtained, after 4 h at reflux and column chromatography (petroleum ether), 0.16 g (71%) of JWH-393 as a clear oil: 1H NMR (500 MHz, CDCl3) δ 0.83 (t, J = 7.1 Hz, 3H), 1.01–1.12 (m, 5H), 1.13–1.29 (m, 10H), 1.39 (s, 3H), 1.48–1.54 (m, 2H), 1.72 (s, 3H), 1.73–1.83 (m, 1H), 1.83–1.92 (m, 2H), 2.11–2.22 (m, 1H), 2.68 (td, J = 4.4, 10.7 Hz, 1H), 3.46 (dd, J = 3.6, 16.5 Hz, 1H), 5.44 (s, 1H), 6.72 (d, J = 1.8 Hz, 1H), 7.06 (d, J = 1.8 Hz, 1H); 13C NMR (125.8 MHz, CDCl3) δ 14.1, 18.1, 22.5, 23.4, 24.2, 27.4, 28.3, 28.6, 32.4, 35.2, 36.3, 37.4, 44.2, 46.4, 76.8, 115.0, 119.5, 122.6, 123.1, 123.5, 134.7, 150.6, 154.5; GC/MS (EI) m/z (rel intensity) 420 (100), 418 (97), 377 (55), 375 (47), 350 (94), 348 (83), 337 (86), 335 (78), 281 (30), 279 (32), 270 (19), 253 (12), 251 (13), 225 (17), 213 (11), 185 (15), 118 (11), 91 (12), 71 (12); HRMS: m/z calcd for C24H35BrO: 418.1871; found: 418.1876; [α]D 20 −277° (c 0.11, CHCl3).
4.16 1-Bromo-1-deoxy-3-(1′,1′-dimethylheptyl)-Δ8-tetrahydrocannabinol (11, JWH-383)
JWH-383 was prepared using the procedure used for the synthesis of JWH-460. From 0.20 g (0.42 mmol) of dioxaborolane (24) and 0.29 g (1.3 mmol) of copper (II) bromide there was obtained, after 4 h at reflux and column chromatography (petroleum ether), 0.12 g (66%) of JWH-383 as a viscous light yellow oil: 1H NMR (500 MHz, CDCl3) δ 0.85 (t, J = 6.4 Hz, 3H), 1.01–1.12 (m, 5H), 1.14–1.29 (m, 12H), 1.39 (s, 3H), 1.47–1.54 (m, 2H), 1.72 (s, 3H), 1.73–1.83 (m, 1H), 1.83–1.92 (m, 2H), 2.11–2.22 (m, 1H), 2.68 (td, J = 4.0, 10.4 Hz, 1H), 3.46 (dd, J = 3.6, 17.0 Hz, 1H), 5.44 (s, 1H), 6.72 (s, 1H), 7.05 (s, 1H); 13C NMR (125.8 MHz, CDCl3) δ 14.1, 18.1, 22.6, 23.4, 24.5, 27.4, 28.3, 28.6 × 2, 29.9, 31.7, 35.2, 36.3, 37.4, 44.3, 46.4, 76.8, 115.0, 119.5, 122.6, 123.1, 123.5, 134.8, 150.7, 154.5; GC/MS (EI) m/z (rel intensity) 434 (93), 432 (95), 391 (39), 389 (37), 352 (10), 351 (57), 350 (59), 349 (100), 348 (49), 347 (51), 279 (15), 121 (10), 85 (18), 71 (28); HRMS: m/z calcd for C25H37BrO: 432.2028; found: 432.2028; [α]D 20 − 268° (c 0.168, CHCl3).
4.17 Receptor Binding Experiments
4.17.1 Materials
Frozen whole brains of male Sprague-Dawley rats were obtained from Harlan (Dublin, VA). CP-55,940 was provided by Pfizer (Groton, CT). [3H]CP-55,940 (168 Ci/mmol) was purchased from NEN Life Science Products, Inc. (Boston, MA). Lipofectamine reagent was purchased from Life Technologies (Gaithersburg, MD). Human CB2 cDNA was provided by Dr. Sean Munro (MRC Lab, Cambridge, UK). DMEM and geneticin was purchased from (GIBCO BRL, Grand Island, NY). Fetal clone II was purchased from Hyclone Laboratories, Inc. (Logan, UT). Aquasil was purchased from Pierce (Rockford, IL). GF/C glass-fiber filters (2.4 cm) were purchased from Baxter (McGaw Park, IL). Polyethylenimine and bovine serum albumin were purchased from Sigma Chemical Co. (St. Louis, MO). Scintillation vials and Budget Solve scintillation fluid were purchased from RPI Corp. (Mount Prospect, IL).
4.17.2 Membrane Preparation
Chinese hamster ovary cells stably expressing the human CB2 receptor (CB2-CHO) cells38 were harvested in phosphate-buffered saline containing 1 μM EDTA and centrifuged at 500g. Cell pellets (for CB2) or whole rat brains (for CB1) were homogenized in 10 mL of membrane buffer (50 μM Tris-HCl, 1 μM EDTA, 3 μM MgCl2, pH 7.4). The homogenate was centrifuged at 50,000 × g = for 10 min. The pellet was resuspended in membrane buffer to yield a protein concentration of approximately 1 mg/mL. The tissue preparation was divided into equal aliquots, frozen on dry ice, and stored at −70 °C.
4.18 Competition Binding Assays
4.18.1 Binding Assay Procedure
[3H]CP-55,940 (168 Ci/mmol) binding to membranes prepared from whole rat brain (CB1) or CB2-CHO cells (CB2) was conducted as previously described.35,38,40 Briefly, CP-55,940 and all cannabinoid analogs were prepared by suspension in membrane buffer containing 5 mg/ml bovine serum albumin (assay buffer A) from a 1 mg/mL ethanolic stock (final concentration of no more than 0.4%). Competition curves were generated by incubating membranes with 1 nM of [3H]CP-55,940 with varying concentrations of unlabeled drugs for 1 hr at 30°C. Nonspecific binding was determined in the presence of 1 μM unlabeled CP,55,940. Binding was terminated by rapid filtration under vacuum through GF/B glass fiber filters (pretreated with polyethyleneimine (0.1%) for at least 2 hours), and radioactivity determined by liquid scintillation spectrophotomery. The assays were performed in triplicate, and the results represent the combined data from three individual experiments.
4.18.2 Data Analysis
Competition assays were conducted with 1 nM [3H]CP-55,940 and 6 concentrations (0.1 nM to 10 μM displacing ligands). Displacement IC50 values were originally determined by unweighted least-squares linear regression of log concentration-percent displacement data and then converted to Ki values using the method of Cheng and Prusoff.41 All experiments were performed in triplicate and repeated 3–6 times. All data are reported as mean values ± SEM.
4.19 [34S]GTPγS Binding Assays
4.19.1 Materials
All chemicals were from Sigma (St. Louis, MO) except the following: [34S]GTPγS (1250 Ci/mmol) was purchased from Perkin Elmer Life Sciences(Boston, MA), DMEM/F-12 from Fischer Scientific (Pittsburg, PA), and Whatman GF/B glass fiber filters from Fisher Scientific (Pittsburgh, PA).
4.19.2 Membrane Preparations
CB2-CHO cell membranes were cultured in a 50:50 mixture of DMEM and Ham F-12 supplemented with 100 U/ml penicillin, 100 Bg/ml streptomycin, 0.25 mg/ml G418, and 5% fetal calf serum. Cells were harvested by replacement of the media with cold phosphate-buffered saline containing 0.4% EDTA followed by agitation. Membranes were prepared by homogenization of cells in 50 μM Tris-HCl, 3 μM MgCl2, 1 μM EGTA, pH 7.4, centrifugation at 50,000 × g for 10 minutes at 4 °C, and resuspension in the same buffer at 1.5 mg/ml. Membranes were stored at −80 °C until use.
4.20.3 [34S]GTPγS Binding
Assays were conducted as previously described.38 Prior to assays, samples were thawed on ice, centrifuged at 50,000 × g for 10 minutes at 4 °C, and resuspended in Assay Buffer B (50 μM Tris-HCl (pH 7.4), 3 μM MgCl2, 0.2 μM EGTA, and 100 μM NaCl). Reactions containing 10 μg of membrane protein were incubated for 1.5 hr at 30°C in Assay Buffer B containing 10 μM GDP, 0.1 nM [34S]GTPγS, 1 mg/mL bovine serum albumin, and various concentrations of drugs. Nonspecific binding was determined in the presence of 20 M unlabeled GTPγS. Reactions were terminated by rapid vacuum filtration through GF/B glass fiber filters, and radioactivity was measured by liquid scintillation spectrophotometry.
4.20.4 Data Analysis
All data are reported as the means ± SEM of at least three experiments, each performed in triplicate. Nonlinear regression analysis was conducted by iterative fitting using JMP (SAS for Macintosh). Nonspecific [34S]GTPγS binding was subtracted from all data. Basal [34S]GTPγS binding is defined as specific [34S]GTPγS binding in the absence of drug. Net-stimulated [34S]GTPγS binding is defined as [34S]GTPγS binding in the presence of drug minus basal. Percent of CP55,940-stimulated binding is expressed as (net stimulated [34S]GTPγS binding by drug/net stimulated [34S]GTPγS binding by 3 μM CP55,940) × 100%.
Acknowledgments
The work at Clemson was supported by grant DA-03590 to JWH and that at Virginia Commonwealth University by grants DA-03672 to JLW and DA-026593 to DES all from the National Institute on Drug Abuse.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Gaoni Y, Mechoulam R. J Am Chem Soc. 1964;86:1648. [Google Scholar]
- 2.Razdan RK. Pharmacol Rev. 1986;38:75. [PubMed] [Google Scholar]
- 3.Rapaka RS, Makriyannis A. NIDA Research Monograph. Vol. 79. National Institute on Drug Abuse; Rockville, MD: 1987. Structure-Activity Relationships of the Cannabinoids. [Google Scholar]
- 4.Mechoulam R, Devane WA, Glaser R. In: Marijuana/Cannabinoids: Neurobiology and Neurophysiology. Murphy L, Bartke A, editors. CRC Press; Boca Raton: 1992. pp. 1–33. [Google Scholar]
- 5.Devane WA, Dysarz FA, Johnson MR, Melvin LS, Howlett AC. Mol Pharmacol. 1988;34:605. [PubMed] [Google Scholar]
- 6.Matsuda LA, Lolait SJ, Brownstein MJ, Young AC. Nature (London) 1990;346:561. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 7.Gérard CM, Mollereau C, Vassart G, Parmentier M. Biochem J. 1991;279:129. doi: 10.1042/bj2790129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Munro S, Thomas KL, Abu-Shar M. Nature (London) 1993;365:61. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 10.Klein TW, Friedman H, Specter S. J Neuroimmunol. 1998;83:102. doi: 10.1016/s0165-5728(97)00226-9. [DOI] [PubMed] [Google Scholar]
- 11.Galiègue S, Mary S, Marchand J, Dussossoy D, Carrière D, Carayon P, Bouaboula M, Shire D, Le Fur G, Casellas P. Eur J Biochem. 1995;232:54. doi: 10.1111/j.1432-1033.1995.tb20780.x. [DOI] [PubMed] [Google Scholar]
- 12.Condie R, Herring A, Koh WS, Lee M, Kaminski NE. J Biol Chem. 1996;271:13175. doi: 10.1074/jbc.271.22.13175. [DOI] [PubMed] [Google Scholar]
- 13.Pettit DA, Anders DL, Harrison MP, Cabral GA. Adv Exp Med Biol. 1996;402:119–129. doi: 10.1007/978-1-4613-0407-4_17. [DOI] [PubMed] [Google Scholar]
- 14.Schatz AR, Lee M, Condie RB, Pulaski JT, Kaminski NE. Toxicol Appl Pharmacol. 1997;142:278. doi: 10.1006/taap.1996.8034. [DOI] [PubMed] [Google Scholar]
- 15.Buckley NE, McCoy KL, Mezey E, Bonner T, Zimmer A, Felder CC, Glass M, Zimmer A. Eur J Pharmacol. 2000;396:141. doi: 10.1016/s0014-2999(00)00211-9. [DOI] [PubMed] [Google Scholar]
- 16.Bifulco M, DiMarzo V. Nature Med. 2002;8:547. doi: 10.1038/nm0602-547. [DOI] [PubMed] [Google Scholar]
- 17.Guzman M. Nature Rev. 2003;3:745. doi: 10.1038/nrc1188. [DOI] [PubMed] [Google Scholar]
- 18.Hanus L, Breuer A, Tchilibon S, Shiloah S, Goldenberg D, Horowitz M, Pertwee RG, Ross RA, Mechoulam R, Fride E. Proc Natl Acad Sci USA. 1999;96:14228. doi: 10.1073/pnas.96.25.14228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Conti S, Costa B, Colleoni M, Parolaro D, Giagnoni G. Br J Pharmacol. 2002;135:181. doi: 10.1038/sj.bjp.0704466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Malan TP, Ibrahim MM, Vanderah TW, Makriyannis A, Porreca F. Chem Phys Lipids. 2002;121:191. doi: 10.1016/s0009-3084(02)00155-x. [DOI] [PubMed] [Google Scholar]; (b) Malan TP, Ibrahim MM, Deng H, Liu Q, Main HP, Vanderah T, Porreca F, Makriyannis A. Pain. 2001;93:239. doi: 10.1016/S0304-3959(01)00321-9. [DOI] [PubMed] [Google Scholar]
- 21.Ibrahim MM, Deng H, Zvonok A, Cockayne DA, Kwan J, Mata HP, Vanderah TW, Lal J, Porreca F, Makriyannis AM, Malan TP. Proc Natl Acad Sci USA. 2003;100:10529. doi: 10.1073/pnas.1834309100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang J, Hoffert C, Vu HK, Groblewski T, Ahmad S, O’Donnell D. Eur J Neurosci. 2003:2750. doi: 10.1046/j.1460-9568.2003.02704.x. [DOI] [PubMed] [Google Scholar]
- 23.Kehl LJ, Hamamoto DT, Wacnik PW, Croft DL, Norsted BD, Wilcox GL, Simone DA. Pain. 2003;103:175. doi: 10.1016/s0304-3959(02)00450-5. [DOI] [PubMed] [Google Scholar]
- 24.Clayton N, Marshall FH, Bountra C, O’Shaughnessy CT. Pain. 2002;96:253. doi: 10.1016/S0304-3959(01)00454-7. [DOI] [PubMed] [Google Scholar]
- 25.Giblin GMP, O’Shaughnessy CT, Naylor A, Mitchell WL, Eatherton AJ, Slingsby BP, Rawlings DA, Goldsmith P, Brown AJ, Haslam CP, Clayton NM, Wilson AW, Chessel IP, Wittington AR, Green R. J Med Chem. 2007;50:2597. doi: 10.1021/jm061195+. [DOI] [PubMed] [Google Scholar]
- 26.Mackie K. Annu Rev Pharmacol Toxicol. 2006;46:101. doi: 10.1146/annurev.pharmtox.46.120604.141254. [DOI] [PubMed] [Google Scholar]
- 27.Pacher P, Batkai S, Kunos G. Pharmacol Rev. 2006;58:389. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huffman JW, Yu S, Showallter V, Abood ME, Wiley JL, Compton DR, Martin BR, Bramblett RD, Reggio PH. J Med Chem. 1996;39:3875. doi: 10.1021/jm960394y. [DOI] [PubMed] [Google Scholar]
- 29.Gareau Y, Dufresne C, Gallant M, Rochette C, Sawyer N, Slipetz DM, Tremblay N, Weech PK, Metters KM, Labelle M. Bioorg Med Chem Lett. 1996;6:189. [Google Scholar]
- 30.Huffman JW, Liddle J, Yu S, Aung MM, Abood ME, Wiley JL, Martin BR. Bioorg Med Chem. 1999;7:2905. doi: 10.1016/s0968-0896(99)00219-9. [DOI] [PubMed] [Google Scholar]
- 31.Huffman JW, Bushell SM, Miller JRA, Wiley JL, Martin BR. Bioorg Med Chem. 2002;10:4119. doi: 10.1016/s0968-0896(02)00331-0. [DOI] [PubMed] [Google Scholar]
- 32.Thompson ALS, Kabalka GW, Akula MR, Huffman JW. Synthesis. 2005:547. [Google Scholar]
- 33.Huffman JW, Miller JRA, Liddle J, Yu S, Thomas BF, Wiley JL, Martin BR. Bioorg Med Chem. 2003;11:1397–1410. doi: 10.1016/s0968-0896(02)00649-1. [DOI] [PubMed] [Google Scholar]
- 34.Nesmejanow AN, Perewalowa EG, Jurjewa LP. Chem Ber. 1960:2729. [Google Scholar]
- 35.Compton DR, Rice KC, De Costa BR, Razdan RK, Melvin LS, Johnson MR, Martin BR. J Pharmacol Exp Ther. 1993;265:218. [PubMed] [Google Scholar]
- 36.Showalter VM, Compton DR, Martin BR, Abood ME. J Pharmacol Exp Ther. 1996;278:989. [PubMed] [Google Scholar]
- 37.Selley DE, Stark S, Sim LJ, Childers SR. Life Sci. 1996;59:659. doi: 10.1016/0024-3205(96)00347-5. [DOI] [PubMed] [Google Scholar]
- 38.Huffman JW, Zengin G, Wu MJ, Lu J, Hynd G, Bushell K, Thompson ALS, Bushell S, Tartal C, Hurst DP, Reggio PH, Selley DE, Cassidy MP, Wiley JL, Martin BR. Bioorg Med Chem. 2005;13:89. doi: 10.1016/j.bmc.2004.09.050. [DOI] [PubMed] [Google Scholar]
- 39.Eliel EL, Willen SH, Mander LN. Stereochemistry of Carbon Compounds. John Wiley and Sons; New York: 1993. p. 1144. [Google Scholar]
- 40.Martin BR, Compton DR, Thomas BF, Prescott WR, Little PJ, Razdan RK, Johnson MR, Melvin LS, Mechoulam R, Ward SJ. Pharmacol Biochem Behav. 1991;40:471. doi: 10.1016/0091-3057(91)90349-7. [DOI] [PubMed] [Google Scholar]
- 41.Cheng YC, Prusoff WH. Biochem Pharmacol. 1973;22:309. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]