

Abstract
The 14 Å resolution structure of the 2.3 MDa Ca2+ release channel (also known as RyR1) was determined by electron cryomicroscopy and single particle reconstruction. This structure was produced using collected data used for our previous published structures at 22–30 Å resolution, but now taking advantage of recent algorithmic improvements in the EMAN software suite. This improved map clearly exhibits more structural detail and allows better defined docking of computationally predicted structural domain folds. Using sequence-based fold recognition, the N-terminal region of RyR1, residues 216–572, was predicted to have significant structural similarity with the IP3-binding core region of the type 1 IP3R. This putative structure was computationally localized to the clamp-shaped region of RyR1, which has been implicated to have a regulatory role in the channel activity.
Keywords: type 1 ryanodine receptor, electron cryomicroscopy, single particle reconstruction
The type 1 ryanodine receptor (RyR1), the main player in excitation–contraction coupling in skeletal muscle, represents an intracellular Ca2+ release channel located in the sarcoplasmic reticulum (SR) membrane. Each subunit of RyR1 is composed of 5037 (or 5032) amino acid residues with predicted Mr of 565 kDa. Four RyR1 monomers assemble as a homotetrameric complex with a molecular mass of ~2.3 MDa, making it one of the largest known ion channels. RyR1 functions as a cation-specific ligand-gated channel which can be modulated by a variety of physiological ligands including Ca2+, Mg2+, calmodulin, FKBP12, and ATP. In skeletal muscle, activation of RyR1 occurs through interaction with the dihydropyridine receptor (DHPR), located in the transverse tubular membrane, directly opposite RyR1 in the SR membrane. The RyR1 structure combines large size with complex allosteric modulation of channel gating, producing conformational variability in the sample corresponding to a mixture of different functional states in a reaction mixture. Due to the large number of potential modulators, producing a structurally homogeneous population for high-resolution imaging is quite challenging.
Nonetheless, this particle was among the first non-icosahedral structures to be solved using electron cryomicroscopy and single particle analysis at low resolution. The overall structure of RyR1 has been determined at resolutions of 22–30 Å from studies of frozen-hydrated receptors, providing a quaternary structure of this complex.1–3 Further experiments exhibited morphological changes associated with the channel’s open and closed states,4,5 as well as localization of cellular regulator binding sites6–8 and of the N-terminal region.3,9 Membrane proteins are among the most difficult targets for single particle reconstruction for numerous reasons, including possible structural flexibility of the channel, possible heterogeneity in the transmembrane domain due to varying amounts of detergent, and poor image contrast due both to the presence of detergent and the use of a continuous carbon substrate for adhering particles to the EM grid. Despite the considerable effort spent to extend the resolution of these reconstructions over the past decade, little success has been achieved, and low-resolution structures have remained the basis for many speculations on the detailed structure-functional relationships in this channel.
Through improvements to several image processing algorithms in the EMAN software suite,10 we have now achieved a substantial improvement to 14 Å resolution. The reconstruction was performed using a larger subset of the same data used for our latest 22 Å structure of the closed state of the channel.3 Our 14 Å map clearly elucidates substructures within both the cytoplasmic and trans-membrane regions of the channel (Figure 1). As expected, the overall morphology of the channel remains the same:2,3 the channel structure exhibits a characteristic square mushroom shape with a bulky cytoplasmic region and the membrane-spanning stem. The cytoplasmic assembly extends over 100 Å from the membrane, providing the possibility for a physical link between RyR1 and DHPR. Among the structural details revealed is the complex structure of the clamp-shaped regions at the four corners with many grooves with numerous intervening cavities (Figure 1(a)) that appear suitable for inter-digitating with neighboring molecules of RyR1 as seen in situ11 or with modulatory auxiliary proteins. The clamp-shaped regions may be those interacting with DHPRs.12,13
Figure 1.
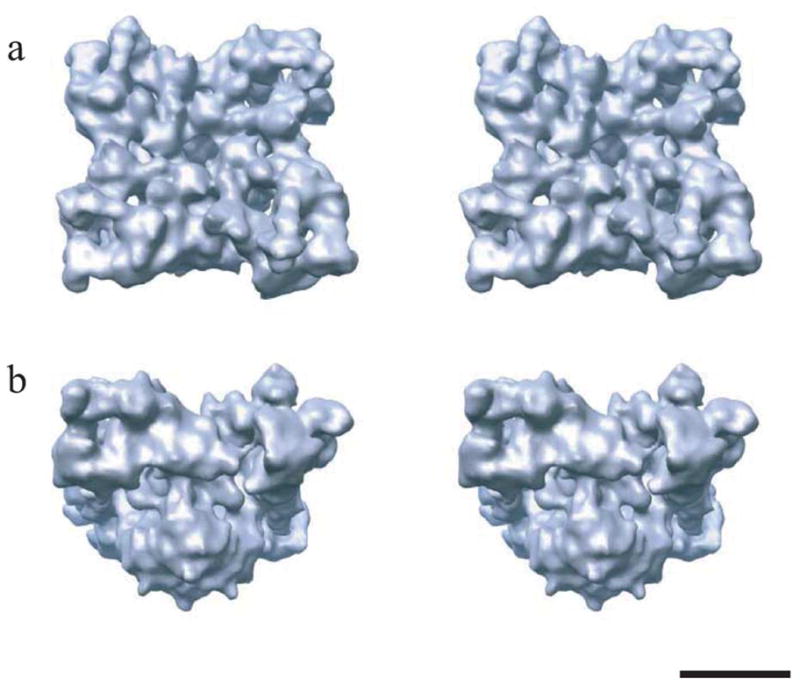
Stereo views of the 3D structure of RyR1 at 14 Å resolution. The structure is shown at an oblique angle as viewed from the cytoplasm (a) and in a side view (b) with the cytoplasmic side facing upward. The reconstruction was generated using ~22,000 individual particle images collected on a JEOL 1200EX electron cryomicroscope, equipped with a tungsten filament and operated at 100 kV under minimal dose conditions (5–7 e/Å2) and at the defocus range of 0.9–4.1 μm.3 The closed channel conformation was obtained by the depletion of Ca2+ with 1 mM EGTA.2,3 The threshold level corresponds to an enclosed protein mass of 2.5 MDa assuming a protein density of 1.35 g/cm3. The scale bar represents 100 Å.
The stem region is comprised of four high density column domains attached to a square-shaped structure spanning the SR membrane (Figure 1(b)). Viewed from the cytoplasmic side, there is a hollow in the center of the transmembrane domain, which, at high isosurface thresholds can even appear as a hole through the structure. At this resolution, no definitive statements can be made about the location of the actual pore, but this region seems a strong candidate. In addition, for the first time, some clear density is observed on the inside of the SR membrane. While the transmembrane region is better defined in this structure than the previous structures, the number of transmembrane helices still cannot be resolved. This is largely due to uncertainties about the amount of disordered detergent expected to exist around the transmembrane domain. Several models have been proposed for the transmembrane region, which predict four,14 eight16 and twelve15 transmembrane helices, respectively. In the current structure, none of these models can be decisively eliminated. It is unlikely that this issue will be resolved until a structure of at least 8–10 Å resolution is available and α-helices can be more clearly resolved (Figure 2).
Figure 2.

Fourier Shell Correlation (FSC) curve for the reconstruction as produced by the EMAN program eotest. To avoid any possibility of initial model bias, no reference was made to any previous model during refinement. The structure was refined from an initial model generated from automatically generated top and side views of the particle with 4-fold symmetry enforced. The overall refinement methodology was the same as a typical iterative EMAN refinement: projection-based particle classification: generation of a contrast transfer function (CTF)-corrected average for each class followed by reconstruction of a new 3D model from the class-averages.29 This refinement is iterated until convergence is achieved. The algorithm improvements which allowed this resolution improvement are described in detail elsewhere.10 First, the precision of the 2D alignment routine was improved, producing more accurate classifications. Second, classification accuracy was also enhanced through use of a new similarity criterion incorporating a true per-particle Wiener filter. Third, the resolution of the individual class-averages was improved through gradual reduction of the iterative self-alignment process used to produce class-averages. Resolution in the final reconstruction was determined using the standard technique of splitting the data into even and odd halves and calculating a FSC curve. In this case the most conservative, 0.5 threshold value was used. Since the determined 14 Å resolution is already approaching the traditional sampling limit of 3× oversampling, claiming any resolution beyond this point would not be justified.
Localization of the N-terminal region of RyR1
In a large molecule like RyR1, it is reasonable to expect that multiple structural domains will exist, and that these domains may correlate with known sequences or folds from other structures. This is a two-stage process. First, homologous folds are predicted using one of many available databases as described below. Once a homologous domain has been predicted, the corresponding 3D fold is converted into an electron density map at an appropriate resolution and docked into the cryo-EM reconstruction. The first step is predictive in nature and its accuracy may vary depending on the algorithm and the scope of the database used in the homology search. The reliability of the second step depends on the size of the homologous structure compared to the resolution of the cryo-EM reconstruction as well as the topological complexity of the features being docked. Nevertheless both procedures have substantially improved over recent years.
In our earlier studies using the UCLA-DOE Fold recognition server,17 we found residues 41–420 of RyR1 to be homologous to phosphorylated isocitrate dehydrogenase, 4ICD.3 However, this fold is no longer found by the current version of this server. This appears to be due to changes made to the homology determination algorithms used on this server. This fold is also not found by any of the newer homology servers available through the Structure Prediction Meta-Server.18
However, homology prediction has been an active field in recent years, and substantial improvements have been made. Using the 3D-PSSM fold recognition server,19 we now find that the N-terminal region of RyR1, residues 216–572, has significant structural similarity with the IP3-binding core region (residues 226–576) of the type 1 IP3R.20 The E-value for this prediction was found to be 2.54×10−27, corresponding to a 95% confidence level. The two sequences have a sequence identity of 20%. This prediction was further confirmed using multiple servers through the Structure Prediction Meta-Server. All servers ranked this as the most homologous fold to RyR1, with the exception of the UCLA-DOE server, which does not contain this fold. Furthermore, at the time of our previous publication, the IP3R crystal structure had not yet been published. This consensus of multiple algorithms strongly supports the hypothesis that this prediction represents a true homology.
To localize the predicted homologous IP3R domain within the 3D structure of RyR1, a homology model for the N-terminal region of RyR1 was constructed based on the X-ray structure of the IP3-binding core region20 and docked into the 14 Å density map using Foldhunter.21 The best fit for this domain was found to be in the clamp-shaped region of the RyR1 structure (Figure 3). Using a new option for ascertaining fit quality, this fit was found to have a confidence level greater than 95%. This assignment of the RyR1 N-terminal region also agrees well with previous assignments based on experimental evidence.3,9
Figure 3.
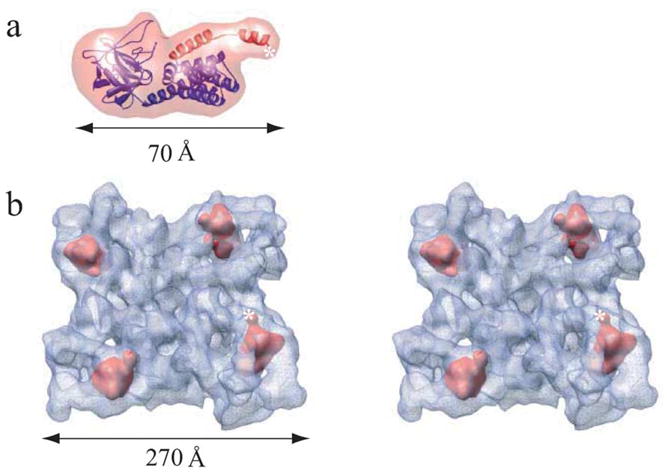
Localization of the IP3-like region of RyR1. (a) Homology model for the InP3-like RyR1 domain (blue/red ribbon) and corresponding 14 Å density map (transparent red) generated using EMAN. The sequence corresponding to the LZ1 region residues 541–572 is shown with red ribbon. A homology model was generated by mapping of the coordinates of the X-ray structure of the IP3-binding core region (1N4K)20 and the sequence residues 216–572 of RyR1 aligned to them. The model was further enhanced by the addition of side-chains using the SCWL algorithm.30 Sequence alignment, secondary structure prediction and homology modeling were performed using 3D-PSSM Fold Recognition server. (b) Fitting of 14 Å electron density map of the IP3-like domain within RyR1 tetramer shown in tilted stereo top-view. Assignment of mass densities for the IP3-like domain was performed using 6D fitting program Foldhunter as previously described.3 The top four correlation peaks with local correlation coefficients of about 0.91 (Foldhunter score) are found in the clamp-shaped domain of each of the four monomers in the RyR1 reconstruction. This fit had a z-score of 1.97. z-Score is a statistical measure of the number of standard deviations from the mean, which, in this case, corresponds a confidence level greater than 95%. A star marks the C terminus of the modeled region.
In an additional test, we found that IP3 has no effect on [3H]ryanodine binding to SR membranes (data not shown). Again, this evidence is not conclusive, since the lack of functional effect may be caused by the mechanism that this structural motif of RyR1 is used for. It is known that similar domain folds may exist in different proteins, and serve very different functions. For example, the seven-bladed beta-propeller fold has been found in proteins from multiple superfamilies with totally different functions.22–25 Sequence analysis shows that the IP3-binding-like region of RyR1 includes much of the region responsible for genetic diseases (MH/CCD in residues 35–614), and the Leucine Zipper region (LZ1, residues 541–593). This latter region has been implicated as a protein–protein interaction motif with anchoring proteins that in turn bind protein kinase A and protein phosphatase 1 to regulate the RyR1 activity.26 It is also worth mentioning that the putative structure corresponding to the LZ1 region is located in close proximity to the region where the FKBP12 binding site was previously localized.6
The clamp-shaped domains of RyR1 have been shown in our previous studies to undergo conformational changes between open and closed states of the channel,4,5 and structural heterogeneity in these regions can be observed even within data collected ostensibly in a single functional state (S.J.L., unpublished results). Remarkably the peripheral regions of the cytoplasmic assembly of IP3R1, in which we have putatively localized the IP3-binding domains,27 also undergo a substantial Ca2+-induced structural change.28 Based on these findings and the relatively high conservation of similar sequence motifs in the N-terminal domains of RyRs and IP3Rs, we propose that the clamp-shaped domains of RyR1 and the peripheral domains in the cytoplasmic region of IP3R1 are the most likely regions for channel-specific modulators. Therefore, they must be allosterically coupled to the trans-membrane domains in order to induce the opening of the Ca2+ permeable pore.
This structure takes a substantial step towards subnanometer resolution on this important macromolecule. Our future work will focus on improvements to the experimental conditions and collection of a larger data set on a higher resolution electron cryomicroscope.
Acknowledgments
We thank Drs Wen Jiang and Matthew L. Baker (Baylor College of Medicine) for improving Foldhunter to provide a quantitative measure of level of confidence. This research was supported by NIH grants (P41RR02250, P01GM99116, R01AR44864 and RO1AR41729), and a grant from the MDA.
Abbreviations used
- RyR1
the type 1 ryanodine receptor
- DHPR
dihydropyridine receptor
References
- 1.Radermacher M, Wagenknecht T, Grassucci R, Frank J, Inui M, Chadwick C, Fleischer S. Cryo-EM of the native structure of the calcium release channel/ryanodine receptor from sarcoplasmic reticulum. Biophys J. 1992;61:936–940. doi: 10.1016/S0006-3495(92)81900-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Serysheva II, Orlova EV, Chiu W, Sherman MB, Hamilton SL, Van Heel M. Electron cryomicroscopy and angular reconstitution used to visualize the skeletal muscle calcium release channel. Nature Struct Biol. 1995;2:18–24. doi: 10.1038/nsb0195-18. [DOI] [PubMed] [Google Scholar]
- 3.Baker ML, Serysheva II, Sencer S, Wu Y, Ludtke SJ, Jiang W, et al. The skeletal muscle Ca2+ release channel has an oxidoreductase-like domain. Proc Natl Acad Sci USA. 2002;99:12155–12160. doi: 10.1073/pnas.182058899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Orlova EV, Serysheva II, van Heel M, Hamilton SL, Chiu W. Two structural configurations of the skeletal muscle calcium release channel. Nature Struct Biol. 1996;3:547–552. doi: 10.1038/nsb0696-547. [DOI] [PubMed] [Google Scholar]
- 5.Serysheva II, Schatz M, van Heel M, Chiu W, Hamilton SL. Structure of the skeletal muscle calcium release channel activated with Ca2+ and AMP-PCP. Biophys J. 1999;77:1936–1944. doi: 10.1016/S0006-3495(99)77035-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wagenknecht T, Radermacher M, Grassucci R, Berkowitz J, Xin HB, Fleischer S. Locations of calmodulin and FK506-binding protein on the three- dimensional architecture of the skeletal muscle ryanodine receptor. J Biol Chem. 1997;272:32463–32471. doi: 10.1074/jbc.272.51.32463. [DOI] [PubMed] [Google Scholar]
- 7.Zhang J, Liu Z, Masumiya H, Wang R, Jiang D, Li F, et al. Three-dimensional localization of divergent region 3 of the ryanodine receptor to the clamp-shaped structures adjacent to the FKBP binding sites. J Biol Chem. 2003;278:14211–14218. doi: 10.1074/jbc.M213164200. [DOI] [PubMed] [Google Scholar]
- 8.Samso M, Wagenknecht T. Apocalmodulin and Ca2+-calmodulin bind to neighboring locations on the ryanodine receptor. J Biol Chem. 2002;277:1349–1353. doi: 10.1074/jbc.M109196200. [DOI] [PubMed] [Google Scholar]
- 9.Liu Z, Zhang J, Sharma MR, Li P, Chen SR, Wagenknecht T. Three-dimensional reconstruction of the recombinant type 3 ryanodine receptor and localization of its amino terminus. Proc Natl Acad Sci USA. 2001;98:6104–6109. doi: 10.1073/pnas.111382798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ludtke SJ, Chen DH, Song JL, Chuang DT, Chiu W. Seeing GroEL at 6 Å resolution by single particle electron cryomicroscopy. Structure (Camb) 2004;12:1129–1136. doi: 10.1016/j.str.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 11.Block BA, Imagawa T, Campbell KP, Franzini-Armstrong C. Structural evidence for direct interaction between the molecular components of the transverse tubule/sarcoplasmic reticulum junction in skeletal muscle. J Cell Biol. 1988;107:2587–2600. doi: 10.1083/jcb.107.6.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Protasi F, Franzini-Armstrong C, Flucher BE. Coordinated incorporation of skeletal muscle dihydropyridine receptors and ryanodine receptors in peripheral couplings of BC3H1 cells. J Cell Biol. 1997;137:859–870. doi: 10.1083/jcb.137.4.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Serysheva II, Ludtke SJ, Baker MR, Chiu W, Hamilton SL. Structure of the voltage-gated L-type Ca2+ channel by electron cryomicroscopy. Proc Natl Acad Sci USA. 2002;99:10370–10375. doi: 10.1073/pnas.162363499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takeshima H, Nishimura S, Matsumoto T, Ishida H, Kangawa K, Minamino N, et al. Primary structure and expression from complementary DNA of skeletal muscle ryanodine receptor. Nature. 1989;339:439–445. doi: 10.1038/339439a0. [DOI] [PubMed] [Google Scholar]
- 15.Zorzato F, Fujii J, Otsu K, Phillips M, Green NM, Lai FA, et al. Molecular cloning of cDNA encoding human and rabbit forms of the Ca2+ release channel (ryanodine receptor) of skeletal muscle sarcoplasmic reticulum. J Biol Chem. 1990;265:2244–2256. [PubMed] [Google Scholar]
- 16.Du GG, Sandhu B, Khanna VK, Guo XH, MacLennan DH. Topology of the Ca2+ release channel of skeletal muscle sarcoplasmic reticulum (RyR1) Proc Natl Acad Sci USA. 2002;99:16725–16730. doi: 10.1073/pnas.012688999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fischer D, Eisenberg D. Protein fold recognition using sequence-derived predictions. Protein Sci. 1996;5:947–955. doi: 10.1002/pro.5560050516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ginalski K, Elofsson A, Fischer D, Rychlewski L. 3D-Jury: a simple approach to improve protein structure predictions. Bioinformatics. 2003;19:1015–1018. doi: 10.1093/bioinformatics/btg124. [DOI] [PubMed] [Google Scholar]
- 19.Kelley LA, MacCallum RM, Sternberg MJ. Enhanced genome annotation using structural profiles in the program 3D-PSSM. J Mol Biol. 2000;299:499–520. doi: 10.1006/jmbi.2000.3741. [DOI] [PubMed] [Google Scholar]
- 20.Bosanac I, Alattia JR, Mal TK, Chan J, Talarico S, Tong FK, et al. Structure of the inositol 1,4,5-trisphosphate receptor binding core in complex with its ligand. Nature. 2002;420:696–700. doi: 10.1038/nature01268. [DOI] [PubMed] [Google Scholar]
- 21.Jiang W, Baker ML, Ludtke SJ, Chiu W. Bridging the information gap: computational tools for intermediate resolution structure interpretation. J Mol Biol. 2001;308:1033–1044. doi: 10.1006/jmbi.2001.4633. [DOI] [PubMed] [Google Scholar]
- 22.Ito N, Phillips SE, Stevens C, Ogel ZB, McPherson MJ, Keen JN, et al. Novel thioether bond revealed by a 1.7 Å crystal structure of galactose oxidase. Nature. 1991;350:87–90. doi: 10.1038/350087a0. [DOI] [PubMed] [Google Scholar]
- 23.Gaudet R, Bohm A, Sigler PB. Crystal structure at 2.4 Å resolution of the complex of transducin betagamma and its regulator, phosducin. Cell. 1996;87:577–588. doi: 10.1016/s0092-8674(00)81376-8. [DOI] [PubMed] [Google Scholar]
- 24.Chen L, Doi M, Durley RC, Chistoserdov AY, Lidstrom ME, Davidson VL, Mathews FS. Refined crystal structure of methylamine dehydrogenase from Paracoccus denitrificans at 1.75 Å resolution. J Mol Biol. 1998;276:131–149. doi: 10.1006/jmbi.1997.1511. [DOI] [PubMed] [Google Scholar]
- 25.Mohri K, Vorobiev S, Fedorov AA, Almo SC, Ono S. Identification of functional residues on Caenorhabditis elegans actin-interacting protein 1 (UNC-78) for disassembly of actin depolymerizing factor/cofilin-bound actin filaments. J Biol Chem. 2004;279:31697–31707. doi: 10.1074/jbc.M403351200. [DOI] [PubMed] [Google Scholar]
- 26.Marx SO, Reiken S, Hisamatsu Y, Gaburjakova M, Gaburjakova J, Yang YM, et al. Phosphorylation-dependent regulation of ryanodine receptors: a novel role for leucine/isoleucine zippers. J Cell Biol. 2001;153:699–708. doi: 10.1083/jcb.153.4.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Serysheva II, Bare DJ, Ludtke SJ, Kettlun CS, Chiu W, Mignery GA. Structure of the type 1 inositol 1,4,5-trisphosphate receptor revealed by electron cryomicroscopy. J Biol Chem. 2003;278:21319–21322. doi: 10.1074/jbc.C300148200. [DOI] [PubMed] [Google Scholar]
- 28.Hamada K, Miyata T, Mayanagi K, Hirota J, Mikoshiba K. Two-state conformational changes in inositol 1,4,5-trisphosphate receptor regulated by calcium. J Biol Chem. 2002;277:21115–21118. doi: 10.1074/jbc.C200244200. [DOI] [PubMed] [Google Scholar]
- 29.Ludtke SJ, Jakana J, Song J, Chuang DT, Chiu W. An 11.5 Å single particle reconstruction of GroEL using EMAN. J Mol Biol. 2001;314:241–250. doi: 10.1006/jmbi.2001.5133. [DOI] [PubMed] [Google Scholar]
- 30.Bower MJ, Cohen FE, Dunbrack RL., Jr Prediction of protein side-chain rotamers from a backbone-dependent rotamer library: a new homology modeling tool. J Mol Biol. 1997;267:1268–1282. doi: 10.1006/jmbi.1997.0926. [DOI] [PubMed] [Google Scholar]