

Abstract
Pif1p is the prototype member of a family of helicases that is highly conserved from yeast to humans. In yeast, Pif1p is involved in many aspects of the preservation of genome stability. In particular, Pif1p is involved in the maintenance of mitochondrial DNA and in the direct inhibition of telomerase at telomeres and double-stranded breaks. Here we describe methods to purify Pif1p and study in vitro its enzymatic properties and functional interaction with telomerase.
Keywords: Yeast, Pif1p, helicase, telomerase, oligonucleotide substrate-based radiometric assays
1. Introduction
This chapter focuses on the characterization of the Saccharomyces cerevisiae Pif1p helicase. Pif1p is the prototype member of the PIF1 family of helicases, which is conserved from yeast to human (1, 2). Two isoforms of the enzyme are expressed in yeast, owing to alternative usage of two start codons from the same mRNA. Translation from the first start codon leads to the synthesis of a mitochondria-directed isoform, while translation from the second AUG codon leads to the synthesis of the nuclear isoform (3). Genetic studies have shown that the nuclear form of Pif1p plays an important role in counteracting the activity of telomerase, the specialized reverse transcriptase that elongates the end of eukaryotic chromosomes. Through this activity, Pif1p prevents gross chromosomal rearrangements that are due to the addition of telomerase-mediated de novo telomere addition at double strand breaks (4). In vivo and in vitro data suggest that this action is achieved through a direct interaction between Pif1p and telomerase (5, 6). Using oligonucleotide-based radiometric assays, Pif1p has been shown to unwind preferentially RNA–DNA hybrids over DNA substrates (7). This preference suggests that Pif1p inhibits telomerase by unwinding the RNA–DNA substrate formed by the telomerase RNA, TLC1, and the telomeric DNA end. Importantly, the interaction between Pif1p and telomerase is conserved in evolution, since Pif1p has been shown to interact with mouse and human telomerase (8–10). This chapter focuses on in vitro methods to purify recombinant yeast Pif1p and to characterize its activity by classical oligonucletide substrate-based radiometric assays. We also describe methods to study in vitro its functional interaction with yeast telomerase.
2. Materials
2.1. Overexpression of Recombinant His-Tagged Pif1p in Bacteria
Luria Bertani (LB) (per liter): 10 g Bacto-tryptone, 5 g yeast extract, 10 g NaCl. Adjust pH to 7.5 with 1 N NaOH. Autoclave.
Isopropyl-β-thiogalactoside (IPTG): 1 M solution in ddH2O. Filter-sterilize and store at −20°C in 1 mL aliquots.
Kanamycin sulfate stock solution: make stock at 50 mg/mL in ddH2O. Filter sterilize. Store at 4°C.
Chloramphenicol stock solution: make stock at 50 mg/mL in 100% ethanol. Store at −20°C.
2.2. Purification of Recombinant His-Tagged Pif1p
2.2.1. Affinity Chromatogaphy
Buffer A: 30.5 mM Na2HPO4, 19.5 mM NaH2PO4, 300 mM NaCl, pH 7.0. Filter sterilize.
1 M imidazole: pH 7.0. Adjust pH with 1 N NaOH and filter sterilize.
Protease inhibitor cocktail tablets, EDTA free (Roche Applied Science).
45 μM sterile syringe filter; 10 mL sterile plastic syringe.
Sonicator and thin probe.
Talon polyhistidine-Tag purification resin (Clontech) (see Note 1).
Low pressure chromatography column (5 mL capacity or above, e.g., Millipore Vantage-L chromatography column or equivalent).
Peristaltic pump (Gilson Minipuls, or equivalent) and silicone tubing.
Low protein binding tubes (e.g., Nunc MiniSorp).
Dialysis tubing (e.g., Pierce Snakeskin, 10 kD cut off) and clips (Pierce).
Anti-His-Tag monoclonal antibody (Novagen).
2.2.2. Cation Exchange Chromatography
Buffer A2: 45.5 mM Na-acetate, 4.5 mM acetic acid, 50 mM (NH4)2SO4, 50 mM Mg-acetate, 200 mM NaCl, 5% glycerol, pH 5.6. Filter sterilize.
Buffer B2: Same composition as buffer A2 but containing NaCl 1 M, pH 5.6. Filter sterilize.
Buffer C: HEPES 50 mM pH 7.8, (NH4)2SO4 50 mM, Mg-acetate 50 mM, 200 mM NaCl, DTT 1 mM, glycerol 5%. Filter sterilize.
Pif1p storage buffer: 0.5 × buffer C containing 50% glycerol.
FPLC system (GE healthcare Akta system or equivalent).
Centrifugal filter concentrator, 10 kD cut off (Millipore Centricon YM-10 or equivalent).
2.3. Preparation of Telomerase Activity from Yeast Protein Extracts
2.3.1. Overexpression of Telomerase Core Subunits
Complete SCGLA medium (per liter): 6.7 g of yeast nitrogen base without amino acids, 1 g glucose, 30 g glycerol, 20 g lactic acid, 20 mg of each adenine, uracil, histidine, tryptophan, proline, arginine, and methionine; 30 mg of each leucine, isoleucine, tyrosine, and lysine. Adjust pH to 5–6 with NaOH 10 N and autoclave.
YPGLA medium (per liter): 10 g yeast extract, 20 g peptone, 2 g glucose, 30 g glycerol, 20 g lactic acid, 20 mg adenine. Adjust to pH 5–6 and autoclave.
D(+)-galactose (Sigma).
Diethylpyrocarbonate (DEPC)-treated ddH2O. Add 0.5 mL DEPC to 500 mL ddH2O. Mix well. Incubate at 37°C overnight. Sterilize by autoclaving.
Buffer L: 40 mM Tris–HCl, pH 8.0, 500 mM sodium-acetate, 2.2 mM MgCl2, 0.2 mM EDTA, 0.2% Triton X-100, 0.4% Igepal CA-630, 20% glycerol. Prepare using RNase-free reagents and glassware (see Note 2).
2.3.2. Telomerase Activity Fractionation and Storage
As telomerase activity is RNase-sensitive, all reagents, solutions, and equipment must be handled in an RNase-free environment (see Note 2).
Automated mortar and Pestle (e.g., Retsch RM100).
RNaseZAP solution (Ambion).
50 mL sterile conical polypropylene tubes.
Sterile single-usage plastic pipettes.
RNase inhibitor (e.g., Promega RNAsin).
DEAE sepharose fast flow (GE healthcare).
TMG-500 buffer: 10 mM Tris–HCl, pH 8.0, 1.1 mM MgCl2, 500 mM Na-Acetate, 0.1 mM EDTA, 0.1% Triton X-100, 0.2% Igepal CA-630, 10% glycerol, 0.1 mM phenyl-methanesulphonylfluoride (PMSF).
TMG-900 buffer: same as above, but containing 900 mM Na-Acetate.
TMG-30 buffer: same as above, but containing 30 mM Na-Acetate.
PD-10 desalting column (GE healthcare).
Centrifugal filter concentrator (e.g., 5 mL centricon YM-30, Millipore).
Glycerol (Sigma).
2.4. Characterization of Pif1p Activity Using Radiolabeled Oligonucleotide Substrates
2.4.1. Preparation of Oligonucleotide Substrates
T4 polynucleotide kinase (New England Biolabs).
[γ-32P]-ATP (>5000 Ci/mmol).
Annealing buffer (5 ×): 10 mM Tris–HCl, pH 7.5, 10 mM MgCl2.
Ficoll loading buffer (6 ×): 17% Ficoll, 0.05% bromophenol blue, 0.05% xylene cyanol.
Polyacrylamide gel electrophoresis (PAGE)-purified oligonucleotides.
Microspin G-25 columns (GE healthcare).
12% polyacrylamide gel (20:1 acrylamide:bis-acrylamide ratio): To prepare 100 mL of gel, mix 28.5 mL 19:1 acrylamide:bis-acrylamide 40% solution (Bio-Rad), 1.45 mL acrylamide 40% solution (Bio-Rad), 10 mL TBE 10 ×, 60 mL ddH2O. Filter and degas. Per mini-gel, mix 10 mL of gel mix with 100 μL of APS 10% and 10 μL of N,N,N′,N′-tetramethylethylenediamine (TEMED). Poor the solution to make a 1.5-mm thick 10 × 8 cm mini-gel.
Microcon Ultrafree-MC centrifugal filters (0.22 μm, Millipore).
D-Tube midi Dialyzer tubes (Novagen).
Microcon YM-10 centrifugal filter unit (Millipore).
Horizontal agarose gel slab unit and power supply.
Sterile razor blades.
Kodak autoradiography film and cassette.
Scintillation cocktail and 20 mL scintillation vials.
2.4.2. Pif1p Activity Assay
Helicase reaction buffer (5 ×): 100 mM Tris, pH 7.5, 200 mM NaCl, 500 μg/mL BSA, 10 mM dithiotreitol (DTT).
Helicase stop/load buffer: Ficoll loading buffer (see Section 2.4.1) supplemented with 50 mM EDTA and 1 μM single stranded DNA oligonucleotide (see Note 3).
12% polyacrylamide gel (20:1 acrylamide:bis-acrylamide ratio): Per gel, mix 40 mL of gel mix with 0.2 mL of APS 10% and 40 μL of TEMED. Poor the solution to make a 1.5-mm thick 18 × 16 cm gel.
Vertical slab gel electrophoresis unit and power supply.
Kodak autoradiography film (BioMax MR) and film cassette.
Radiolabeled oligonucleotide helicase substrate (see Section 2.4.1).
3 MM and DE81 chromatography papers (Whatman).
2.5. Functional Interaction Assay Between Pif1p and Yeast Telomerase
2.5.1. Telomerase Assay
Telomerase Reaction (TR) buffer (10 ×): 200 mM Tris, pH 8.0, 200 mM NaCl, 10 mM DTT, 10 mM spermidine.
50 mM MgCl2.
dNTP mix: 0.5 mM of each dATP, dGTP, and dCTP, 50 μM dTTP, 40 mM ATP.
[α-32P]-TTP (>5000 Ci/mmol).
TR stop buffer: 20 mM Tris, pH 8.0, 1 mM EDTA, 0.5% SDS, 250 mg/mL PCR-grade proteinase K, 50 pM [γ-32P]-labeled control oligonucleotide (see Note 4).
Glycogen.
(NH4)-Acetate 4 M. Filter sterilize.
Formamide loading buffer: 10 mM NaOH, 95% formamide, 0.05% bromphenol blue, 0.05% xylene cyanole.
2.5.2. Electrophoresis and Detection of Reaction Products
16% urea-polyacrylamide sequencing gel. To prepare 1 L of gel mix, add successively in a beaker 100 mL TBE 10 ×, 400 mL 19:1 acrylamide:bis-acrylamide 40% solution (Bio-Rad) and 420 g urea. Mix by stirring on medium heat until urea is dissolved. Let cool to room temperature, poor into a 1 L graduated cylinder, and complete to 1 L with ddH2O. Mix, filter through a paper filter and degas. To prepare one gel, mix 100 mL of 16% gel mix, 0.5 mL APS 10%, and 100 μL TEMED.
Nucleic acid sequencing unit (Sigma IBI model STS-45 is recommended) and power supply.
2.5.3. Telomerase Displacement Assay
All buffers and material should be RNase-free (see Note 2).
Dynabeads M-280 streptavidin magnetic beads (Invitrogen).
PAGE-purified biotinylated telomeric oligonucleotide.
Magnet.
Buffer S1: 0.1 M NaOH, 0.05 M NaCl.
0.1 M NaCl.
Buffer B/W: 10 mM Tris–HCl (pH 7.5), 1 mM EDTA, 2 M NaCl.
TR buffer: see Section 2.5.1.
3. Methods
3.1. Overexpression of Recombinant Pif1p in Bacteria
These instructions assume the use of a bacterial strain expressing the Pif1p nuclear isoform (amino acids 40–859) fused to a 6-histidine tag. As an example, we use the BL21(DE3) derivative strain Rosetta (Novagen) expressing the PIF1 ORF (minus the first 117 base pairs) cloned into the pET28(b) vector (Novagen), in order to express, upon IPTG induction, Pif1p fused at its N-terminus to a 6-histidine tag (see Note 5). Fresh colonies are grown overnight on a LB plate containing 30 μg/mL kanamycin and 34 μg/mL chloramphenicol. Inoculate a colony in 50 mL LB supplemented with kanamycin and chloramphenicol and grow at 37°C overnight with agitation (150 rpm). Inoculate 5 mL of the overnight culture in 1 L of LB and grow cells at 37°C with agitation until OD600 reaches 0.6–0.8 (see Note 6). Cool the bacterial culture for 30 min by placing it in an ice bucket. After the culture has cooled down, add IPTG at 1 mM final concentration and incubate the culture at 18–23°C with agitation for another 15 h (see Note 7). Pellet the bacteria by centrifugation at 4000 ×g and freeze the pellet at −80°C, or keep on ice and proceed to purification (see Section 3.2).
3.2. Purification of Recombinant Pif1p
3.2.1. Affinity Chromatography
All steps should be performed at 4°C.
Resuspend cell pellet in 1/20th culture volume of cold buffer A containing protease inhibitors (one Complete EDTA-free protease inhibitor tablet per 30 mL buffer A). Cells are broken by a single passage in a French press at 10,000 PSI. The lysate is subsequently cleared by centrifugation for 30 min at 16,000 ×g at 4°C. At this stage, the supernatant is still viscous due to presence of bacterial nucleic acids. The supernatant is transferred to a beaker and sonicated on ice using a thin probe at the following settings: 40% amplitude, pulse 40% (see Note 8). Continue until the lysate is no longer viscous. Filter the supernatant through a 45-μM syringe filter to remove remaining aggregates.
Load the supernatant on a column containing 5 mL packed Talon resin equilibrated in buffer A using a peristaltic pump (see Note 9). Load at a constant flow rate of about 3 mL/min or slower. Faster flow rates tend to decrease binding of recombinant Pif1p to the resin. After loading the supernatant, wash the column successively with 3 volumes of buffer A supplemented with 30 mM imidazole and 3 volumes of buffer A. Recombinant Pif1p is eluted from the Talon column with 3 column volumes buffer A supplemented with 200 mM imidazole (see Note 10). Collect fractions from flow through, washes and elution steps in low protein-binding tubes (e.g., MiniSorp tubes) for analysis. Most of recombinant Pif1 should elute between one and two column volumes.
The elution profile of Pif1p should be monitored by analyzing aliquots of the colleted fractions by SDS-polyacrylamide gel electrophoresis followed by Western blotting using an anti His-tag antibody and/or Coomassie staining (An example is shown in Fig. 25.1).
Pool fractions containing Pif1p and proceed to cation exchange chromatography.
Fig. 25.1.
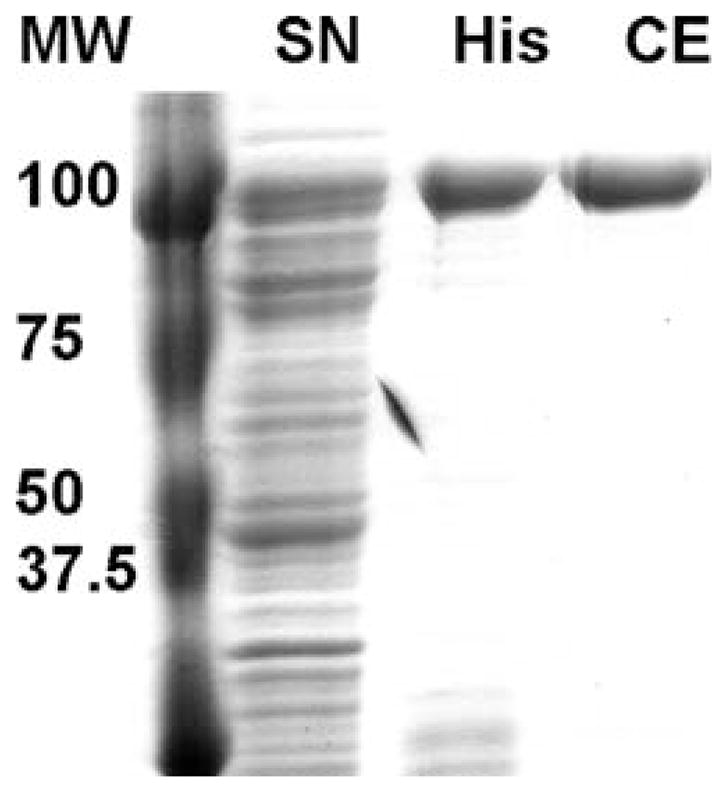
Pif1p purification. Coomassie staining of an 8% SDS-PAGE gel showing the supernatant after IPTG induction (SN), the protein pool after elution from the Talon resin (His), or the protein pool after cation exchange (CE) chromatography.
3.2.2. Cation Exchange Chromatography
The pooled fractions from the affinity purification are poured into dialysis tubing (10 kD cut off) and dialyzed against 1 L buffer A2 at 4°C overnight. Provide gentle agitation using a stirring bar.
Remove sample from dialysis tubing. The sample usually contains some precipitate. Pellet precipitate briefly at 4°C at 10,000 ×g and filter the supernatant through a 0.2-μm filter fitted on a 10-mL syringe. The volume of the sample can be reduced if desired using centrifugal concentrators.
The following steps assume the use of a FPLC system with gradient elution capabilities, to which is connected a 2-mL cation exchange column. The column is first rinsed with water and then equilibrated in buffer A2 until the OD280 and the conductivity signals are stable. The sample injection loop should also be rinsed with buffer A2 during this step. Load the sample on the column at a rate of 1–1.5 mL/min and then wash the column with 5 volumes of buffer A2. Start collecting 2-mL fractions and elute proteins from the column with a 0–100% gradient in buffer B2 (the gradient should be 10 column volumes in length). Using these conditions, Pif1p will elute around 300 mM NaCl. Check elution fractions by analyzing 20-μL aliquots by SDS-PAGE followed by Coomassie staining. Pool fractions containing pure Pif1p based on visual estimation of the stained gel.
Concentrate pooled fractions using a centrifugal concentrator with a cut of 10 kD to a volume of around 500 μL. Add 4 mL of cold buffer C and concentrate the sample again. Repeat this step three times and concentrate the sample to less than 500 μL. Transfer the concentrated protein to a clean eppendorf tube and place on ice. Add 0.8 volume of glycerol, mix well, and distribute in aliquots of 10–50 μL (or desired volume). The aliquots are then flash frozen in liquid N2 and stored at −80°C (see Note 11).
3.3. Preparation of Telomerase Activity from Yeast Protein Extracts
3.3.1. Overexpression of Telomerase Core Subunits
This protocol assumes the use of a yeast strain containing a 2-μm plasmid allowing the expression of EST2 and TLC1 ORFs under the control of GAL promoters. For example, we use a pESC plasmid (stratagene) containing the EST2 and TLC1 genes placed under the control of he GAL1 and GAL10 promoters, respectively. The yeast strain used for expression was the protease deficient strain BCY123 est1Δ type II survivor strain (described in (5)).
Starting from a fresh colony on plate, grow a 50-mL culture in SCGL media lacking appropriate amino acids to saturation. Use this culture to inoculate 1 L of SCGL media (minus appropriate amino-acids) and grow at 30°C with agitation to an OD600 of ~1. Add 1 L of YPGLA media and incubate at 30°C with agitation for another 3 h. Add 20 g galactose (2% final concentration) to induce expression of EST2 and TLC1 and incubate for another 16 h at 30°C.
Pellet cells by centrifugation at 3000 ×g. Resuspend the pellet in 1 volume L buffer and pour slowly in liquid N2. Keep frozen noodles at −80°C or proceed directly to telomerase fractionation.
3.3.2. Telomerase Activity Fractionation and Storage
Since telomerase is a ribonucleoprotein, all steps of the purification should be performed at 4°C and in an RNase-free environment (see Note 2). With these considerations in mind, telomerase activity can be fractionated from yeast cells using the following method, adapted from (8).
This protocol assumes the use of a RM100 automated mortar and pestle. Breakage of the cells is performed while cells are still frozen with liquid N2 (see Note 12). Pre-cool the mortar and pestle with liquid N2. To avoid hazardous splashes, pour only little amount of liquid N2 at a time. The mortar should be cold during the entire procedure. Add the frozen noodles obtained as described in Section 3.3 to the mortar and grind them at the finest setting of the RM100 for approximately 15 min. The cell lysate will appear as a fine white powder. Add a small amount of liquid N2 every 2–3 min to ensure that the cells stay frozen. After 15 min, check a small amount of the frozen powder under a microscope to estimate roughly lysis efficiency. Most of the cells should appear as dark debris (a lysis efficiency of about 70% or above can be easily achieved using this technique). Continue grinding if necessary.
Add the frozen powder to a clean 50 mL polypropylene tube and add 120 units RNase inhibitor (e.g., RNAsin, Promega) and one protease inhibitor tablet. Thaw the lysate at 4°C (see Note 13). Pour the lysate in DEPC-treated corex tubes and centrifuge at 4°C at 10,000 ×g for 30 min. Transfer supernatant in a fresh tube and place on ice.
Incubate the supernatant with DEAE sepharose pre-equilibrated in TMG-500 buffer at 4°C for 30 min on a rotating wheel. Use 1 mL bed volume for 10 mL supernatant.
Pellet resin by centrifugation at 800 ×g for 1 min. Discard supernatant and replace with the same volume of TMG-500 buffer. Resuspend by gentle pipetting and gently rotate 10 min at 4°C. Repeat three times.
Telomerase is eluted by incubation with the high salt TMG-900 buffer. After the last wash, add 1 mL TMG-900 per mL of resin and incubate 15 min on the rotating wheel at 4°C. Pellet resin by centrifugation at 3000 ×g for 10 min and save the supernatant containing telomerase at 4°C.
The elution fraction is then desalted by passage through a PD-10 column equilibrated in TMG-30 buffer according to the manufacturer’s instructions. Concentrate the eluate using a centrifugal concentrator (10 kD cut off) to a volume of approximately 100 μL or less. Add an equal volume of glycerol. Mix well by pipetting, distribute in aliquots of 10 μL RNase-free microfuge tubes and freeze in liquid N2. Store at −80°C.
3.4. Characterization of Pif1p Activity Using Radiolabeled Oligonucleotide Substrates
3.4.1. Preparation of Oligonucleotide Substrates
Detailed methodological reviews describing radiometric assays and their application to helicase mechanistic studies exist (9–11). A favorite general reference that details synthesis of radiolabeled nucleic acids substrates and gel-based analysis of reaction products can be found in this book series (11). However, since optimal assay conditions vary among different helicases, we will briefly describe the assay system that we developed to analyze Pif1p helicase enzymatic properties using radiometric assays (5, 7).
Radiolabel the top (helicase-displaced)-strand oligonucleotide by adding the following to a microfuge tube: 1 μL 10 × T4 polynucleotide kinase buffer, 10 pmol of oligonucleotide (5 μL, 2 μM), 3 μL [γ-32P]-ATP, 1 μL T4 polynucleotide kinase. Incubate the mixture for 1 h at 37°C. Inactivate the kinase by heating 5 min at 95°C. Place on ice for 5 min and spin down briefly.
For the annealing reaction, add directly to the previous reaction: 5 μL of complementary oligonucleotide (loading strand), 2 μL 10 × annealing buffer, 3 μL ddH2O. Place on a 95°C heating block for 5 min, then remove the block from the thermostat and allow cooling to room temperature over a period of 2–3 h.
To remove unincorporated [γ-32P]-ATP, load the 20 μL reaction on a prespun MicroSpin G-25 column. Centrifuge at 735 ×g for 1 min and collect the eluate.
Measure carefully the volume of the eluate and calculate the specific activity assuming 95% recovery. Add 6 × Ficoll loading buffer to a final 1 × concentration and load substrate on a 12% non denaturing mini-gel set up at 4°C. Electrophorese at 5 V/cm until the bromophenol blue dye reaches two-thirds of the gel (or appropriate time). Disassemble and wrap gel in saran wrap. Expose briefly to an autoradiography film, develop, and cut out the gel area containing the substrate with a sterile razor blade.
To electroelute the substrate, place the gel piece in a dialysis midi D-Tube, fill with 1 × TBE, close the tube, and place it in an electrophoresis tank containing cold 1 × TBE (most horizontal agarose gel slabs will be convenient for this). Electroelute the substrate for 1 h at 80 V at 4°C.
Invert current for 40 s, remove power supply, and disassemble. Filter the content of the dialysis tube through an Ultra-free-MC centrifugal filter and concentrate the eluate to approximately 30 μL. Measure the activity of the sample, and calculate the concentration of the radiolabeled substrate according to the previously determined specific activity. Prepare a dilution of the substrate at 10 fmol/μL and store at −20°C behind a shield until use.
3.4.2. Pif1p Activity Assay
Reaction mixtures are set up in 10 μL aliquots. Place a microfuge tube on ice and add in the following order: 2 μL of 5 × helicase reaction buffer, 1 μL 50 mM Mg2+, 10 fmol radiolabeled DNA substrate (1 μL, 10 fmol/μL), 4 μL H2O, and 1 μL of Pif1p dilution in Pif1p storage buffer (see Note 14). Place the tube at 35°C (or desired temperature). Start the reaction by adding 1 μL of 40 mM ATP and incubate for 15 min (or desired time) (see Note 15).
Stop the reaction by adding 2 μL of helicase stop buffer.
Load the reaction products on a 13 × 18 cm non-denaturing 12% polyacrylamide gel (20:1 acrylamide:bis-acrylamide ratio). Run the gel by electrophoresis at 150 V for 2 h or appropriate time at 4°C (see Note 16).
Disassemble the gel tray and recover the gel on DEAE paper (see Note 17). It is convenient to double the DEAE paper with a piece of 3 MM paper in order to ease handling the gel. Cover with plastic wrap and expose against an autoradiography film at −80°C.
Detection and quantification are best carried out on a dried gel using phosphorimaging systems, such as the Molecular Dynamics Storm device, and the Image quant software (GE healthcare).
3.5. Functional Interaction Assay Between Pif1p and Yeast Telomerase
3.5.1. Telomerase Assay
Yeast telomerase activity can be monitored with an oligonucleotide extension assay, using an oligonucleotide whose sequence mimicks the end of a yeast telomere. Similarly to results from other labs, we find that telomerase extends efficiently short oligonucleotides (around 15 nucleotides in size) but displays poor polymerization efficiency on longer oligonucleotides (above 30 nucleotides). It is also advantageous that the 3′ end of the telomeric primer has with a unique sequence complementary to TLC1 RNA. We had the best success with the following sequence: TEL15: 5′-TGTGGTGTGTGTGGG-3′, which anneals on TLC1 at the template position 475C (8).
Enzymes and solutions should be kept cold during the assembly of the reaction.
Prepare a dilution of Pif1p in Pif1p storage buffer such that 1 μL will contain the desired amount of enzyme.
Assemble the telomerase reaction by adding to a microfuge tube, in the following order: 1 μL 10 × TR buffer, 1 μL Telomeric primer at 1 μM, 1 μL dNTP mix, 1 μL 40 mM ATP, 2 μL [α-32P]-dTTP, and 0.3 μL RNAsin. Place the tube in a water bath at 30°C for 2 min to equilibrate the reaction in temperature, and then add 1 μL Pif1p dilution and 3 μL telomerase extract. Incubate the tube for 45 min or desired time.
Stop the reaction by adding 200 μL TR stop buffer containing the radiolabeled control oligonucleotide that will serve as a quantitative loading control. Prepare enough TR stop buffer containing the loading control to distribute to all samples from the same stock. Incubate for another 45 min at 30°C.
Extract the reaction twice with phenol/chloroform.
Precipitate the reaction products by adding 1 volume of 4 M (NH4)2-Acetate, 30 μg glycogen, and 2.5 volumes of cold 100% Ethanol. Place the tubes at −80°C for 30 min. Centrifuge at 20,000 ×g for 30 min at 4°C.
Due to the presence of glycogen, a clear white pellet should be visible at the bottom of the tube after centrifugation. Remove supernatant carefully and add 300 μL of 70% ethanol. Centrifuge again, and air-dry the pellet. Add 5 μL of formamide loading buffer and heat denature the samples at 96°C for 5 min. Telomerase reaction products are now ready to be separated on a sequencing gel.
3.5.2. Electrophoresis and Detection of Reaction Products
The telomerase reaction products are best resolved on a polyacrylamide-urea sequencing gel. We had the best results with 16% polyacryamide gels run on a STS45 IBI sequencing gel unit, but the method can be adapted for other equipment.
Before pouring the gel, plates need to be cleaned with a water soluble detergent and rinsed extensively with distilled water. Plates are then rinsed briefly with ethanol 95% and air-dried. Avoid touching the surface of the plates with hands after cleaning. Plates should then be coated with a glass plate coating solution (e.g., gel slick solution, FMC bioproducts) to facilitate removing of the gel. Distribute the coating solution evenly using a paper towel on both plates, air dry and assemble the gel unit. Prepare the 100 mL of sequencing-gel mix and pour promptly into the plates. Insert the comb (see Note 18) and let the gel polymerize for an hour. Assemble the gel unit, and pre-run the gel in 1 × TBE for 1 h at 75 W constant power setting (around 1600 V on the STS45 unit).
Disconnect the power supply and load the samples, leaving the wells on the side of the gel empty if possible. Reconnect the power supply and run the gel until the bromophenol blue dye has reached four-fifths of the gel.
Disassemble the gel unit and delicately remove one plate, leaving the gel stuck to the bottom plate. Apply two sheets of 3 MM paper to the gel and carefully remove the gel from the glass plate by having it stick to the paper, starting from a corner. Cover with plastic wrap and expose at −80°C against an autoradiography film. A 24-h exposure is usually sufficient. An example of the signal observed is shown in Fig. 25.2.
If quantitation is desired, the gel can be exposed against a phosphor screen to be scanned on a phosphorImager. Sixteen percent urea-gel cannot be dried before exposure. Therefore, to expose the gel against a phosphor screen, thaw the gel at room temperature for 10 min and place it in a phosphorImager cassette. Cover with a clean plastic wrap (to prevent the phosphor screen from contact with the wet gel) and expose for 4–5 h. Longer exposure is not recommended, as the gel likely will start deteriorating.
Given the unique alignment between TLC1 and the TEL15 oligonucleotide, the sequence of the extension products is predicted to be 5′-TGTGGTG (8). Using the ImageQuant software, measure the intensity of each addition product as well as the intensity of the loading control. If several lanes are to be compared, the intensity of the signal given by the loading control with serve as a reference for normalization of the signal observed in each lane.
-
Calculate the percentage of each addition product synthesized during the reaction according to the following rule, which takes into account the number of radiolabeled nucleotide for each product size:
The total amount of products being synthetized (in arbitrary units) is given by the following formula: T=I1+I2+I3/2+I4/2+I5/2+I6/3+I7/3 where In is the quantification of band at position +n. Therefore, the percentage of product synthesized of a specific size (+n) is given by the ratio In/T, divided by the number of radiolabeled nucleotide in the product of this size (e.g., %(+5) = I+5/T/2) (see Notes 19 and 20).
Fig. 25.2.
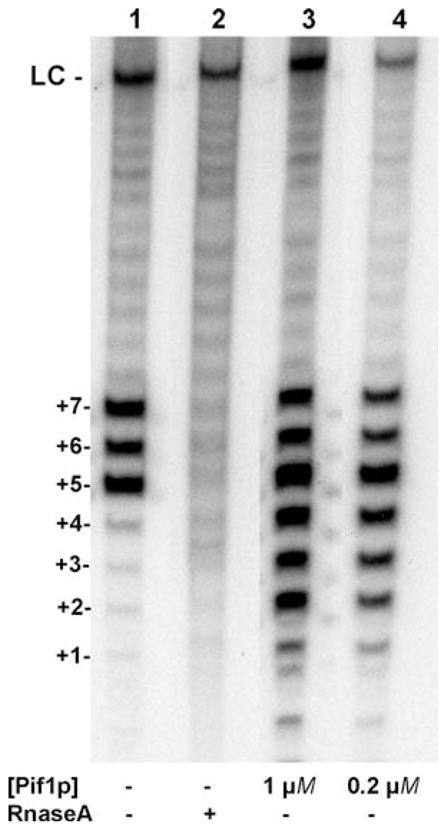
Effects of Pif1p on telomerase activity in vitro. Autoradiograph of a sequencing gel showing telomerase extension products of a TEL15 oligonucleotide in the presence of telomerase alone (lane 1), in presence of RNase A (lane 2), or two concentrations of Pif1p (lane 3, 1 μM Pif1p; lane 4, 0.2 μM Pif1p). LC: loading control.
3.5.3. Telomerase Displacement Assay
Prepare 100 μL (or the desired volume) of M-280 Streptavidin beads by washing them successively in 5 volumes buffer S1, 5 volumes 0.1 M NaCl, and 5 volumes buffer W/B. Resuspend the beads in one volume buffer W/B. To remove buffer between each step, place tube on magnet for 1–2 min to separate beads from buffer and remove buffer by gentle pipetting.
Using a 100 μM stock of 5′-biotinylated TEL15 telomeric oligonucleotide (or the desired oligonucleotide), add the amount of oligonucleotide to the equilibrated beads to give a final oligonucleotide concentration of 1 μM. incubate at room temperature for 15 min with occasional mixing by gently taping the tube. Remove buffer and wash the beads 2–3 times with 1 × buffer W/B and resuspend beads in 1 volume 1 × TR buffer.
Assemble on ice the following reaction in a microfuge tube: 3 μL of beads (coated with the telomeric oligonucleotide), 4 μL of 1 × TR buffer, and 3 μL of telomerase extract. Incubate at room temperature for 10 min. In the meantime, assemble in a separate tube on ice the following helicase mix: 1 μL 10 × TR buffer, 1 μL ATP (40 mM stock), 1 μL Pif1p (at a concentration of 10 μM if possible), 1 μL non-biotinylated TEL15 oligonucleotide (10 μM stock), and 6 μL H2O. Separate beads from reaction mix containing unbound telomerase using a magnet and replace with the Helicase mix. Incubate the reaction at 30 °C for 10 min (or desired time).
Again, separate beads from supernatant and carefully remove supernatant by pipetting. Wash beads by gentle pipetting with 100 μL 1 × TR buffer and resuspend beads in 10 μL 1 × TR buffer. Add to both tubes (beads and reaction supernatant) 4 μL of 3 × Laemmli buffer and boil for 5 min. Centrifuge briefly and load on an 8% SDS-PAGE protein gel.
Following electrophoresis, reveal bound fraction (bead fraction) and unbound fraction (supernatant) by Western blotting using an anti-Est2p antibody (or other appropriate antibody if using, for example, a tagged version of Est2p) (see Note 21).
Acknowledgments
This work was supported by grants from the National Insitutes of Health to VAZ.
Footnotes
Other type of IMAC resins can be adapted to fit this protocol, although binding, wash, and elution buffer components (pH, salt, and imidazole concentration) should be optimized for the specific resin.
To prevent RNase contamination, wear latex gloves at all times and change them regularly. All glassware should be DEPC-treated. Work surfaces should be cleaned with an RNase inhibitor solution, e.g., RNaseZAP (Ambion). All buffers should be DEPC-treated and autoclaved prior to use. Tris-containing buffers can not be DEPC-treated as DEPC will react with primary amines. Therefore, RNase-free Tris and DEPC-treated stock solutions should be used to make these buffers.
The presence of excess unlabeled oligonucleotide prevents reannealing of the unwound strand to its complementary strand.
To prevent confusion between the loading control and the addition products, we use a 60-mer single-stranded oligonucleotide of random sequence as a loading control.
Although the pET system is convenient and has become a widely used standard, other inducible systems for heterologous expression in bacteria can be used.
Given the low level of Pif1p overexpression, we usually induce large volumes (5–10 L). The volume of LB in each flask should be no more than a third of the flask volume. For example, use a 6 L flask for a 2 L culture.
Induction at lower temperatures greatly increases Pif1p solubility. The optimal induction temperature and time will depend on the expression system and should be determined experimentally.
To prevent heating of the supernatant, sonication should be paused every minute for 30 s to cool the probe. One round of 100 pulses is usually enough to sonicate 100 mL of supernatant.
Optimal column volume will depend on the culture volume and on the level of Pif1p expression in the system used. We find that 5-mL columns give reproducible yields and quality in Pif1p purified from 10-L cultures.
As a rule of thumb, 5 column volumes of washing buffer should be enough to remove proteins interacting non-specifically with the Talon resin. If another resin is used for affinity chromatography, optimal volumes for washing the column before elution should be determined experimentally.
We find that the enzyme is stable at −80°C for a year. It is not recommended to freeze/thaw the enzyme as it makes the helicase activity decrease rapidly. If an aliquot is thawed, it can be kept at −20°C for 2–3 months without a significant drop in activity. However, we have observed precipitation at −20°C. Therefore, aliquots kept at −20°C should be checked for precipitation and protein concentration should be recalculated before each use.
Since liquid N2 can cause serious burns, safety glasses and protective gloves should be worn at all times during this procedure.
Frozen lysates can take a long time to thaw (> 30 min for a 20-mL lysate). Thawing can be initiated by warming the lysate between the hands and then on a rotating wheel at 4°C until thawing is complete.
Similarly to what has been reported for other helicases, we find that the Pif1p concentration necessary to observed efficient unwinding exceeds several fold the concentration of the substrate. We routinely perform Pif1p helicase assays using 100 nM enzyme and 1 nM substrate.
For a control reaction, set up a reaction using Pif1p storage buffer instead of the helicase. This is the “no enzyme” control. Another control is the heat denatured substrate, which is achieved by heating the reaction mix containing the labeled substrate and no enzyme at 95°C for 2 min.
The final polyacrylamide percentage of the gel and the optimal electrophoresis time depends on the sizes of the intact nucleic acids substrate and the unwound radiolabeled product. These conditions should be optimized depending on the size of the substrate used.
Use of DEAE paper is recommended when the gel is going to be dried before quantification, since, unlike 3 MM paper, it will bind and retain small nucleic acids.
Use a 5-mm well-dented comb, not a sequencing shark-tooth comb.
To visualize the displacement of telomerase, a variation of this protocol can be performed. During the course of the reaction, an excess of a “chasing” telomeric oligonucleotide of different size is incorporated in the reaction. For example, we use a 30-mer oligonucleotide containing the TEL15 sequence extended by 15 random nucleotides from its 5′ end. This oligonucleotide is utilized more efficiently than a 30-mer oligonucleotide containing only telomeric sequence, as discussed in Section 3.5.1. The reaction is started as described, but a 10-fold excess chasing oligonucleotide is added after 15 min into the reaction. Since yeast telomerase stays associated with its product (12), telomerase will only elongate the chasing oligonucleotide if telomerase is released from its elongation product.
The effect of Pif1p on telomerase activity can be calculated in term of telomerase nucleotide processivity, defined as the probability P with which telomerase adds more than one nucleotide without dissociating from its product. Telomerase nucleotide processivity is then defined for each product of size +n by P+n = (Σ(x > n) Ix)/T. This calculation assumes that in the presence of Pif1p, an already elongated product is not re-elongated by secondary association with telomerase, provided that TEL15 primer is present in large excess compared to telomerase.
Alternatively, quantification of telomerase core enzyme in each fraction can be achieved by detection of the TLC1 RNA, either by qRT-PCR or Northern blotting.
References
- 1.Bessler JB, Torres JZ, Zakian VA. The Pif1p subfamily of helicases: region specific DNA helicases. Trends Cell Biol. 2001;11:60–65. doi: 10.1016/s0962-8924(00)01877-8. [DOI] [PubMed] [Google Scholar]
- 2.Boulé J-B, Zakian VA. Roles of Pif1-like helicases in the maintenance of genomic stability. Nucleic Acids Res. 2006;34:4147–4153. doi: 10.1093/nar/gkl561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schulz VP, Zakian VA. The Saccharomyces PIF1 DNA helicase inhibits telomere elongation and de novo telomere formation. Cell. 1994;76:145–155. doi: 10.1016/0092-8674(94)90179-1. [DOI] [PubMed] [Google Scholar]
- 4.Myung K, Chen C, Kolodner RD. Multiple pathways cooperate in the suppression of genome instability in Saccharomyces cerevisiae. Nature. 2001;411:1073–1076. doi: 10.1038/35082608. [DOI] [PubMed] [Google Scholar]
- 5.Boulé J-B, Vega LR, Zakian VA. The yeast Pif1p helicase removes telomerase from telomeric DNA. Nature. 2005;438:57–61. doi: 10.1038/nature04091. [DOI] [PubMed] [Google Scholar]
- 6.Zhou JQ, Monson EM, Teng SC, Schulz VP, Zakian VA. The Pif1p helicase, a catalytic inhibitor of telomerase lengthening of yeast telomeres. Science. 2000;289:771–774. doi: 10.1126/science.289.5480.771. [DOI] [PubMed] [Google Scholar]
- 7.Boulé J-B, Zakian VA. The yeast Pif1p DNA helicase preferentially unwinds RNA DNA substrates. Nucleic Acids Res. 2007;35:5809–5818. doi: 10.1093/nar/gkm613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Forstemann K, Lingner J. Molecular basis for telomere repeat divergence in budding yeast. Mol Cell Biol. 2001;21:7277–7286. doi: 10.1128/MCB.21.21.7277-7286.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bachrati CZ, Hickson ID. Analysis of the DNA unwinding activity of RecQ family helicases. Methods Enzymol. 2006;409:86–100. doi: 10.1016/S0076-6879(05)09005-1. [DOI] [PubMed] [Google Scholar]
- 10.Brosh RM, Jr, Opresko PL, Bohr VA. Enzymatic mechanism of the WRN helicase/nuclease. Methods Enzymol. 2006;409:52–85. doi: 10.1016/S0076-6879(05)09004-X. [DOI] [PubMed] [Google Scholar]
- 11.Brosh RM, Jr, Sharma S. Biochemical assays for the characterization of DNA helicases. Methods Mol Biol. 2006;314:397–415. doi: 10.1385/1-59259-973-7:397. [DOI] [PubMed] [Google Scholar]
- 12.Prescott J, Blackburn EH. Functionally interacting telomerase RNAs in the yeast telomerase complex. Genes Dev. 1997;11:2790–2800. doi: 10.1101/gad.11.21.2790. [DOI] [PMC free article] [PubMed] [Google Scholar]