

Abstract
Axin is a negative regulator of Wnt/β-catenin signaling via regulating the level of β-catenin, which is a key effector molecule. Therefore, controlling the level of Axin is a critical step for the regulation of Wnt/β-catenin signaling. It has been shown that ubiquitination-mediated proteasomal degradation may play a critical role in the regulation of Axin; however, the E3 ubiquitin ligase(s), which attaches ubiquitin to a target protein in combination with an E2 ubiquitin-conjugating enzyme, for Axin has not yet been identified. Here, we show that Smurf2 is an E3 ubiquitin ligase for Axin. Transient expression of Smurf2 down-regulated the level of Axin and increased the ubiquitination of Axin. Conversely, shRNA specific to Smurf2 blocked Axin ubiquitination. Essential domains of Axin responsible for Smurf2 interaction as well as Smurf2-mediated down-regulation and ubiquitination were identified. In vitro ubiquitination assays followed by analysis using mass spectroscopy revealed that Smurf2 specifically ubiquitinylated Lys505 of Axin and that the Axin(K505R) mutant resisted degradation. Knockdown of endogenous Smurf2 increased the level of endogenous Axin and resulted in reduced β-catenin/Tcf reporter activity. Overall, our data strongly suggest that Smurf2 is a genuine E3 ligase for Axin.
Keywords: Glycogen Synthase Kinase 3, Protein Degradation, Signal Transduction, Ubiquitin Ligase, Wnt Pathway, Axin, Smurf2
Introduction
Axin plays a crucial role in embryonic development and tumorigenesis (1). Duplication of the embryonic axis in Axin−/Axin− mouse embryos and blocked embryonic axis formation by ectopic expression of Axin in Xenopus embryos suggest that Axin is a key regulator of embryonic axis formation via regulation of the Wnt pathway, which has been shown to play a major role in embryogenesis (2). In this pathway, Axin forms a complex with proteins, including adenomatous polyposis coli protein, glycogen synthase kinase 3β (GSK3β),3 β-catenin, protein phosphatase type 2A, and others, to control the level of β-catenin. In the absence of Wnt signaling, Axin facilitates phosphorylation of β-catenin by GSK3β, after which phosphorylated β-catenin is recognized by β-transducin repeat-containing protein, leading to ubiquitination and proteasomal degradation. Cellular stimulation with Wnt, such as Wnt3a, disintegrates the Axin complex and stabilizes β-catenin. The accumulated β-catenin then migrates into nuclei, where it interacts with T cell factor (Tcf)/lymphoid enhancer factor-1 and controls the expression of target genes involved in the regulation of diverse biological processes (3). In addition to Wnt signaling, Axin has been shown to participate in a variety of other critical signaling cascades, including those for p53 (4), TGF-β (5, 6), and MAPK JNK (7).
The level of Axin is very low compared with other Wnt signaling components in Xenopus cell extracts (8). Therefore, it was suggested that controlling the level of Axin is a critical step in the regulation of Wnt/β-catenin signaling (9). Loss of Axin leads to increased levels of β-catenin, as shown in many cancer cell lines (10), whereas too much Axin expression enhances the in vivo level of β-catenin (8). This suggests that tight regulation of Axin expression is essential. Ubiquitination-mediated proteasomal degradation may play a critical role in the regulation of Axin (11); however, E3 ligase(s) for the degradation of Axin has not been identified.
Ubiquitination occurs by sequential reactions including at least three types of enzymes. The C-terminal glycine of ubiquitin forms a thiol ester bond with an E1 ubiquitin-activating enzyme in an ATP-dependent manner. This ubiquitin is then transferred to E2 ubiquitin-conjugating enzymes. Finally, the transfer of E2-conjugated ubiquitin to the substrate is catalyzed by E3 ubiquitin ligase, which determines the substrate specificity. Two types of E3 ligases, the HECT (homologous to the E6-AP carboxyl terminus) domain and RING finger, have been identified. HECT domain E3 enzymes form a thiol ester intermediate with ubiquitin, followed by the transfer of ubiquitin to the substrate, whereas RING finger E3 enzymes catalyze the direct transfer of ubiquitin from E2 to the substrate (12). Smurf2 (Smad ubiquitination regulatory factor 2) is a HECT domain E3 ligase involved in the ubiquitination of several proteins, including Smad proteins, which are involved in TGF-β signaling (13).
Smurf2 can induce ubiquitination/proteasomal degradation of target proteins via interactions with other proteins. For example, Smurf2 induces the degradation of the TGF receptor or β-catenin by interacting with Smad7 (14, 15). In the absence of TGF-β signaling, Smad3 strongly interacts with Axin (5, 6). Interestingly, Smurf2 interacts with Smad3 but does not induce the degradation of Smad3 (13). These results prompted us to test whether or not Smurf2 acts as an E3 ubiquitin ligase for Axin, and we present data in strong support of such a possibility.
EXPERIMENTAL PROCEDURES
Plasmids and shRNAs
The coding region of human Smurf2 was inserted into the pCS2-HA3 vector (constructed in our laboratory) and the pFLAG-CMV4 vector using a general PCR method. Smurf2(C716G) (ligase-dead mutant) and WW1 domain-deleted constructs were generated by PCR. For bacterial expression of GST-fused Smurf2 proteins, Smurf2 and Smurf2(C716G) coding sequences were subcloned into a pGEX4T-3 vector. Full-length cDNAs encoding human UbcH2, UbcH5A, UbcH5C, UbcH6, UbcH7, UbcH9, and UbcH10 were obtained by PCR and inserted into a pGEX vector (GE Healthcare). pCS2-HA-ubiquitin was generated by subcloning HA-ubiquitin cDNA into a pCS2-HA3 vector. pCS-FLAG-ubiquitin was provided by Dr. Ro (National Institutes of Health). HA-Nedd4 cDNA was a kind gift from Dr. McDonald (Victoria University, Wellington, New Zealand). Cloning of pCS2-Myc-Axin, pBI-Myc-Axin, and many deletion constructs of Axin was described previously (16, 17). Additional deletion constructs were generated by PCR and a general molecular cloning technique. For bacterial expression of His tag-fused Axin protein, Myc-Axin-(1–686) coding sequence was subcloned into a pET32b vector. shRNA targeting sequences against both human and mouse Smurf2 were designed using the web tools from Dharmacon and Promega. Sense (5′-GATCCCCGGAAGAATCCAGTATCTAATTCAAGAGATTAGATACTGGATTCTTCCTTTTTGGAAA-3′) and antisense (5′-AGCTTTTCCAAAAAGGAAGAATCCAGTATCTAATCTCTTGAATTAGATACTGGATTCTTCCGGG-3′) oligonucleotides were annealed and then ligated into the BglΙΙ and HindΙΙΙ sites of the pSUPER.retro.puro vector (Oligoengine).
Cell Culture, Transfection, and Stable Cell Lines
HEK293T, HEK293, HeLa, L929, Caco-2, SNU475, SNU449, and DLD1 cells were cultured in DMEM supplemented with 10% FBS (Invitrogen) and 1× antibiotic/antimycotic (Invitrogen) and maintained at 37 °C in a humidified 5% CO2 incubator. Transient transfection in HEK293T, HEK293, and Caco-2 cells was performed using the calcium phosphate method. For preparation of shRNA stable cell lines, HEK293T cells were transfected with pSUPER.retro.puro-shGFP or pSUPER.retro.puro-shSmurf2 using the calcium phosphate method. HeLa, DLD1, and SNU449 stable cell lines expressing GFP or Smurf2 shRNA were generated via retroviral infection. Retroviruses were produced in GP2-293 cell lines (Clontech) by transfection with pSUPER.retro.puro-shGFP or pSUPER.retro.puro-shSmurf2. The medium containing retroviruses was collected, filtered, concentrated, and mixed with 8 μg/ml Polybrene (Sigma) before infection of cells. Stable cell populations were selected with 2 μg/ml puromycin (Sigma).
Antibodies
Rabbit anti-Axin antibody was kindly provided by Dr. Virshup (University of Utah). Anti-HA (F-7), anti-α-tubulin (TU-02), anti-Smurf2 (H-50), and anti-ubiquitin (P4D1) antibodies were purchased from Santa Cruz Biotechnology. Anti-β-catenin antibody was purchased from BD Bioscience. Anti-FLAG and anti-β-actin antibodies were purchased from Sigma. Anti-Myc antibody (9E10) was obtained from hybridoma cell lines (Developmental Studies Hybridoma Bank, University of Iowa).
Immunoprecipitation and Western Blotting
Cells were lysed in lysis buffer (20 mm Tris-HCl (pH 7.5), 150 mm NaCl, 0.5% Triton X-100, 50 mm NaF, 2 mm EDTA, 100 μm sodium orthovanadate, 1 mm PMSF, 5 μg/ml leupeptin, and 1 μm pepstatin A). The lysates were centrifuged at 13,000 rpm for 15 min at 4 °C, and the supernatant was collected and used for immunoprecipitation and Western blotting. For immunoprecipitation, 200∼800 μg of cell lysate was incubated with an appropriate antibody in lysis buffer (RIPA buffer (50 mm Tris-Cl (pH 7.5), 150 mm NaCl, 0.5% sodium deoxycholate, 1% Nonidet P-40, and 0.1% SDS) was used for in vivo ubiquitination assay) for >2 h at 4 °C with rotation. 20 μl of protein A/G Plus-agarose (Santa Cruz Biotechnology) was added and continuously incubated for >1 h. Immunoprecipitates were washed three times with lysis buffer or RIPA buffer, resolved by SDS-PAGE, and analyzed by Western blotting.
Preparation of Recombinant Proteins
pGEX-Smurf2, pGEX-Smurf2(C716G), and pGEX-UbcH were transformed in Escherichia coli BL21(DE3). For the induction of GST fusion proteins, 0.5 mm isopropyl β-d-thiogalactopyranoside (Calbiochem) was added in yeast extract/Tryptone medium. Purification of GST fusion proteins was performed according to standard procedures (GE Healthcare). To remove GST, GST fusion proteins were incubated overnight with biotinylated thrombin (Novagen) at 4 °C. The biotinylated thrombin was then removed by streptavidin-agarose (Novagen). S·His-tagged Axin proteins were expressed in E. coli and purified according to standard methods (Novagen).
In Vitro Ubiquitination Assay
The reaction was carried out for 2 h at 37 °C in 25 μl of reaction buffer (50 mm Tris-HCl (pH 7.5), 10 mm MgCl2, and 50 μm dithiothreitol supplemented with 5 mm ATP) containing 10 μg of ubiquitin (Boston Biochem), 200 ng of E1 (Boston Biochem), and purified UbcH5C, Smurf2, and S·His-Axin-(1–686) proteins. The reaction samples were incubated with 30 μl of S-protein-agarose (Novagen) in RIPA buffer and washed three times with RIPA buffer. The pulled down samples were resolved by SDS-PAGE, and polyubiquitinated Axin was detected by Western blotting with anti-ubiquitin antibody P4D1. [35S]Met-labeled Myc-Axin-(1–832), that was translated in vitro using the TnT system (Promega), was incubated with ubiquitin, E1, UbcH5A, and Smurf2 in reaction buffer. The samples were resolved by SDS-PAGE and analyzed by autoradiography.
Mass Spectrometry
The regions for analysis were excised from SDS-polyacrylamide gels. The proteins were reduced, alkylated, and then digested overnight with a sequencing grade modified trypsin (12.5 ng/μl; Promega) at 37 °C, followed by endoproteinase Glu-C (Roche Applied Science) digestion. The digested peptides were extracted three times with 5% formic acid in 50% acetonitrile solution at room temperature for 20 min and desalted using ZipTipC18 (Millipore) before MS analysis. The proteolytic peptides were loaded onto a fused silica microcapillary column (12 cm × 75 μm) packed with the C18 reversed-phase resin (5 μm, 200 Å). LC separation was conducted under a linear gradient as follows: a 3–40% solvent B (0.1% formic acid in 100% acetonitrile) gradient at a flow rate of 250 nl/min for 60 min. The column was directly connected to an LTQ linear ion trap mass spectrometer (Thermo Finnigan) equipped with a nanoelectrospray ion source. The electrospray voltage was set at 1.95 kV, and the threshold for switching from MS to MS/MS was 500. The normalized collision energy for MS/MS was 35% of the main radio frequency amplitude, and the duration of activation was 30 ms. All spectra were acquired in data-dependent scan mode. Each full MS scan was followed by five MS/MS scans corresponding to the most intense to the fifth intense peaks of the full MS scan. The repeat count of the peak for dynamic exclusion was 1, and its repeat duration was 30 s. The dynamic exclusion duration was set for 180 s, and the width of the exclusion mass was ±1.5 Da. The list size of dynamic exclusion was 50. The collected MS/MS spectra were searched using the SEQUEST program (Thermo Finnigan) with the selected criteria of oxidation at Met (+16 Da), carboxyamidomethylation at Cys (+57 Da), and ubiquitination at Lys (+114 Da) as variable modifications.
Luciferase Reporter Assay
Cells were cotransfected with SuperTOPFlash (kindly provided by Dr. Moon, University of Washington), pRL-TK (Promega), and the plasmids indicated in the figures. The Dual-Luciferase assay was performed according to standard procedures using a GloMAX 20/20 luminometer (Promega).
RESULTS
Interaction of Axin with Smurf2
Axin is polyubiquitinated at steady state, but its level is further decreased in the presence of Wnt signaling via proteasomal degradation (11, 18). To examine whether or not Smurf2 is an E3 ubiquitin ligase for Axin, we tested the interaction between Axin and Smurf2. HEK293T cells were transfected with Myc-tagged Axin and HA-tagged Smurf2(C716G), which is a ligase-dead mutant of Smurf2 in which the cysteine responsible for the conjugation of ubiquitin is changed to glycine (13). Because we found that ectopic expression of Smurf2, but not Smurf2(C716G) (indicated as CG in the figures), caused down-regulation of Axin (see Fig. 2A), Smurf2(C716G) was used to examine the interaction of Axin with Smurf2. Immunoprecipitation analysis showed that both exogenously expressed and endogenous Axin interacted with Smurf2 (Fig. 1, A and B). To determine which Axin domain is necessary for the interaction, deletion constructs of Axin and HA-tagged Smurf2(C716G) were transfected into HEK293T cells, and co-immunoprecipitation experiments were performed (Fig. 1, C and D). Deletion of several single domains resulted in reduced interaction between Axin and Smurf2. However, none of them completely blocked the interaction (for example, AxinΔ209–407 and AxinΔ508–711, etc.) (Fig. 1D). These results suggest that multiple domains containing amino acids 228–354, 508–711, and 745–826 of Axin are responsible for the interaction with Smurf2.
FIGURE 2.

Axin is down-regulated by ectopic expression of Smurf2, but not Smurf2(C716G), through enhanced polyubiquitination. A, Myc-Axin, HA-ubiquitin, and Myc-EGFP were cotransfected with increasing amounts of FLAG-Smurf2 or FLAG-Smurf2(C716G) into HEK293T cells. Myc-EGFP was used to show that the degradation of Axin induced by Smurf2 was specific and that the cells were transfected with an equal amount of plasmids. B, left panel, HEK293 cell lysates were subjected to immunoprecipitation (IP) with anti-Myc antibody, followed by immunoblotting (IB) with anti-HA antibody. Polyubiquitinated species of Axin are indicated as Myc-Axin-(HA-Ub)n. The arrow indicates the location of unubiquitinated Axin. Whole cell lysates (WCL) were immunoblotted to show the proper expression of transfected plasmids. Right panel, the immunoprecipitated products with anti-Myc antibody in the left panel were boiled in 1% SDS to elute Myc-Axin from the beads and to break any protein-protein interactions. The supernatant was then collected and diluted 10 times in RIPA buffer, followed by immunoprecipitation with anti-Myc antibody and immunoblotting to detect Axin ubiquitination. C, Myc-Axin and FLAG-ubiquitin were cotransfected with empty vector, HA-Smurf2, or HA-Nedd4 into HEK293T cells. The cells were then treated with 10 μm MG132 for 5 h before harvesting, and cell lysates were subjected to immunoprecipitation, followed by immunoblotting. D and E, the Axin construct that did not interact with Smurf2 was neither down-regulated nor ubiquitinylated by Smurf2. Myc-Axin or Axin(Δ228–354 and Δ508–711) with HA-ubiquitin was cotransfected with or without FLAG-Smurf2 into HEK293T (D) or HEK293 (E) cells. Western blotting using whole cell lysates with the indicated antibodies (D) and immunoprecipitation with anti-Myc antibody followed by Western blotting (E) were performed. FL, full-length.
FIGURE 1.

Axin interacts with Smurf2. A, HEK293T cells were transfected with plasmids for Myc-tagged Axin and HA-tagged Smurf2(C716G) (inactive mutant of Smurf2) either alone or together. Cell lysates were then subjected to immunoprecipitation (IP), followed by immunoblotting (IB) with the antibodies indicated. Whole cell lysates (WCL) were immunoblotted with anti-HA antibody. B, HEK293T cells were transfected with a plasmid for FLAG-Smurf2(C716G), and the cell lysates were subjected to immunoprecipitation with rabbit anti-Axin antibody or rabbit serum, followed by immunoblotting with anti-FLAG and anti-Axin antibodies. The asterisk and arrow indicate nonspecific and FLAG-Smurf2(C716G) (Flag-CG) bands, respectively. DIX, dishevelled homologous domain. C, shown is a schematic diagram of deletion constructs of Axin. a.a., amino acids; APC, adenomatous polyposis coli; β-cat, β-catenin; PP2Ac, protein phosphatase type 2A catalytic subunit. D, the plasmids indicated were transfected into HEK293T cells, followed by immunoprecipitation and immunoblotting experiments. Amino acids 228–354, 508–711, and 745–826 of Axin were responsible for the interaction with Smurf2. FL, full-length.
Down-regulation of Axin by Smurf2 via Polyubiquitination
The ectopic expression of Smurf2, but not the Smurf2(C716G) mutant, reduced the level of cotransfected Axin (Fig. 2A). However, the level of EGFP, which was cotransfected as a control for the equal transfection of all constructs, remained unchanged. These results suggest that Smurf2 specifically down-regulated the level of Axin (Fig. 2A). We then tested whether or not the ectopic expression of Smurf2 could enhance polyubiquitination of Axin. Cotransfection of Smurf2, but not Smurf2(C716G), enhanced Axin ubiquitination (Fig. 2B, left panel). To exclude the possibility that the polyubiquitination signal from the above experiment originated from Axin-associated proteins, double immunoprecipitation experiments were performed as shown in Fig. 2B. The polyubiquitination signal was detected only in the sample containing Axin and Smurf2 (Fig. 2B, right panel), which strongly suggests that Smurf2 ubiquitinylates Axin. Nedd4 is a HECT-type E3 ligase with very similar domains to Smurf2 (19). However, cotransfection of Nedd4 did not enhance polyubiquitination of Axin (Fig. 2C), suggesting that Smurf2 specifically ubiquitinylates Axin. The Axin construct deleted of amino acids 228–354 and 508–711 did not interact with Smurf2 (Fig. 1D). Thus, we tested whether or not the interaction between Axin and Smurf2 is necessary for the degradation and polyubiquitination of Axin. When AxinΔ228–354 and AxinΔ508–711 was transfected with Smurf2, down-regulation and polyubiquitination of Axin were severely blocked (Fig. 2, D and E). These findings indicate that the interaction between Axin and Smurf2 is necessary for the Smurf2-mediated ubiquitination and down-regulation of Axin.
In Vitro Ubiquitination of Axin by Smurf2
To determine whether Smurf2 directly ubiquitinylates Axin, in vitro ubiquitination assays were performed. UbcH5A and UbcH5C were used in subsequent experiments because the in vitro ubiquitination assay using various E2 enzymes showed that UbcH5A, UbcH5C, and UbcH7 were all capable of auto-ubiquitination of Smurf2 (supplemental Fig. 1) (20). Because bacterially produced E2 enzymes and Smurf2 did not exhibit full activity upon fusing with GST, thrombin was used to remove the GST tag. However, an in vitro ubiquitination assay using these proteins caused nonspecific degradation of Axin. To overcome this problem, biotinylated thrombin was used, and additional purification with streptavidin was performed, which prevented the nonspecific degradation of Axin that occurred during the in vitro ubiquitination assays. Therefore, it seems that Axin is a very labile protein and that residual thrombin might cause nonspecific degradation of Axin. Using the purified components shown in Fig. 3A, an in vitro ubiquitination assay was performed. The incubation of Smurf2, but not the Smurf2(C716G) mutant, with ubiquitin, E1, and UbcH5C caused ubiquitination of Axin (Fig. 3A, lanes 7 and 8). The reaction without Axin also showed a weak ubiquitination signal (Fig. 3A, lane 2). This signal seems to have been due to the auto-ubiquitination of Smurf2, which was nonspecifically pulled down by S-protein-agarose (supplemental Fig. 2).
FIGURE 3.

In vitro ubiquitination of Axin by Smurf2. A, S·His-Axin or S·His (as a control; lane 2) was incubated at 37 °C for 2 h with or without purified ubiquitin, E1, E2 (UbcH5C), and E3 (Smurf2 or Smurf2(C716G)) as indicated. The reaction samples were subjected to pull-down analysis using S-protein-agarose, and polyubiquitinated Axin was detected by immunoblotting with anti-ubiquitin antibody. WB, Western blot. B, [35S]Met-labeled Axin was incubated with the components indicated. Polyubiquitinated [35S]Met-labeled Axin was detected by autoradiography. C, S·His-Axin-(1–686) was incubated in the presence or absence of a purified mutant form of ubiquitin whose seven lysine residues were changed to arginine, E1, E2 (UbcH5C), and Smurf2 for 2 h at 37 °C. The reaction samples were pulled down using S-protein-agarose, resolved by SDS-PAGE, and stained with Coomassie Brilliant Blue. The areas marked with rectangles were excised, and MS/MS spectrometric analysis was performed as described under “Experimental Procedures.” D and E, shown are MS/MS spectra for Axin and mono-ubiquitinated Axin, respectively. F, Myc-Axin or Myc-Axin(K505R) with HA-ubiquitin and Myc-EGFP were cotransfected with or without FLAG-Smurf2 into HEK293T cells, followed by immunoblotting.
Because Axin interacted with Smurf2 (Fig. 1), we suspected that the stronger ubiquitination signal shown in Fig. 3A (lane 7) resulted from the auto-ubiquitination of Smurf2 because Smurf2 could be pulled down more in the presence of S·His-Axin. To circumvent this, an in vitro ubiquitination assay using Axin translated in vitro with [35S]methionine was performed (Fig. 3B). Mixtures of rabbit reticulocyte lysates containing [35S]methionine-labeled Axin were incubated with the purified proteins indicated in Fig. 3B. The reaction mixtures were separated by SDS-PAGE, and polyubiquitination of Axin was detected by autoradiography (Fig. 3B). The incubation of Smurf2, but not the Smurf2(C716G) mutant, with ubiquitin, E1, and UbcH5A produced polyubiquitination signals (Fig. 3B, lanes 6 and 7). Polyubiquitination of Axin was also detected in the reaction mixture incubated without E1 (Fig. 3B, lane 3). Because the E1 enzyme does not have any target specificity (21), endogenous E1 enzyme from rabbit reticulocyte lysates might be used in the reaction.
Axin was mono-ubiquitinylated in vitro by Smurf2 using a mutant form of ubiquitin, and the putative ubiquitinylated Axin band was excised, followed by mass spectrometric analysis to determine the ubiquitination site(s) of Axin (Fig. 3C). Trypsin digestion of unubiquitinated Axin produced a peptide with a Lys505 terminal residue (Fig. 3D), whereas the putative ubiquitinylated Axin bands indicated a peptide that terminated with the next lysine due to mono-ubiquitination of Lys505 (Fig. 3E). In addition, the longer peptide produced peaks that were 114 Da larger in mass than the expected peaks in the LC-MS/MS analysis, which was due to the branching of two additional glycine residues leftover from ubiquitin after trypsin digestion (Fig. 3, D and E). This result confirms that Smurf2 ubiquitinylates Axin in vitro and suggests that Lys505 is a potential in vivo ubiquitination site of Smurf2. Axin(K505R) resisted degradation upon cotransfection with Smurf2 (Fig. 3F). In addition, knockdown of Smurf2 enhanced the level of wild-type Axin, but not Axin(K505R) (supplemental Fig. 3). Furthermore, the level of Axin(K505R) was maintained at a higher level compared with wild-type Axin in cell lines expressing GFP shRNA (supplemental Fig. 3, lanes 1 and 3). These results indicate that Lys505 is the ubiquitination site responsible for Axin degradation by Smurf2 in vivo. Overall, the data from LC-MS/MS analysis and the results with Axin(K505R) strongly suggest that Smurf2 specifically ubiquitinylates Axin at Lys505 both in vitro and in vivo.
Knockdown of Endogenous Smurf2 Increases the Axin Level
Consistent with the enhanced ubiquitination and down-regulation of Axin by the ectopic expression of Smurf2, knockdown of endogenous Smurf2 increased the level of endogenous Axin (Fig. 4, A and B). Knockdown of Smurf2 by shRNA also reduced the ubiquitination of Axin, which was rescued by the ectopic expression of Smurf2ΔWW1 (Fig. 4, A and C). Smurf2ΔWW1, a mutant form of Smurf2 lacking the shRNA targeting sequence (Fig. 4A), interacted and ubiquitinylated Axin similar to wild-type Smurf2 (data not shown). As Axin negatively regulates Wnt/β-catenin signaling, we tested whether or not knockdown of Smurf2 could inhibit β-catenin/Tcf-mediated reporter activity by increasing Axin expression. The reporter activity was reduced by the expression of Smurf2 shRNA, but this reduction was rescued by the coexpression of Axin shRNA (Fig. 4D). Knockdown of Smurf2 in HeLa and Caco-2 cells, but not in SNU475 cells that do not express Axin due to mutation (10), resulted in reduced reporter activity (Fig. 4E). These results suggest that the inhibition of β-catenin/Tcf reporter activity by Smurf2 shRNA is mediated by the regulation of Axin.
FIGURE 4.

Down-regulation of Smurf2 increases Axin levels via reduction of Axin ubiquitination. A, shown is schematic diagram of full-length Smurf2 and Smurf2ΔWW1. The arrow indicates the targeting region of shRNA against Smurf2. Smurf2ΔWW1 is a mutant form that does not include the Smurf2 shRNA targeting sequence. B, HeLa cells were infected with retroviruses expressing GFP shRNA or Smurf2 shRNA and then selected using puromycin. The pool of cells selected with puromycin was harvested, and Western blotting was performed. C, HEK293T cells stably expressing GFP or Smurf2 shRNA were transfected with plasmids. The transfected cells were treated with 25 μm MG132 for 4 h before harvesting. Cell lysates were subjected to immunoprecipitation (IP) with anti-Axin antibody, followed by immunoblotting with anti-HA antibody. HA-Ub, HA-ubiquitin; WCL, whole cell lysate. D, SuperTOPFlash and Renilla luciferase were cotransfected with plasmids expressing shRNA for Smurf2 (shSmurf2) or Axin (shAxin) into HEK293T cells, after which the relative luciferase activity was measured. This experiment was performed in triplicate. E, luciferase assays were performed as described for D using HeLa, Caco-2, and SNU475 cell lines.
In addition, we found that HeLa and DLD1 cells expressing Smurf2 shRNA showed slower growth rates than GFP shRNA-expressing control cells (supplemental Fig. 4). Because Smurf2 is involved in the regulation of various signaling pathways, the reduced growth rate of Smurf2 knockdown cells may not be due only to the inhibition of β-catenin/Tcf signaling. However, it is interesting that SNU449 cells, which have a mutation of β-catenin and in which the level of β-catenin is not regulated by Axin (10), did not show slower growth when Smurf2 was knocked down (supplemental Fig. 4). In summary, the results of the knockdown of endogenous Smurf2 support the idea that Smurf2 is an E3 ligase for the ubiquitination and down-regulation of Axin.
DISCUSSION
Controlling the level of Axin is a critical step in Wnt signaling. As such, Axin is considered as a potential therapeutic target; therefore, identification of small molecules that control Axin expression has received increasing attention (22, 23). The ubiquitin/proteasomal pathway is involved in Axin regulation; however, the E3 ligase responsible for this process has not been identified due to the extremely low expression and instability of Axin.
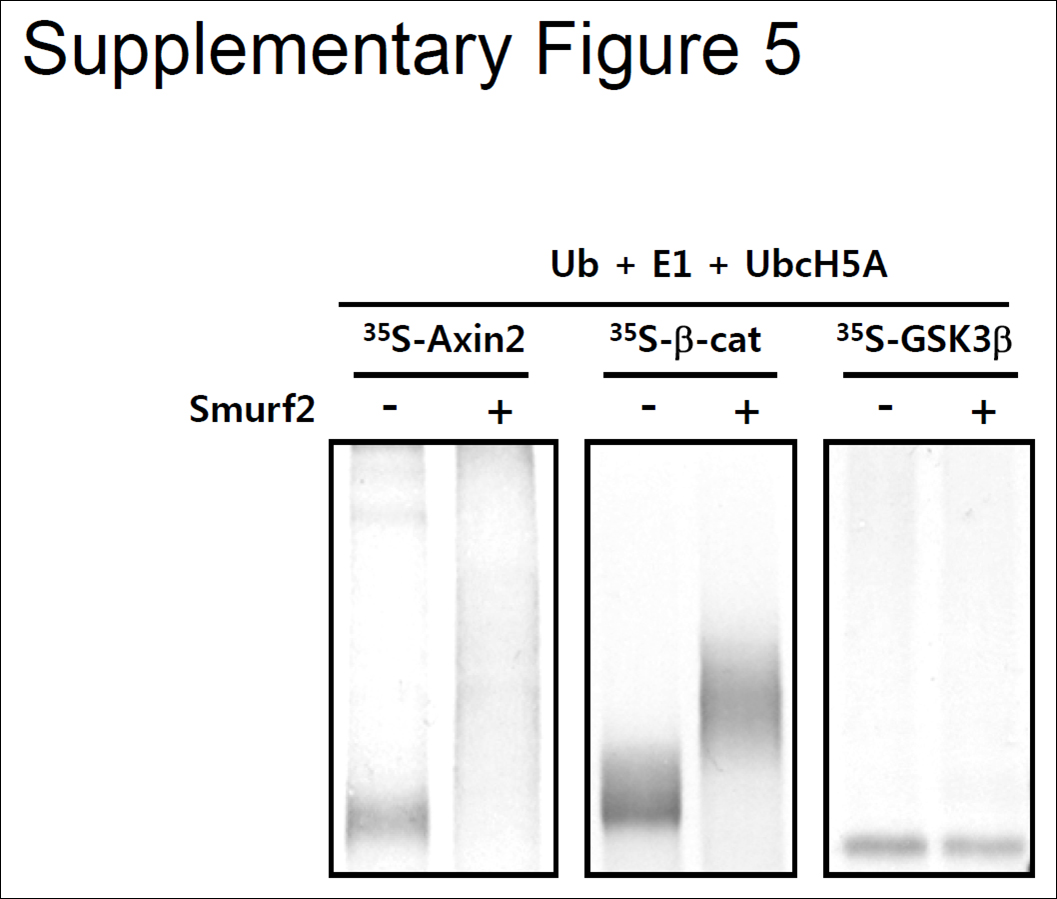
It was found previously that Smurf2 enhances the ubiquitination of Prickle via interaction with Dvl, β-catenin, and GSK3β (15, 24, 25). However, our in vitro ubiquitination analysis suggests that GSK3β is not a direct substrate of Smurf2; rather, it may require extra modification before ubiquitination by Smurf2 (supplemental Fig. 5). In a study by Wu et al. (25), this result was not examined by in vitro ubiquitination analysis. In addition, it seems unusual that Smurf2 would act as an E3 ligase for both Axin and β-catenin if we consider that Axin negatively regulates the level of β-catenin. Interestingly, Han et al. (15) showed that Smad7 is necessary for the Smurf2-mediated ubiquitination of β-catenin. In their study, they could not detect β-transducin repeat-containing protein, a previously identified E3 ligase involved in Axin/GSK3β-mediated β-catenin degradation, in Smad7/Smurf2 immunoprecipitates. These results suggest that Smurf2 may ubiquitinylate different substrates in a context-dependent manner. We found that knockdown of endogenous Smurf2 increased the Axin level and reduced β-catenin/Tcf reporter activity (Fig. 4, B–E), which suggests that Smurf2 works mainly as an E3 ligase for Axin in HEK293T, HeLa, and Caco-2 cells.
MS/MS spectrometric analysis suggested that Smurf2 ubiquitinylates Lys505 of Axin in vitro. However, it is still possible that Smurf2 ubiquitinylates other sites besides Lys505, as two shifted bands were detected in the Coomassie Blue-stained gel (Fig. 3C). However, based on the result that Axin(K505R) was resistant to degradation by cotransfection with Smurf2 (Fig. 3F), it seems that Lys505 is a critical site for the degradation of Axin by Smurf2.
Knockdown of Smurf2 reduced the growth of HeLa and DLD1 cells, but not SNU449 cells (supplemental Fig. 4). Smurf2 controls the levels of proteins such as the TGF-β receptor (14), Smad1 (13), and SnoN (26). Thus, the reasons for the reduced growth rate upon knockdown of Smurf2 are uncertain. However, the following evidence supports the idea that the reduced growth is due, at least partially, to the inhibition of β-catenin/Tcf signaling via increasing Axin expression. (i) Ectopic expression of Axin and stabilization of Axin upon treatment with XAV959 cause reduced cell growth (23, 27). (ii) SNU449 cells, in which the level of β-catenin is not regulated by Axin due to mutation of β-catenin (10), did not show reduced growth upon knockdown of Smurf2. (iii) Knockdown of Smurf2 in HEK293T and HEK293 cells did not cause any changes in the reporter assays that measure TGF-β signaling activity (supplemental Fig. 6).
Overall, our data strongly suggest that Smurf2 is an E3 ligase for Axin. Further investigation of the regulation of Smurf2 activity for Axin degradation will provide new ways of modulating Wnt/β-catenin signaling, thus leading to treatment of diseases related to that signaling.
Supplementary Material
This work was supported in part by Grants 2006-2004046 and R01–2007-000-20944-0 from the National Research Foundation of Korea, funded by the Ministry of Education, Science, and Technology.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–6.
- GSK3β
- glycogen synthase kinase 3β
- Tcf
- T cell factor
- RIPA
- radioimmune precipitation assay
- EGFP
- enhanced GFP.
REFERENCES
- 1.Klaus A., Birchmeier W. (2008) Nat. Rev. Cancer 8, 387–398 [DOI] [PubMed] [Google Scholar]
- 2.Zeng L., Fagotto F., Zhang T., Hsu W., Vasicek T. J., Perry W. L., 3rd, Lee J. J., Tilghman S. M., Gumbiner B. M., Costantini F. (1997) Cell 90, 181–192 [DOI] [PubMed] [Google Scholar]
- 3.Polakis P. (2007) Curr. Opin. Genet. Dev. 17, 45–51 [DOI] [PubMed] [Google Scholar]
- 4.Rui Y., Xu Z., Lin S., Li Q., Rui H., Luo W., Zhou H. M., Cheung P. Y., Wu Z., Ye Z., Li P., Han J., Lin S. C. (2004) EMBO J. 23, 4583–4594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Furuhashi M., Yagi K., Yamamoto H., Furukawa Y., Shimada S., Nakamura Y., Kikuchi A., Miyazono K., Kato M. (2001) Mol. Cell. Biol. 21, 5132–5141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu W., Rui H., Wang J., Lin S., He Y., Chen M., Li Q., Ye Z., Zhang S., Chan S. C., Chen Y. G., Han J., Lin S. C. (2006) EMBO J. 25, 1646–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rui Y., Xu Z., Xiong B., Cao Y., Lin S., Zhang M., Chan S. C., Luo W., Han Y., Lu Z., Ye Z., Zhou H. M., Han J., Meng A., Lin S. C. (2007) Dev. Cell 13, 268–282 [DOI] [PubMed] [Google Scholar]
- 8.Lee E., Salic A., Krüger R., Heinrich R., Kirschner M. W. (2003) PLoS Biol. 1, E10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tolwinski N. S., Wieschaus E. (2004) Trends Genet. 20, 177–181 [DOI] [PubMed] [Google Scholar]
- 10.Satoh S., Daigo Y., Furukawa Y., Kato T., Miwa N., Nishiwaki T., Kawasoe T., Ishiguro H., Fujita M., Tokino T., Sasaki Y., Imaoka S., Murata M., Shimano T., Yamaoka Y., Nakamura Y. (2000) Nat. Genet. 24, 245–250 [DOI] [PubMed] [Google Scholar]
- 11.Kim M. J., Chia I. V., Costantini F. (2008) FASEB J. 22, 3785–3794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pickart C. M. (2001) Annu. Rev. Biochem. 70, 503–533 [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y., Chang C., Gehling D. J., Hemmati-Brivanlou A., Derynck R. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 974–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kavsak P., Rasmussen R. K., Causing C. G., Bonni S., Zhu H., Thomsen G. H., Wrana J. L. (2000) Mol. Cell 6, 1365–1375 [DOI] [PubMed] [Google Scholar]
- 15.Han G., Li A. G., Liang Y. Y., Owens P., He W., Lu S., Yoshimatsu Y., Wang D., Ten Dijke P., Lin X., Wang X. J. (2006) Dev. Cell 11, 301–312 [DOI] [PubMed] [Google Scholar]
- 16.Fagotto F., Jho E., Zeng L., Kurth T., Joos T., Kaufmann C., Costantini F. (1999) J. Cell Biol. 145, 741–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lyu J., Costantini F., Jho E. H., Joo C. K. (2003) J. Biol. Chem. 278, 13487–13495 [DOI] [PubMed] [Google Scholar]
- 18.Willert K., Shibamoto S., Nusse R. (1999) Genes Dev. 13, 1768–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen C., Matesic L. E. (2007) Cancer Metastasis Rev. 26, 587–604 [DOI] [PubMed] [Google Scholar]
- 20.Ogunjimi A. A., Briant D. J., Pece-Barbara N., Le Roy C., Di Guglielmo G. M., Kavsak P., Rasmussen R. K., Seet B. T., Sicheri F., Wrana J. L. (2005) Mol. Cell 19, 297–308 [DOI] [PubMed] [Google Scholar]
- 21.Haas A. L., Bright P. M. (1988) J. Biol. Chem. 263, 13258–13267 [PubMed] [Google Scholar]
- 22.Chen B., Dodge M. E., Tang W., Lu J., Ma Z., Fan C. W., Wei S., Hao W., Kilgore J., Williams N. S., Roth M. G., Amatruda J. F., Chen C., Lum L. (2009) Nat. Chem. Biol. 5, 100–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang S. M., Mishina Y. M., Liu S., Cheung A., Stegmeier F., Michaud G. A., Charlat O., Wiellette E., Zhang Y., Wiessner S., Hild M., Shi X., Wilson C. J., Mickanin C., Myer V., Fazal A., Tomlinson R., Serluca F., Shao W., Cheng H., Shultz M., Rau C., Schirle M., Schlegl J., Ghidelli S., Fawell S., Lu C., Curtis D., Kirschner M. W., Lengauer C., Finan P. M., Tallarico J. A., Bouwmeester T., Porter J. A., Bauer A., Cong F. (2009) Nature 461, 614–620 [DOI] [PubMed] [Google Scholar]
- 24.Narimatsu M., Bose R., Pye M., Zhang L., Miller B., Ching P., Sakuma R., Luga V., Roncari L., Attisano L., Wrana J. L. (2009) Cell 137, 295–307 [DOI] [PubMed] [Google Scholar]
- 25.Wu Q., Huang J. H., Sampson E. R., Kim K. O., Zuscik M. J., O'Keefe R. J., Chen D., Rosier R. N. (2009) Exp. Cell Res. 315, 2386–2398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bonni S., Wang H. R., Causing C. G., Kavsak P., Stroschein S. L., Luo K., Wrana J. L. (2001) Nat. Cell Biol. 3, 587–595 [DOI] [PubMed] [Google Scholar]
- 27.Jeon S. H., Yoon J. Y., Park Y. N., Jeong W. J., Kim S., Jho E. H., Surh Y. J., Choi K. Y. (2007) J. Biol. Chem. 282, 14482–14492 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}