

Abstract
Phospholipases are critical enzyme mediators participating in many aspects of cellular function through modulating the generation of lipid 2nd messengers, membrane physical properties, and cellular bioenergetics. Here, we demonstrate that mice null for calcium-independent phospholipase A2γ (iPLA2γ−/−) are completely resistant to high fat diet-induced weight gain, adipocyte hypertrophy, hyperinsulinemia, and insulin resistance, which occur in iPLA2γ+/+ mice after high fat feeding. Notably, iPLA2γ−/− mice were lean, demonstrated abdominal lipodystrophy, and remained insulin-sensitive despite having a marked impairment in glucose-stimulated insulin secretion after high fat feeding. Respirometry of adipocyte explants from iPLA2γ−/− mice identified increased rates of oxidation of multiple different substrates in comparison with adipocyte explants from wild-type littermates. Shotgun lipidomics of adipose tissue from wild-type mice demonstrated the anticipated 2-fold increase in triglyceride content after high fat feeding. In sharp contrast, the adipocyte triglyceride content was identical in iPLA2γ−/− mice fed either a standard diet or a high fat diet. Respirometry of skeletal muscle mitochondria from iPLA2γ−/− mice demonstrated marked decreases in state 3 respiration using multiple substrates whose metabolism was uncoupled from ATP production. Shotgun lipidomics of skeletal muscle revealed a decreased content of cardiolipin with an altered molecular species composition thereby identifying the mechanism underlying mitochondrial uncoupling in the iPLA2γ−/− mouse. Collectively, these results identify iPLA2γ as an obligatory upstream enzyme that is necessary for efficient electron transport chain coupling and energy production through its participation in the alterations of cellular bioenergetics that promote the development of the metabolic syndrome.
Keywords: Adipose Tissue Metabolism, Bioenergetics, Diabetes, Metabolic Syndrome, Skeletal Muscle Metabolism, Lipidomics, Respirometry
Introduction
High fat (HF)3 diets, in conjunction with caloric excess and a sedentary lifestyle, result in increased white adipose tissue mass and insulin resistance that represent the biochemical progenitors of the metabolic syndrome (1, 2). A core component of the metabolic syndrome is a pathologic increase in white adipose tissue mass that results from either an increased number of adipocytes (hyperplasia) or the accumulation of increased lipid within an adipocyte (hypertrophy) or from combinations of both (3). Although obesity, insulin resistance, and the downstream sequelae of the metabolic syndrome are the major causes of mortality in industrialized nations, the molecular mechanisms underlying the maladaptive alterations that occur during prolonged and excessive HF diets are incompletely understood.
Metabolic flexibility is dependent on the efficient and coordinated transitions of metabolic flux that occur during changes in substrate utilization (e.g. glucose versus fatty acid) or changes in energy demand (e.g. exercise). Many of the adverse sequelae of obesity and type 2 diabetes are thought to result from disruption in the efficiency of these transitions, yet the biochemical mechanisms responsible for the integration of multiple diverse responses that occur in a cell type-specific manner are at their earliest stages of understanding (4).
Mitochondrial and peroxisomal phospholipases are important participants in the regulation of cellular bioenergetics and signaling (5–10). For example, mitochondrial phospholipases catalyze the production of nonesterified fatty acids that regulate uncoupling protein function, release lipid 2nd messengers of signal transduction (e.g. eicosanoids and lysolipids), and modulate mitochondrial membrane molecular dynamics that influence electron transport chain efficiency (11). Similarly, peroxisomal enzymes facilitate the synthesis of triglycerides for energy storage by provision of metabolic precursors for their synthesis during times of lipid excess and can also effectively relieve cells of their lipid burden through inefficient fatty acid oxidation with concomitant heat production (12–14).
Previously, we cloned and characterized a novel enzyme termed iPLA2γ (GenBankTM accession number NM_015723) (also known as patatin-like phospholipase 8 (PNPLA8; HUGO nomenclature)) that was remarkable for the presence of multiple translation initiation sites that encoded distinct protein products containing either mitochondrial or peroxisomal localization signals (or both) (15). The presence of dual mitochondrial and peroxisomal localization signals encoded in a single gene in conjunction with the complexity of its mRNA processing (leading to over 25 splice variants) and alternative translation initiation sites suggested the importance of this enzyme in regulating cellular bioenergetics in a cell type-specific manner (7). Although peroxisomes and mitochondria both have anabolic and catabolic functions, lipids that enter mitochondria are predominantly destined for oxidation and energy production, whereas lipids that enter the peroxisomes are directed to either anabolic metabolism (e.g. metabolic precursors of triglycerides, ether lipid biosynthesis, etc.) or to inefficient fatty acid oxidation leading to heat production. In a previous study, we overexpressed iPLA2γ in a cardiac myocyte-specific manner and identified the dramatic accumulation of triglycerides in transgenic myocardium after fasting (7). In contrast, genetic ablation of iPLA2γ demonstrated this enzyme is an important mediator of myocardial bioenergetics because its ablation led to defects in myocardial mitochondrial complex IV function and to increased mortality after cardiac stress (16).
In this study, we investigated the biochemical mechanisms underlying the effects of iPLA2γ ablation on cellular bioenergetics during HF feeding. Remarkably, iPLA2γ−/− mice were completely resistant to HF diet-induced obesity and the subsequent development of insulin resistance. The results demonstrate an unanticipated increase in adipocyte oxidation of fatty acids that occurred in concert with the expansion of adipocyte mitochondrial mass, increased adipocyte mitochondrial oxygen consumption, and elevated levels of UCP3 mRNA. Moreover, multidimensional mass spectrometry-based shotgun lipidomics of adipocyte lipids demonstrated a marked reduction in adipocyte triglyceride content that was accompanied by a shift to shorter chain length molecular species. Conversely, skeletal muscle mitochondrial function was concurrently compromised by a decreased respiratory capacity that was accompanied by increased uncoupling. Collectively, the results demonstrate the prominent roles of iPLA2γ in whole animal bioenergetics through modulation of adipocyte and skeletal muscle metabolism, insulin release and insulin sensitivity, and cellular mitochondrial function that collectively integrate organismal bioenergetics.
EXPERIMENTAL PROCEDURES
Materials
Standard and high fat diets were obtained from Purina (St. Louis, MO) and Bio-Serv (F3282; Frenchtown, NJ), respectively. The standard diet contained 6% fat, whereas the HF diet contained 35.5% fat. On a caloric basis, this represented 13% of the calories from fat in the standard diet and 59% of calories from fat in the HF diet. The standard and HF diets contained 0.93 and 14.2% saturated fat, respectively. Mass spectrometric analyses of the diet confirmed the dietary composition. Mice (beginning at 5 weeks of age) were fed a high fat diet to induce obesity with body weights measured weekly. Radiolabeled nucleotides were purchased from PerkinElmer Life Sciences. ECL reagents were purchased from GE Healthcare. Serum leptin and resistin levels were measured using a Leptin ELISA kit (Cayman Chemical) or a Resistin Quantikine kit (R & D Systems) according to the manufacturer's instructions. Western analyses of tissues were performed as described previously (17). Rabbit polyclonal antibodies for Western analyses were purchased as follows: HSL, pAKT, AKT, pMAPK, MAPK kinase, and acetyl-CoA carboxylase (ACC) were obtained from Cell Signaling (Danvers, MA); PPARγ was from Santa Cruz Biotechnology (Santa Cruz, CA); and FAS was from Abcam (Cambridge, MA). Synthetic phospholipids used as internal standards in mass spectrometric analyses were purchased from either Avanti Polar Lipids (Alabaster, AL) or Nu-Chek Prep, Inc. (Elysian, MN) and used as described previously (18, 19). Solvents for sample preparation and for mass spectrometric analysis were purchased from Burdick and Jackson (Muskegon, MI). Most other reagents were obtained from Sigma.
Animal Diets and Study Protocols
Mice were maintained and used in strict accordance with the National Institutes of Health guidelines for humane treatment of animals. All protocols were reviewed and approved by the Animal Care Committee of Washington University. Following euthanasia, tissues were dissected from male mice, weighed, and expressed as a fraction of total body weight. For food intake studies, ad libitum food consumption of individually housed mice was measured at 1, 1.5, 4, and 9 months of age (n = 4) over a consecutive 3–5-day period essentially as described previously (16).
Determination of Adipocyte Size
Adipocyte size was assessed as described previously (20) utilizing the AdCount program from the Biomedical Imaging Resource at Mayo Clinic (Mayo Clinic, Rochester, MN). In brief, isolated fat pads were digested with 1 mg/ml collagenase for 10–20 min followed by isolation of adipocytes, centrifugation at 300 × g, nuclear staining with 0.2% methylene blue/HEPES, and automated digital measurement of the cell diameter by AdCount analyses of at least 300 fat cells. Total adipocyte numbers in adipose tissue were estimated by manual counting of cells on H&E slides from at least three 20× fields of three mice per genotype and approximated assuming cubic packing. Adipocyte cell diameters of adipose tissue were measured to determine the mean adipocyte diameter. The mean adipocyte density (cells/unit volume) was next calculated from the mean adipocyte diameter assuming cubic closest packing of adipocytes. The total number of adipocytes in each epididymal fat pad during normal feeding or HF feeding was then calculated from the density of adipocytes (cells/unit volume) and the measured total volume of each epididymal fat pad (Fig. 1C).
FIGURE 1.

Severely reduced growth rate of iPLA2γ−/− mice in comparison with WT controls on standard and HF diets. A, comparisons of weight gain on a standard (Std) versus a HF diet. Total average body weight in grams was measured and plotted versus age (in days) for male WT and iPLA2γ−/− (KO) mice. The results represent 55 measurements of males between the ages of 28–350 days. Values represent the mean ± S.E. for each data point. iPLA2γ−/− mice fed a standard diet (dashed lines) had significantly decreased growth (†, p < 0.05) as early as day 28 (by the Student's t test) in comparison with WT littermates (solid lines). iPLA2γ−/− animals on the HF diet demonstrated severe growth retardation compared with wild-type animals on the HF diet after approximately day 60 (**, p < 0.05) with very little weight gain through day 350. In contrast, WT animals on the HF diet showed significantly greater body mass compared with WT animals on the standard diet by day 200 (*, p < 0.05). Symbols represent the earliest time points at which a significant difference was observed in comparisons of WT versus KO on the standard diet (†), WT versus KO on the HF fat diet (**), and standard diet WT versus HF diet WT (*). B, DEXA analysis determinations of the percent body fat in 4- and 9-month (mo)-old iPLA2γ−/− (KO, open bars) and wild-type (WT, closed bars) littermate mice on a standard (Std) versus a high fat (HF) diet. Total body fat to body weight (% fat) was lower (p < 0.001) in iPLA2γ−/− mice when compared with WT mice at 9 mo on the HF diet. All data are reported as means ± S.E. At least nine animals per group were analyzed. C, organ to body weight ratio comparisons of WT and iPLA2γ−/− mice after 10 months on a HF diet. A 6-fold decrease in epididymal fat (white adipose tissue (WAT)) to body weight and a 3-fold decrease in brown adipose tissue (BAT) to body weight were observed. Data are presented as means ± S.E. Statistical significance was performed using the Student's t test, and significant differences are indicated by brackets. Pan, pancreas. n = 3–6 per group.
In Vivo Intestinal Triglyceride (TAG) Absorption
To determine whether iPLA2γ−/− mice had abnormal intestinal lipid absorption, overnight fasted mice were injected with Triton WR-X-100 (500 mg/kg) and then given olive oil (8.5 μl/g) via intragastric gavage. Blood was sampled at 0–5 h and subjected to centrifugation at 4,000 rpm for 20 min, and TAG content was measured enzymatically.
Dual-energy X-ray Absorptiometry
Dual-energy x-ray absorptiometry (DEXA) was used to analyze total body mass, body fat mass, percent fat, fat-free body mass, and percent lean mass using a PIXIMUS small animal densitometer and PIXIMUS software (PIXIMUS Lunar GE, Madison, WI). All DEXA measurements were obtained with the skull excluded and tail included to increase the accuracy of the measurements as recommended by the manufacturer. Animals were lightly anesthetized using an intraperitoneal injection of a ketamine-HCl (100 mg/ml)/xylazine (20 mg/ml) mixture at 0.1 ml per 10 g of body weight. Quality controls included a phantom mouse as a calibration standard.
Glucose and Insulin Tolerance Studies
Mice (8–10 months old) were fasted overnight (16–18 h) on wood chip bedding with ad libitum access to water. Glucose tolerance tests were initiated by the intraperitoneal injection of glucose (2 mg/g body weight). Blood was drawn from the tail vein before and 30, 60, and 120 min after glucose administration. For insulin tolerance studies, mice were fasted for 1 h in cages without bedding and with free access to water prior to receiving an intraperitoneal injection of human regular insulin (Lilly) at a dose of 0.75 units/kg body weight. Blood glucose levels were measured from tail blood at 0, 30, 60, and 90 min. Glucose levels were measured using a Glucometer Elite (Bayer Corp., Elkhart, IN) and a Glucose Analyzer (Analox GM7 Analyser, Analox Instruments Inc., Lunenberg, MA). Integration of the area under each curve during glucose tolerance testing was quantified using Kodak ID 3.5 software.
Glucose-induced Insulin Release
Mice on the standard and HF diets were fasted overnight and challenged with glucose as in the glucose tolerance test above. Insulin levels in mice on standard and HF diets were measured 10 min after glucose challenge utilizing an ELISA kit according to the manufacturer's instructions (Crystal Chem).
Insulin-stimulated 2-Deoxyglucose Uptake
Adipocytes were isolated as described above, and an aliquot of suspended cells (30% v/v in KRH buffer with 1% BSA) was incubated in the presence of 200 μm [3H]deoxyglucose (0.1 μCi) (PerkinElmer Life Sciences) under basal conditions or after supplementation with 100 nm insulin (Humalog, Lilly). The reaction was terminated by washing the cells first with cold KRH and then subsequently three times using a manifold filtration device (Hoefer FH 225V, San Francisco) equipped with 0.22-μm filters (Millipore). The glucose uptake rate was determined from a linear regression analysis of the time course for uptake (10–40 min) using GraphPad Prism software (San Diego). The number of cells was calculated by dividing the packed adipocyte volume of cell suspension (obtained by centrifugation in a microcapillary centrifuge) by the individual adipocyte volume determined from measurements of the average adipocyte diameter.
Measurement of Stool Triglycerides
Levels of fecal TAG were determined from mice fed a standard diet and fed ad libitum. TAGs were extracted as described above and quantitated using the triglyceride reagent (Thermo Electron Corp., Pittsburgh, PA) and standard curves were generated using glycerol standards (Sigma). Serum glucose was measured as described above.
Adipose Tissue Lipolysis
Lipolysis studies were performed in explants of epididymal fat pads (≈100 mg) as described previously (21, 22). In brief, adipose tissue was excised and divided into equal slices. Tissue was added to Krebs-Ringer buffer (12 mm HEPES, 121 mm NaCl, 4.9 mm KCl, 1.2 mm MgSO4, and 0.33 mm CaCl2) with 3.5% fatty acid-free BSA and 0.1% glucose for 30 min supplemented with or without 200 nm isoproterenol (Sigma). Aliquots of 100 μl were collected at 1-, 2-, and 4-h time points, and released glycerol was measured in duplicate samples utilizing the “Free Glycerol Reagent” according to the manufacturer's instructions (Sigma).
Fatty Acid Oxidation Studies
Adipose explants were washed four times in buffer containing 1% albumin prior to measurement of fatty acid oxidation. Fatty acid oxidation was measured using 1-[14C] palmitic acid (American Radiolabeled Chemicals, Inc., St. Louis, MO) by measuring 14CO2 production as described previously (23).
Quantitative PCR Analyses
Quantitative PCR (qPCR) analyses of RNA message from white adipose tissues were performed as described previously (16, 23, 24). In brief, RNA was isolated utilizing TRIzol reagent (Invitrogen). Amplification was performed utilizing SYBR Green chemistry on an ABI 7500 qPCR cycler (Applied Biosystems, Foster City, CA). In some studies, PCR reagents were purchased from Applied Biosystems (Foster City, CA) and used with an ABI GeneAmp PCR System 9700 thermocycler. Acidic ribosomal phosphoprotein (36B4) RNA was used for normalization of expression. To quantify mitochondrial DNA copy number per nuclear genome, genomic DNA was first isolated from adipose tissue with a DNeasy tissue kit (Qiagen, Valencia, CA). The ratio of mitochondrial cytochrome c oxidase subunit II gene copies to nuclear 36B4 gene copies was then determined by qPCR. Primers for quantitative PCR analyses are listed in Table 1.
TABLE 1.
Primers for qPCR
Forward and reverse primer sequences (5′ to 3′) utilized for qPCR analyses are shown. Abbreviations used are as follows: 36B4, acidic ribosomal phosphoprotein P0; ACO, acyl-CoA oxidase; CCOII (for mDNA and RNA analyses), cytochrome oxidase II; CD36, cluster of determination 36; FABP3, fatty acid-binding protein; FOXO1, forkhead box 1; PGC1α, peroxisome proliferator-activated receptor γ coactivator 1α; PNPLA, patatin-like phospholipase; SCD1, stearoyl-CoA desaturase 1; and UCP, uncoupling protein.
| Gene | Forward | Reverse |
|---|---|---|
| 36B4 | GCAGACAACGTGGGCTCCAAGCAGAT | GGTCCTCCTTGGTGAACACGAAGCCC |
| ACC | CTAAACCAGCACTCCCGATT | ACTAGGTGCAAGCCAGACAT |
| ACO | GTATAAACTCTTCCCGCTCCTG | CCAGGTAGTAAAAGCCTTCAGC |
| Adiponectin | AAGGACAAGGCCGTTCTCT | TATGGGTAGTTGCAGTCAGTTGG |
| aP2 | GACGACAGGAAGGTGAAGAG | ACATTCCACCACCAGCTTGT |
| CCOII | CTGGTGAACTACGACTGCTAGA | GGCCATAGAATAACCCTGGTC |
| CD36 | CAAGCTCCTTGGCATGGTAGA | TGGATTTGCAAGCACAATATGAA |
| FABP3 | GGAAGCTAGTGGACAGCAAGA | CTGTCACCTCGTCGAACTCTAT |
| FAS | GTCTGGAAAGCTGAAGGATCTC | TGCCTCTGAACCACTCACAC |
| FOXO1 | CTGGGTGTCAGGCTAAGAGT | GGGGTGAAGGGCATCTTT |
| PGC1α | CGGAAATCATATCCAACCAG | TGAGGACCGCTAGCAAGTTTG |
| PNPLA1 | ACTGAATGCAGCGTACCTTGACT | GGCGACCTCTATCTGGCAGTATAC |
| PNPLA2 | GCCACAGCGCTGGTCACT | CCTCCTTGGACACCTCAATAATG |
| PNPLA3 | ACTGCACGCGGTCACTT | CACGAGGTCCATGAGGATCTC |
| PNPLA5 | GAGCGCCTCGCCTCATC | AGCCGCTCCACAAGTTTCA |
| PNPLA6 | CAGCCGAACTAAACA | TCCTGGAAGACACGGTGGA |
| PNPLA7 | AGCGGAGCTACAGCCAAACA | GGTCCAGTATAGTCTTCATCATTGATG |
| PNPLA9 | CCTTCCATTACGCTGTGCAA | GAGTCAGCCCTTGGTTGTT |
| Porin | GAGTATGGGCTGACGTTTACAG | CCCTCTTGTACCCTGTCTTGAT |
| PPARα | CCCTGTTTGTGGCTGCTAT | TCGGACTCGGTCTTGTTGAT |
| PPARδ | GCTGCTGCAGAAGATGGCA | CACTGCATCATCTGGGCATG |
| PPARγ | GCCCTTTGGTGACTTTATGG | GTCATCTTCTGGAGCACCTTG |
| SCD1 | CCGGAGACCCCTTAGATCGA | TAGCCTGTAAAAGATTTCTGCAAACC |
| UCP1 | CTGGGCTTAACGGGTCCTC | CTGGGCTAGGTAGTGCCAGTG |
| UCP2 | TCCACGCAGCCTCTACAAT | GACCTTTACCACATCTGTAGGC |
| UCP3 | CAGAGGGACTATGGATGCCTAC | AGGTGAGACTCCAGCAACTTCT |
Mass Spectrometry
Lipid analyses were performed essentially as described previously (19, 25). In brief, individual lipid extracts were reconstituted with 1:1 (v/v) CHCl3/CH3OH, flushed with nitrogen, and stored at −20 °C prior to electrospray ionization-MS. A TSQ Quantum Ultra Plus triple-quadrupole mass spectrometer (Thermo Fisher Scientific, San Jose, CA) equipped with an automated nanospray apparatus (i.e. Nanomate HD, Advion Bioscience Ltd., Ithaca, NY) and Xcalibur system software was utilized (26). For analyses of anionic lipids, ethanolamine phospholipids, choline phospholipids, and TAG, ionization voltages of −1.1, −0.95, and +1.2 kV and gas pressures of 0.3, 0.15, and 0.3 p.s.i. were employed, respectively, under control of ChipSoft 8.1.0 software. Each lipid extract solution was diluted to <50 pmol of total lipids/μl with CHCl3/CH3OH/isopropyl alcohol (1:2:4, v/v/v) prior to infusion into the mass spectrometer using the Nanomate apparatus. For all analyses, the first and third quadrupoles were used as independent mass analyzers with mass resolution settings of 0.7 Thomson, and the second quadrupole served as a collision cell for tandem mass spectrometry. Typically, spectra were acquired using a 1–2-min period of signal averaging in the profile mode for each full MS scan. For tandem mass spectrometry, a collision gas pressure was set at 1.0 millitorr, but the collision energy varied with the classes of lipids as indicated or as described previously (27, 28). For each tandem MS mass spectrum, a 2–5-min period of signal averaging in the profile mode was employed. Enhanced shotgun lipidomics analyses of cardiolipins (CL) were performed with a mass resolution setting of 0.3 Thomson as described previously in detail (29). All full mass scans and tandem MS scans were automatically acquired using a customized sequence subroutine operated under Xcalibur software. Data from biologic samples were normalized to the protein content, and all data are presented as the means ± S.E.
Mitochondrial Respirometry
Mice were sacrificed, and tissues were immediately dissected on ice. Isolation of tissue and mitochondria were performed in a cold room at 4 °C. The use of adipose tissue explants for mitochondrial studies has previously been described in detail (30). For the current studies, epididymal adipose tissue was removed and placed in mitochondrial isolation buffer (MIB; 0.21 m mannitol, 70 mm sucrose, 0.1 mm potassium EDTA, 1 mm EGTA, 10 mm Tris-HCl, 0.5% BSA, pH 7.4). Adipose tissue was dissected into two (40–60 mg) portions and minced into 1-mm3 pieces. Minced adipose tissue was placed in 2 ml of respiration buffer (MiRO5: 110 mm sucrose, 60 mm potassium lactobionate, 20 mm taurine, 20 mm HEPES, 10 mm KH2PO4, 3 mm MgCl2, 0.5 mm EGTA, 1 g/liter BSA (fraction V), pH 7.1) for high resolution respirometry analysis. Muscle mitochondria were isolated from the quadriceps. Briefly, a 50-mg piece of quadriceps muscle was removed and placed in muscle mitochondrial isolation buffer (MMIB: 100 mm sucrose, 100 mm KCl, 50 mm Tris-HCl, 1 mm KH2PO4, 0.1 mm EGTA, 0.2% BSA, pH 7.4). The muscle was minced into 1-mm3 pieces and incubated with 1.5 ml of MMIB buffer containing 0.2 mg/ml bacterial protease (Subtilisin Carlsberg, EC 232-752-2) for 2 min as described previously (31, 32). The sample was then homogenized using a Potter-Elvehjem Teflon-coated pestle homogenizer for an additional 2 min at 180 rpm, followed by dilution of the sample 6-fold with MMIB. Next, the homogenate was centrifuged for 5 min at 850 × g, and the supernatant was collected and diluted 2-fold with MMIB. The supernatant was then centrifuged at 7,500 × g for 10 min, and the mitochondrial pellet was resuspended in 400 μl of MIB without BSA. Mitochondrial protein content was determined using a BCA protein assay (Thermo Fisher Scientific, San Jose, CA).
High resolution respirometry was performed using a 2-ml chamber OROBOROS® Oxygraph 2K (Innsbruck, Austria). Respiratory measurements were performed at 30 °C in MiRO5 buffer with 500 rpm stir bar rotation. The oxygen concentration at air saturation was determined to be 230 nmol of O2 per ml at 105 kilopascal barometric pressure. Oxygen calibration and slope calculations were performed routinely to ensure accuracy of the flux measurements. Oxygen flux was calculated as a time derivative of oxygen concentration using the DatLab4.3 Analysis software (OROBOROS). The same assay protocol was used for muscle mitochondria (200 μg of protein) and adipose tissue (40–60 mg) per assay. Respiration was started with the addition of glutamate (10 mm) or palmitoylcarnitine (20 μm) and malate (5 mm) (state 2), followed by ADP (1.25 mm) (state 3), succinate (5 mm) (state 3 max), rotenone (0.5 μm), oligomycin (1 μm) (state 4), carbonyl cyanide p-(trifluoromethoxy) phenyl-hydrazone (FCCP, 0.5 μm), and antimycin A (1 μm), N,N,N′,N′-tetramethyl-p-phenylenediamine (5 μm) with ascorbate (0.5 mm). This protocol has been used previously to assess mitochondrial respiratory capacity, and all values reflected the subtraction of residual oxygen consumption as determined by antimycin A addition (33). Additionally, for each assay, a 10-μl aliquot was collected from the respirometry chamber, saturated with DMSO, and stored at −80 °C for subsequent determination of ATP utilizing the ENLITEN® detection system (Promega, Madison, WI) according to the manufacturer's instructions.
Miscellaneous Studies
Energy expenditure by wild-type and iPLA2γ −/− mice was evaluated by indirect calorimetry (Oxymax, Columbus Instruments, Columbus, OH). Ambulatory activity was monitored by an infrared motion sensor utilizing an InfraMot apparatus (TSE Systems, Midland, MI) as described previously (34). Stool production was measured in metabolic cages as described previously (34).
Statistical Analysis
Data were analyzed using a two-tailed Student's t test. Differences were regarded as significant at the p < 0.05 level. All data are reported as the means ± S.E. unless otherwise indicated. Some data were presented as “box-and-whisker plots” to identify the median, 25th, and 75th percentiles as well as the extremes.
RESULTS
iPLA2γ−/− Mice Are Completely Resistant to the Development of Obesity on a High Fat Diet
As anticipated, wild-type (WT) mice fed a HF diet exhibited nearly a 50% increase in weight in comparison with WT littermates fed a standard diet. In sharp contrast, iPLA2γ−/− mice fed a HF diet were completely resistant to weight gain (Fig. 1A). iPLA2γ−/− mice fed a HF diet were visually distinguishable from their WT littermates by their thin body habitus and the presence of an oily skin coat (supplemental Fig. 1). After 11 months of HF feeding, WT littermates displayed the anticipated increase of abdominal adiposity at necropsy. Remarkably, visible abdominal adiposity was completely absent in iPLA2γ−/− mice even after 11 months of HF feeding (supplemental Fig. 1B). Quantitation of whole body fat content by DEXA analysis demonstrated that WT mice fed a standard diet contained 22 ± 3% fat, although WT mice fed a HF diet contained 33 ± 2% fat (i.e. a 50% increase in adiposity was present in WT mice after HF feeding) (Fig. 1B). In sharp contrast, DEXA analysis of iPLA2γ−/− mice fed a HF diet demonstrated only 17% whole body fat, which was unchanged from their fat content on a standard diet and was identical to that present in WT mice on a standard diet (Fig. 1B).
Epididymal Fat Pad Weights Are Decreased during High Fat Feeding in iPLA2γ−/− Mice
HF feeding resulted in a nearly 6-fold higher epididymal fat pad/body weight ratio in WT littermates compared with iPLA2γ−/− mice (Fig. 1C). Similarly, the proportion of brown adipose tissue was substantially lower in iPLA2γ−/− mice in comparison with WT littermates after HF feeding. The proportionate weights of liver and pancreas were not significantly changed (Fig. 1C).
Caloric Intake and Triglyceride Absorption Are Normal in the iPLA2γ−/− Mouse
To determine the mechanisms underlying the absence of adiposity during HF feeding in iPLA2γ−/− mice, additional experiments were performed. First, the caloric intake of mice from each group was determined. The absence of weight gain in iPLA2γ−/− mice was not due to a decrease in overall caloric intake, which was similar to that measured in WT littermates. In fact, iPLA2γ−/− mice consumed more food/g body weight than WT littermates during HF feeding (supplemental Fig. 2). Second, fat absorption from the intestine after intragastric administration of olive oil was indistinguishable in iPLA2γ−/− mice in comparison with their WT littermates (supplemental Fig. 3). Third, the fat content of stool samples was similar in iPLA2γ−/− mice in comparison with WT littermates (supplemental Fig. 4). Thus, the absence of weight gain in the iPLA2γ−/− mice did not result from decreased intestinal fat absorption.
Whole Animal Respirometry Demonstrates a Hypometabolic State in iPLA2γ−/− Mice
To determine the global metabolic rate of the iPLA2γ−/− mouse, whole body respirometry was performed in mice maintained on a HF diet. The results demonstrated that HF feeding of iPLA2γ−/− mice resulted in decreased O2 consumption, decreased CO2 production, and less heat generation than in wild-type littermates (Fig. 2). Because the large majority of whole body O2 consumption and CO2 production in mammals resulted from physical activity, we quantified differences in ambulatory activity using InfraMot analyses. The results demonstrated a 65% decrease in locomotor activity in iPLA2γ−/− mice in comparison with WT littermates (supplemental Fig. 5). Collectively, these results demonstrate that the iPLA2γ−/− mouse resists obesity during HF feeding even though it demonstrates a markedly decreased ambulatory activity. We note that if the 24-h O2 consumption is normalized to locomotor activity, then there is a 2-fold increase in O2 consumption/activity in the iPLA2γ−/− mouse.
FIGURE 2.

Indirect calorimetry analysis of wild-type and iPLA2γ−/− mice on the HF diet. Oxygen consumption (VO2), (A), carbon dioxide consumption (VCO2) (B), respiratory exchange ratio (RER) (C), and heat generation (D) were measured in a 24-h period in wild-type (WT) and iPLA2γ−/− mice fed a HF diet from the time of weaning until 12 months of age utilizing an indirect calorimeter (Oxymax) as described under “Experimental Procedures.” n = 5 per group. Shaded regions represent the dark period of the 24-h cycle. WT values are indicated as solid diamonds and solid lines, and iPLA2γ−/− are represented with solid squares and dotted lines. Data are presented as means ± S.E. *, p < 0.05 by the Student's t test.
Near Normal Glucose Tolerance and the Prevention of Insulin Resistance in the iPLA2γ−/− Mouse during High Fat Feeding
To determine whether the lack of weight gain after HF feeding in iPLA2γ−/− mice is associated with protection from insulin resistance and glucose intolerance, glucose tolerance and insulin sensitivity tests were performed. Glucose tolerance tests in iPLA2γ−/− mice fed a standard diet were indistinguishable from those of WT littermate controls (Fig. 3). After 8 months of HF feeding, a modest increase in the glucose level at 30 min of post-glucose challenge was present in iPLA2γ−/− mice (Fig. 3A, right panel). An increased rate of return during the ensuing 90 min in comparison with WT littermates was also present. The area under the glucose curve in the HF fed iPLA2γ−/− mice was only 10% greater than that for their WT littermates. The iPLA2γ−/− mice fed a HF diet exhibited an increased sensitivity to intraperitoneal insulin administration with an increased clearance of serum glucose in comparison with their WT littermates (Fig. 3B). Additionally, Western analyses demonstrated increased pAKT protein mass in iPLA2γ−/− adipose tissue consistent with increased insulin signaling in adipose tissue (supplemental Fig. 6).
FIGURE 3.

Alterations in glucose homeostasis in the iPLA2γ−/− mouse. A, glucose tolerance test of mice on the standard diet (left panel) or HF (right panel) diets. As described under “Experimental Procedures,” d-glucose (2 mg/g) was administered by intraperitoneal injection to WT (solid lines) or iPLA2γ−/− (dotted lines) male mice after 8–10 months fed on either a standard (left panel) or HF (right panel) diet. Blood was collected at base line and at 30, 60, and 120 min after injection for measurement of serum glucose concentrations. Glucose intolerance with an enhanced rate of glucose uptake was observed in the iPLA2γ−/− mice fed a HF diet (*, p < 0.05). Values are displayed as means ± S.E. (n = 10–16 per group). B, increased insulin sensitivity of iPLA2γ−/− mice on a HF diet. An insulin tolerance test was performed as described under “Experimental Procedures” with iPLA2γ−/− (square symbols and dotted lines) and WT (diamond symbols and solid lines) mice. Human regular insulin (0.75 unit/kg) was administered by intraperitoneal injection to a 1-h fasted male mice after 8 months on the HF diet, and blood was collected at base line and at 30, 60, and 90 min after injection to measure glucose concentration as described under “Experimental Procedures.” Values are displayed as means ± S.E. (n = 10–16). *, p < 0.05 by the Student's t test. C, serum blood insulin levels in mice on either the standard or the HF diet. Insulin levels were determined as described under “Experimental Procedures” with measurements taken at time 0 after an overnight fast (pre) and at 10 min following intraperitoneal glucose injection (post). n = 3 per group. WT, solid line; KO, dashed line. All experiments were performed with four WT and iPLA2γ−/− mice between the ages of 8–10 months of age. D, box-and-whisker analysis as described under “Experimental Procedures” of nonfasted (NF) blood insulin of wild-type (WT) and iPLA2γ−/− (KO) mice on the HF diet. Statistical significance was performed using the Student's t test, and significant differences are indicated by brackets. n = 4 WT and 11 KO.
iPLA2γ−/− Mice Exhibit a Severe Defect in Insulin Secretion during Both Standard and High Fat Feeding
To identify the influence of alterations in serum insulin to the development of the observed phenotype of the iPLA2γ−/− mouse, serum insulin levels after fasting, during normal feeding, or after intraperitoneal injection of glucose were measured. Although serum insulin levels in WT and iPLA2γ−/− mice fed a standard diet and fasted overnight were similar before glucose administration, a dramatic defect in glucose-stimulated insulin release was present in iPLA2γ−/− mice. Specifically, WT mice released over 4-fold greater amounts of insulin into the serum after glucose challenge in comparison with iPLA2γ−/− mice fed a standard diet (Fig. 3C, left panel). Moreover, after HF feeding, fasting serum insulin levels were 7-fold higher in WT littermates in comparison with iPLA2γ−/− mice (Fig. 3C, right panel). As anticipated, intraperitoneal injection of glucose into fasted WT mice fed a HF diet resulted in nearly a 3-fold further increase in insulin release (from 0.6 ng/ml to a final level of 1.6 ng/ml) relative to standard diet WT mice. In sharp contrast, fasting serum insulin levels in the HF fed iPLA2γ−/− mice were only minimally increased after glucose challenge (from a level of 0.14 to 0.19 ng/ml). Finally, to assess differences in circulating insulin during normal feeding, we compared serum insulin levels in nonfasting iPLA2γ−/− mice fed a HF diet to their corresponding WT littermates. Remarkably, morning serum insulin levels in nonfasting WT mice were 40-fold higher than those present in iPLA2γ−/− mice (Fig. 3D, note log scale). This massive difference in serum insulin was present despite similar levels of serum glucose in iPLA2γ−/− mice and WT littermates (i.e. 150 versus 180 mg/dl) consistent with the presence of enhanced insulin sensitivity in these mice. In support of these findings, ex vivo studies demonstrated that insulin-stimulated glucose uptake was over 3-fold higher in adipocytes cultured from iPLA2γ−/− mice in comparison with adipocytes from wild-type mice (supplemental Fig. 7). Collectively, these results demonstrate the insulin-sensitive phenotype of the iPLA2γ−/− mouse.
No genotype-specific differences in plasma free fatty acid or triglyceride levels between WT and iPLA2γ−/− mice were detected on either the standard diet or after HF feeding (Table 2). In addition, no evidence of increased reactive oxygen species between WT and iPLA2γ−/− mouse adipose tissue was found as assessed by multiple methods, including mass spectrometry and fluorescent dye approaches (data not shown). As anticipated from the large decrease in adipose tissue mass, serum leptin levels in the iPLA2γ−/− mice were substantially decreased in comparison with their WT littermates (supplemental Fig. 8A). The decreased serum leptin is also consistent with the observed increase in food intake normalized to body weight on the HF diet (supplemental Fig. 2). In sharp contrast, serum resistin levels were not significantly different in the iPLA2γ−/− mice compared with wild-type littermates (supplemental Fig. 8B).
TABLE 2.
Serum levels of FFA and TAG in wild-type and iPLA2γ−/− mice maintained on either the standard or high fat diet
Serum levels of FFA (mmol/liter) and TAG (mg/dl) levels were measured as described under “Experimental Procedures.”
| Standard diet |
High fat diet |
|||
|---|---|---|---|---|
| WT | KO | WT | KO | |
| FFA | 1.06 ± 0.08 | 1.17 ± 0.12 | 1.79 ± 0.08 | 1.64 ± 0.08 |
| TAG | 79.5 ± 5.3 | 83.7 ± 6.2 | 203.3 ± 18.2 | 184.3 ± 10.0 |
iPLA2γ−/− Mouse Is Completely Resistant to Adipocyte Hypertrophy on a High Fat Diet
To determine whether the decreased adiposity resulted from a decrease in adipocyte triglyceride accumulation (hypertrophy) or from a failure of preadipocytes to differentiate during HF feeding (hyperplasia), adipocyte morphology was examined as described under “Experimental Procedures.” Adipocytes from WT mice fed a standard diet possessed an average size of 56 μm, whereas those from iPLA2γ−/− mice fed a normal diet showed a trend to a smaller size (50 μm) that was not statistically significant (Fig. 4A). As expected, during HF feeding, adipocytes from WT mice underwent hypertrophy (mean diameter = 80 μm). In marked contrast, no significant increase in adipocyte size was present after HF feeding of iPLA2γ−/− mice (mean diameter = 53 μm). The total number of adipocytes was estimated from the density of adipocytes and the volume of epididymal fat pads as described under “Experimental Procedures.” A small but not statistically significant decrease in adipocyte number was calculated in epididymal fat pads in iPLA2γ−/− mice (Fig. 4B). Histology confirmed the lack of adipocyte hypertrophy in either the epididymal or inguinal fat pads of the iPLA2γ−/− mouse (Fig. 4, C and D). Collectively, these results demonstrate that the HF diet-induced adipocyte hypertrophy present in WT mice was completely prevented in iPLA2γ−/− mice.
FIGURE 4.

Reduced white adipose adipocyte size in the iPLA2γ−/− mice. A, adipocyte size comparison of WT and iPLA2γ−/− adipocytes from epididymal fat from mice fed a standard diet or HF diet. Adipocyte sizing was performed utilizing the AdCount program as described under “Experimental Procedures.” Male mice (8–10 months of age) were utilized in these studies (n = 3 per group). A box-and-whisker plot (inset) shows the relative distribution of adipocyte sizes (as measured by adipocyte diameter in micrometers) from epididymal adipose tissue. The data illustrate the dramatically restricted distribution of adipocyte sizes in iPLA2γ−/− mice fed either the standard (KO) or HF (HFKO) diets and lack of adipocyte hypertrophy compared with epididymal fat pads from WT animals on either the standard (WT) or the HF (HFWT) diets. Labels used are as follows: WT, WT on the standard diet; KO, iPLA2γ−/− on the standard diet; HFWT, WT on the HF diet; HFKO, KO fed a HF diet. B, number of adipocytes per epididymal fat pad in wild-type (WT) and iPLA2γ−/− (KO) mice on a HF diet for 10 months computed assuming cubic packing as described under “Experimental Procedures.” Data are presented as means ± S.E. n = 4 male mice per group, p < 0.01 by the Student's t test. C, H&E staining of adipocytes from WT and KO epididymal white adipose tissue from animals on the HF diet for 8 months. The images illustrate the differences in adipocyte size in WT versus KO epididymal adipose tissue. D, H&E staining of adipocytes from WT and KO inguinal white adipose tissue from animals on the HF diet for 8 months. The images illustrate the differences in adipocyte size in WT versus KO inguinal adipose tissue. C and D are representative images from three animals per group. Bar, 100 μm.
Transcriptional Regulation of Adipogenesis Is Intact in iPLA2γ−/− Mice
Adipogenesis is under tight control by multiple transcriptional programs. Foremost among these are the activations of metabolic programs regulated by PPARγ, which serves as the master transcriptional regulator of adipogenesis. Surprisingly, the mRNA levels of PPARγ in iPLA2γ−/− mice were 4-fold higher during HF feeding in comparison with WT littermates (Fig. 5). Moreover, the mRNA levels of PPARγ downstream targets, including ACC, FAS, and adipocyte protein 2 (aP2), were also markedly elevated demonstrating an intact functional axis for transcriptional programs to increase adipose cell differentiation and adipogenesis (Fig. 5). Furthermore, Western analysis of FAS and ACC revealed increases in protein mass concordant with mRNA levels (supplemental Fig. 9). A trend toward increased PPARγ protein mass was also observed in iPLA2γ−/− adipose tissue.
FIGURE 5.

Expression of adipogenesis markers in epididymal adipose tissue from wild-type and iPLA2γ−/− mice. Quantitative PCR was utilized to examine expression of PPARγ (A), ACC (B), FAS (C), aP2 (D), adiponectin (E), and SCD1 (F) mRNA in epididymal adipose tissue from male wild-type (WT, closed bars) and iPLA2γ−/− (KO, open bars) mice. Expression levels of genes were measured as described under “Experimental Procedures.” Significant increases in PPARγ, ACC, FAS, aP2, adiponectin, and SCD1 were observed in the iPLA2γ−/− mouse on the HF diet. Male mice 8–10 months of age were utilized in these studies. Values represent arbitrary units (AU) after normalization to a ribosomal 36B4 internal standard. Data are presented as means ± S.E. Statistical significance was performed using the Student's t test, and significant differences indicated by the p values above the brackets. n = 3–4 per group.
Multidimensional Mass Spectrometry-based Shotgun Lipidomics (MDMS-SL) of Adipose Tissue from High Fat Fed WT and iPLA2γ−/− Mice
To further clarify the role of iPLA2γ in mediating alterations in adipocyte metabolism, we quantified triglyceride and phospholipid molecular species in adipose tissue using MDMS-SL. Detailed analyses of TAG molecular species composition in mice fed a standard diet demonstrated a shift to shorter acyl chain length molecular species in iPLA2γ−/− mice in comparison with WT littermates. This is illustrated by comparison of the neutral loss spectra from adipocyte extracts from WT and iPLA2γ−/− mice fed a standard diet (Fig. 6A). After HF feeding, adipocyte TAG content in WT adipocytes increased nearly 2-fold (52.2 ± 0.9 μmol/mg of protein on the standard diet versus 98.9 ± 0.9 μmol/mg of protein on the HF diet) (Fig. 6B). Conversely, no increase in adipocyte TAG content was manifest after HF feeding in iPLA2γ−/− mice (43.0 ± 3.2 μmol/mg of protein on the standard diet versus 42.3 ± 3.8 μmol/mg of protein on the HF diet) (Fig. 6B). It should also be noted that the shift toward shorter acyl chain length molecular species present during standard diet feeding was absent during HF feeding (supplemental Fig. 10). No substantial alterations in choline, ethanolamine, serine, or inositol glycerophospholipid content or individual molecular species distribution in adipocytes from WT littermates or iPLA2γ−/− mice fed either a standard or HF diet were apparent (data not shown).
FIGURE 6.

Alterations in epididymal adipose tissue TAG content. A, two-dimensional electrospray ionization-MS profile of TAG molecular species from mice fed the standard diet. Adipose lipid extracts were analyzed by electrospray ionization-MS as described under “Experimental Procedures.” Spectra for WT (left) and iPLA2γ−/− (KO, right) epididymal adipose tissue TAG illustrate multiple alterations of individual TAG species. Panels are representative mass spectra from extracts of adipose tissue from at least three mice for each condition. Ion intensities for all samples were normalized to a T17:1 TAG internal standard (IS) for quantitation after correction (28). B, comparison of total epididymal TAG from WT and iPLA2γ−/− mice fed either the standard (Std) or the HF diet. Total TAG decreased by ∼15% (p = 0.06) in the KO on the standard diet (left) and 57% (p < 0.001) on the HF diet (right). Male mice 8–10 months of age were utilized in these studies. Data are presented as means ± S.E. and statistical significances are indicated by the p values above the brackets. WT, closed bars; KO, open bars. n = 3–4 per group.
Fatty Acid Oxidation Is Increased in Adipose Tissue from iPLA2γ−/− Mice Fed a High Fat Diet
Despite the increased levels of PPARγ and lipogenic genes in adipose tissue, iPLA2γ−/− mice were unable to increase their triglyceride mass in adipocytes during HF feeding. Accordingly, we examined the ability of adipocytes to oxidize exogenous fatty acid by incubating 1-[14C]palmitic acid with explants of epididymal fat pads from both groups. The rate of fatty acid oxidation in iPLA2γ−/− adipose explants was 2-fold higher in iPLA2γ−/− mice than that manifest in adipose explants from WT littermates (Fig. 7A).
FIGURE 7.

Increased fatty acid oxidation but unaltered lipolysis in epididymal adipose tissue explants from the iPLA2γ−/− mouse on the standard diet. A, measurement of [1-14C]palmitate oxidation in epididymal adipose tissue explants from wild-type (WT) and iPLA2γ−/− (KO) mice was performed as described under “Experimental Procedures.” Epididymal adipose tissue from male mice 6–8 months of age were utilized for these studies. n = 4 per group. Data are presented as means ± S.E. Statistical significance was performed using the Student's t test (p < 0.002) as indicated by the bracket. B, assessment of TAG hydrolysis in epididymal adipose tissue explants by measurement of glycerol released into the media with time under basal conditions or following stimulation with isoproterenol supplementation as described under “Experimental Procedures.” Media glycerol concentrations in explant cultures from wild-type and iPLA2γ−/− mice under basal (WT, squares; KO, diamonds) or isoproterenol-stimulated (WT, triangles; KO, circles) conditions are indicated by solid lines and dashed lines, respectively.
To identify the molecular mechanisms underlying the increased rate of fatty acid oxidation in iPLA2γ−/− adipocytes, additional experiments were performed. First, we measured the rate of [9,10-3H]oleic acid incorporation into adipose cellular polar and nonpolar lipids. The rates of incorporation of oleic acid into adipocyte polar and nonpolar lipids from WT and iPLA2γ−/− mice were indistinguishable (data not shown). Thus, alterations in fatty acid uptake or decreased shunting of lipids into anabolic pathways were not responsible for the increases in adipocyte fatty acid oxidation rates. Next, we examined the rates of lipolysis present in adipose tissue explants obtained from WT and iPLA2γ−/− mice by measuring glycerol release. The basal release of glycerol in adipocyte explants from WT mice fed a HF diet was indistinguishable from that observed in iPLA2γ−/− mice (Fig. 7B). Moreover, isoproterenol stimulated identical amounts of lipolysis in WT and iPLA2γ−/− mice (Fig. 7B). Next, to assess the relative mRNA levels of adipocyte triglyceride lipase expression, qPCR was performed. The results demonstrated a trend toward decreased amounts of PNPLA2 (also known as ATGL or iPLA2ζ) and decreased PNPLA3 (also known as adiponutrin or iPLA2ϵ) in iPLA2γ−/− mice fed a HF diet in comparison with their WT counterparts (Table 3). No significant differences in ATGL or HSL protein content as determined by Western analyses were observed between WT and iPLA2γ−/− mice on either a standard diet or a HF diet (data not shown). Collectively, these results demonstrate that the observed increases in adipocyte fatty acid oxidation result from the direct entry of fatty acid into oxidative processes and do not result from an increase in triglyceride hydrolysis or shunting of fatty acids from anabolic into oxidative pathways in the iPLA2γ−/− mouse adipocytes.
TABLE 3.
qPCR analyses of intracellular phospholipases
Expression of patatin-like intracellular phospholipases in wild-type (WT) and iPLA2γ−/− (KO) epididymal adipose tissue from mice on the standard and HF diets. qPCR analyses were performed as described under “Experimental Procedures” utilizing three male mice per group (ages 6–8 months). PNPLA4 is not present in the mouse genome. The mean values are presented along with standard error, and p values were determined by the Student's t test.
| Gene | Standard diet |
HF diet |
||||
|---|---|---|---|---|---|---|
| WT | KO | p value | WT | KO | p value | |
| PNPLA1 | 0.44 ± 0.24 | 0.40 ± 0.23 | 0.9 | 0.55 ± 0.14 | 3.40 ± 1.10 | 0.004 |
| PNPLA2 | 11.0 ± 0.08 | 2.0 ± 0.16 | 0.01 | 1.20 ± 0.21 | 0.90 ± 0.14 | 0.05 |
| PNPLA3 | 6.5 ± 0.56 | 1.2 ± 0.34 | 0.001 | 1.42 ± 0.31 | 0.24 ± 0.08 | 0.001 |
| PNPLA5 | 0.14 ± 0.07 | 0.07 ± 0.02 | 0.48 | 0.34 ± 0.03 | 0.48 ± 0.19 | 0.61 |
| PNPLA6 | 4.8 ± 2.0 | 3.2 ± 0.7 | 0.2 | 0.74 ± 0.03 | 0.08 ± 0.28 | 0.57 |
| PNPLA7 | 0.8 ± 0.06 | 1.3 ± 0.15 | 0.04 | 0.97 ± 0.03 | 0.83 ± 0.31 | 0.21 |
| PNPLA9 | 0.77 ± 0.14 | 1.30 ± 0.12 | 0.02 | 1.45 ± 0.14 | 0.80 ± 0.12 | 0.02 |
Molecular Mechanisms Underlying Increased Fatty Acid Oxidation in iPLA2γ−/− Adipose Tissue
We next considered whether the increased fatty acid oxidative capacity present in epididymal adipose tissue from iPLA2γ−/− mice was due to increased mitochondrial content. The levels of mitochondrial DNA in epididymal adipose tissue from iPLA2γ −/− mice on the standard or HF diets were 2-fold higher in comparison with their WT littermates (Fig. 8) indicating a marked increase in mitochondrial content.
FIGURE 8.
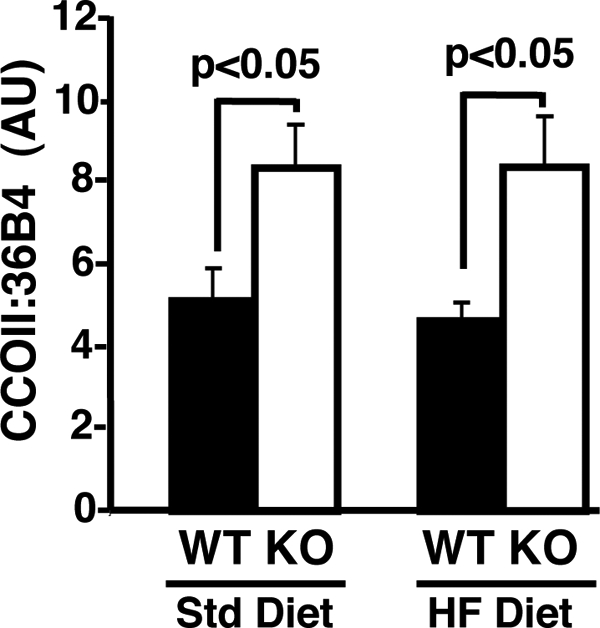
Alterations in mitochondrial DNA in standard and high fat epididymal adipose tissue. Mitochondrial DNA content was assessed by quantitative PCR analyses of the ratio of mitochondrial cytochrome c oxidase subunit II (CCOII) gene copies to nuclear 36B4 gene copies in adipose tissue as described under “Experimental Procedures.” A nearly 2-fold increase (p < 0.05) in mitochondrial DNA was observed in epididymal adipose tissue from iPLA2γ−/− mice on both the standard (Std) and HF diets relative to wild-type littermate controls. In all of the above studies, mouse epididymal adipose tissues were obtained from three 8–10-month old wild-type (WT, closed bars) or iPLA2γ−/− (KO, open bars) mice fed ad libitum on a Std or HF diet. Data are presented as means ± S.E. Statistical significance was performed using the Student's t test, and significant differences are indicated by p values above the brackets. n = 3–4 per group.
To gain insight into adipocyte bioenergetics resulting from the expanded mitochondrial pool in adipose tissue from iPLA2γ−/− mice fed a HF diet, we examined the mRNA expression levels of the three prominent uncoupling proteins UCP1, UCP2, and UCP3. The results demonstrated that although no alterations in UCP1 or UCP2 were manifest, a 4-fold increase in UCP3 was present (p < 0.05) in adipose tissue from iPLA2γ−/− mice fed a HF diet (Table 4). Because UCP3 regulates mitochondrial uncoupling and participates in the regulation of mitochondrial potential, these results suggest that the increased adipocyte β-oxidation present in iPLA2γ−/− mice results from a combination of mitochondrial uncoupling and the increased content of mitochondria in adipocytes of the iPLA2γ−/− mouse.
TABLE 4.
qPCR analyses of UCP1, -2, and -3 mRNA expression in epididymal adipose from wild-type and iPLA2γ−/− mice
qPCR analyses were performed as described under “Experimental Procedures.” Data are expressed as the mean (in arbitrary units) ± S.E. (n = 3 per group). Male mice 8–10 months of age were utilized in these studies. The ratios of iPLA2γ−/− (KO) to wild-type littermate (WT) expression for each gene are presented. Statistical significance was performed using the Student's t test, and significant differences are indicated by their p values.
| WT | KO | KO/WT | p value | |
|---|---|---|---|---|
| UCP1 | 0.006 ± 8E-0.04 | 0.003 ± 9E-0.04 | 0.5 | 0.06 |
| UCP2 | 0.2 ± 0.03 | 0.4 ± 0.08 | 2 | 0.15 |
| UCP3 | 0.01 ± 0.002 | 0.04 ± 0.008 | 4 | 0.02 |
To directly examine the efficiency of adipocyte mitochondrial bioenergetics, we utilized high resolution respirometry with glutamate/malate or palmitoylcarnitine/malate as primary substrates. In the presence of glutamate/malate (state 2), or after addition of ADP (state 3), adipose tissue from iPLA2γ−/− mice demonstrated significantly higher oxygen consumption in comparison with their WT littermates (Fig. 9A). Likewise, increased O2 consumption in adipose explants from iPLA2γ−/− mice was observed following addition of succinate, demonstrating maximal stimulated respiratory capacity using both complex I and II. Increased respiration was also observed under inhibition of complex I (rotenone) and complex V (oligomycin, state 4) as well as under conditions of maximum uncoupling capacity with FCCP (Fig. 9A). Similarly, utilizing palmitoylcarnitine/malate as the initial substrate, substantial increases in respiration were manifest in adipocytes from iPLA2γ−/− mice in the presence of ADP, ADP/succinate, rotenone, and FCCP (Fig. 9B). Thus, the respiratory capacity of iPLA2γ−/− adipose tissue was significantly increased compared with WT adipose tissue utilizing either glutamate/malate or palmitoylcarnitine/malate with no significant change in either the basal leak (state 2/3) or the respiratory control ratio (state 3/4). Furthermore, despite the increased respiration manifest, no significant change in ATP synthesis was observed under these conditions demonstrating the presence of electron transport chain uncoupling. Collectively, these data demonstrate increases in mitochondrial content, fatty acid oxidation, and respiration in adipose tissue from iPLA2γ−/− mice in comparison with their WT littermates that was accompanied by electron transport chain dysfunction.
FIGURE 9.

Increased oxygen consumption in iPLA2γ−/− epididymal adipose tissue. Oxygen consumption in the presence of the indicated mitochondrial substrates and inhibitors was measured in epididymal adipose tissue explants utilizing an Oroboros apparatus as described under “Experimental Procedures.” A, oxygen consumption in the presence of glutamate/malate for wild-type (solid bars) and iPLA2γ−/− (open bars) samples. As a control for differences in mitochondrial number per tissue section, oxygen consumption for each substrate or inhibitor condition was normalized to cytochrome c oxidase oxygen consumption values. Epididymal adipose tissue from the iPLA2γ−/− mice had significantly increased oxygen consumption utilizing either ADP (state 3) or succinate (state 3 max) as substrates and in the presence of inhibitors rotenone (state 3), oligomycin (state 4), and the uncoupler FCCP. B, oxygen consumption in the presence of palmitoylcarnitine/malate. Oxygen consumption was increased in iPLA2γ−/− adipose explants relative to wild-type controls with each substrate or inhibitor addition with statistically significant differences observed in the presence of ADP, succinate, rotenone, and FCCP. C, no significant alterations in state 2/3 were observed in the KO utilizing either glutamate/malate or palmitoylcarnitine/malate. D, no significant alterations in state 3/4 metabolism utilizing either glutamate/malate or palmitoylcarnitine/malate were observed. Male mice 9–11 months of age were utilized. n = 3–4/group. E, ATP synthesis rate in adipose explants. No significant alterations in either glutamate/malate (complex I) or palmitoylcarnitine/malate (complex I) ATP synthesis were observed comparing WT and iPLA2γ−/− adipose explants. Male mice (n = 3–4 per group) on the standard diet were utilized. Data are presented as means ± S.E. Statistical significance was performed using the Student's t test, and significant differences are indicated by brackets and the associated p values. Abbreviations used: palm-carn/malate, palmitoylcarnitine plus malate.
Decreased High Fat Skeletal Muscle Triglyceride Content by Shotgun Lipidomics
To determine the effects of HF feeding on skeletal muscle TAG content in WT and iPLA2γ−/− mice, MDMS-SL was performed. HF feeding of WT mice resulted in a 5-fold increase in TAG content in skeletal muscle (from 106.8 ± 9.2 to 555.7 ± 64 nmol/mg of protein; p < 0.001) (supplemental Fig. 11A). In contrast, skeletal muscle from iPLA2γ−/− mice demonstrated a more modest 2-fold increase in triglycerides after HF feeding (from 124.9 ± 37.2 to 249.7 ± 43.6 nmol/mg; p = 0.06). WT mice contained over twice the amount of triglycerides in skeletal muscle after HF feeding in comparison with skeletal muscle from iPLA2γ−/− mice (p < 0.005) (supplemental Fig. 11A). Comparisons of individual molecular species by MDMS-SL demonstrated no significant differences in skeletal muscle triglyceride molecular species from WT or iPLA2γ−/− mice fed a standard or HF diet (supplemental Fig. 11, B and C).
iPLA2γ−/− Skeletal Muscle Mitochondria Demonstrate Decreased Respiratory Capacity That Is Uncoupled from ATP Production
Assessment of mitochondrial function in iPLA2γ−/− skeletal muscle was performed using high resolution respirometry with mitochondria isolated from skeletal muscle as described under “Experimental Procedures.” Utilizing glutamate/malate or palmitoylcarnitine/malate as substrates, iPLA2γ−/− skeletal muscle mitochondria demonstrated dramatically deficient oxygen consumption following addition of either substrate reflecting multiple complex deficiencies (Fig. 10, A and B). The state 2/3 respiration ratio, reflecting basal uncoupling, was significantly increased in iPLA2γ−/− muscle (Fig. 10C). In contrast, the ratio of state 3/4 (reflecting coupling with ATP production) was significantly reduced using either substrate (Fig. 10D). Supporting this finding, total ATP synthesis was reduced 2-fold in iPLA2γ−/− skeletal muscle mitochondria utilizing either substrate (Fig. 10E). Collectively, these studies demonstrate that iPLA2γ−/− skeletal muscle mitochondria have multiple defects in mitochondrial function, including decreased respiration and severe uncoupling, that collectively result in dramatically reduced ATP generation.
FIGURE 10.

Respiratory studies of skeletal muscle mitochondria from iPLA2γ−/− and wild-type control mice fed the standard diet. Oxygen concentration and flux per volume (respiration) of isolated mitochondria from skeletal muscle was measured per mg of mitochondrial protein utilizing an Oroboros apparatus as described under “Experimental Procedures.” A and B, oxygen consumption in the presence of glutamate/malate (A) and palmitoylcarnitine/malate (B) are shown for wild-type (WT, solid bars) and iPLA2γ−/− (KO, open bars) mitochondria following additions of the indicated substrates and inhibitors. Oxygen consumption was significantly reduced following the addition of each substrate or inhibitor addition in the presence of either glutamate/malate (with the exception of oligomycin) or palmitoylcarnitine/malate in this study. C, significant increase in state 2/3 was observed for the KO utilizing glutamate/malate (p < 0.01), and a smaller but not significant increase was observed for palmitoylcarnitine/malate (p < 0.1). D, bar graphs showing significant reductions in state 3/4 metabolism utilizing glutamate/malate (p < 0.05) and palmitoylcarnitine/malate (p < 0.01). Male mice 9–11 months of age were utilized. n = 3–4/group. p < 0.05. E, ATP synthesis rate for muscle mitochondria. ATP synthesis of mitochondria was determined (mmol/min/mg mitochondrial protein) as described under “Experimental Procedures” for wild-type (WT) and iPLA2γ−/− (KO) samples from approximately 1-year-old male mice (n = 3–4 per group) on the standard diet. 2-Fold reductions in glutamate/malate (p < 0.01, complex I) and palmitoylcarnitine/malate (p < 0.05, complex I) ATP synthesis were observed in the KO. Data are presented as means ± S.E. Statistical significance was performed using the Student's t test, and significant differences are indicated by brackets and the associated p values. Abbreviations: AATA, antimycin A; TMPD, tetramethyl-p-phenylenediamine with ascorbate addition; palm-carn/malate, palmitoylcarnitine plus malate.
Consistent with the notion that decreased mitochondrial oxidative capacity was present in skeletal muscle from iPLA2γ−/− mice, the mRNA levels of multiple markers of fatty acid transport and mitochondrial function (CD36, FABP3, CCOII, FOXOI, ACO, PGC1α, UCP3, porin, PPARα, and PPARδ) were significantly reduced in iPLA2γ−/− muscle in comparison with wild-type littermates (Table 5). Finally, Western analysis demonstrated increased pMAPK protein mass in skeletal muscle from iPLA2γ−/− mice in comparison with WT littermates (supplemental Fig. 12).
TABLE 5.
qPCR analyses from skeletal muscle of wild-type and iPLA2γ−/− mice maintained on the standard diet
qPCR analyses were performed as described under “Experimental Procedures.” Data are presented as the mean (in arbitrary units) ± S.E. (n = 3 per group). Statistical significance was performed using the Student's t test, and significant differences are indicated by their p values. The ratio of iPLA2γ−/− (KO) to wild-type littermate (WT) expression is indicated for each gene.
| Gene | WT | KO | p value | KO/WT |
|---|---|---|---|---|
| CD36 | 0.26 ± 0.05 | 0.10 ± 0.03 | 0.045 | 0.4 |
| FABP3 | 0.09 ± 0.01 | 0.03 ± 0.01 | 0.007 | 0.3 |
| CCOII | 14.43 ± 1.41 | 2.99 ± 0.64 | 0.001 | 0.2 |
| FOXO1 | 0.025 ± 0.003 | 0.012 ± 0.003 | 0.035 | 0.5 |
| ACO | 0.024 ± 0.001 | 0.015 ± 0.002 | 0.014 | 0.6 |
| PGC1α | 0.0216 ± 0.0004 | 0.008 ± 0.002 | 0.001 | 0.4 |
| UCP3 | 0.025 ± 0.004 | 0.004 ± 0.0009 | 0.008 | 0.2 |
| Porin | 4.07 ± 0.24 | 1.74 ± 0.23 | 0.001 | 0.4 |
| PPARα | 0.009 ± 0.001 | 0.004 ± 0.001 | 0.010 | 0.4 |
| PPARγ | 0.0017 ± 0.0004 | 0.0012 ± 0.0004 | 0.386 | 0.7 |
| PPARδ | 0.017 ± 0.002 | 0.0093 ± 0.0005 | 0.020 | 0.5 |
Reduced Skeletal Muscle Cardiolipin Content in iPLA2γ−/− Mice
Previously, the obligatory role of CL in efficient supercomplex formation and optimal electron transport chain efficiency has been demonstrated (35, 36). To determine whether alterations in cardiolipin content or composition contributed to the compromised bioenergetic function in skeletal muscle in iPLA2γ−/− mice, we used enhanced shotgun lipidomics to determine their CL content and molecular species distribution. Total CL content was reduced by 40% (p < 0.05) in iPLA2γ−/− skeletal muscle compared with WT littermates on a standard diet (3.2 ± 0.3 versus 1.9 ± 0.2 nmol/mg of protein; p < 0.05) (Fig. 11, A–C). A smaller decrease in skeletal muscle CL content was present in mice fed a HF diet (Fig. 11C). Comparisons between M + ½ isotopologue profiles identified substantial alterations in the CL molecular species distribution demonstrated by the altered pattern of M + ½ isotopologues present. Reductions in symmetric tetra 18:2 CL were observed on both the standard (0.93 ± 0.12 versus 0.50 ± 0.08 nmol/mg of protein for WT versus iPLA2γ−/−, respectively) as well as the HF diets (0.72 ± 0.14 versus 0.35 ± 0.08 nmol/mg of protein for WT versus iPLA2γ−/−, respectively) (Fig. 11D). In addition, there were marked decreases in the major 22:6 containing CL molecular species (18:2–18:2–18:2–22:6 and 18:1–18:2–18:2–22:6) in the iPLA2γ−/− skeletal muscle in comparison with WT littermates on either the standard diet or the HF diet (Fig. 11E).
FIGURE 11.

Alterations in skeletal CL content. Lipid extracts of wild-type (WT) and iPLA2γ−/− (KO) skeletal muscle from mice on the standard (Std) diet were prepared by a modified Bligh and Dyer procedure. Negative ion electrospray ionization-mass spectra were acquired using a TSQ Quantum Ultra Plus triple-quadrupole mass spectrometer as described under “Experimental Procedures.” Representative spectra for WT (A) and KO (B) were normalized to a T14:0 CL internal standard. The insets show extended mass spectra that display additional internal standards and other major anionic phospholipids. The asterisks indicate the M + ½ isotopologues of the doubly charged CL whose ion peak intensities were utilized to quantify individual CL molecular species as described previously (29). The results illustrate a decrease in total iPLA2γ−/− CL content as well as an altered molecular species distribution. C, quantitation of the total CL content of iPLA2γ−/− skeletal muscle relative to WT control tissue from mice fed either the standard diet (Std Diet) or the HF diets resulted in significant decreases in total CL content in KO muscle on the standard (Std) (p < 0.001) or the HF (p < 0.05) diets. Data analyses combined two separate runs of samples from skeletal muscle (n = 3/group). D, significant decreases in skeletal muscle 18:2–18:2–18:2–18:2 CL ((18:2)4) content in KO relative to WT control tissue were observed on either the standard or the HF diets. E, a significant decrease in the major 22:6 containing CL species (18:2–18:2–18:2–22:6) in the KO was also observed on either the standard or HF diets. WT, solid bars; KO, open bars. Data are presented as means ± S.E. Statistical significance was performed using the Student's t test, and significant differences are indicated by p values above the brackets. n = 3 per group.
Decreased TAG Content in Liver from iPLA2γ−/− Mice
Hepatic TAG content was markedly less in the iPLA2γ−/− mice during the standard diet as well as during HF feeding. Moreover, as observed with skeletal muscle TAG, the increase in hepatic TAG after HF feeding in iPLA2γ−/− mice was substantially less in comparison with WT littermates with little change in molecular species distribution (supplemental Fig. 13). Collectively, these results demonstrate that hepatic TAG content is decreased in iPLA2γ−/− mice and is relatively refractory to increases in TAG accumulation after HF feeding.
Increased Total Cardiolipin Content in iPLA2γ−/− Livers after High Fat Feeding
Total hepatic CL was increased in iPLA2γ−/− mice in comparison with WT liver on either the standard or HF diet, although the distribution of CL species was largely unaltered (supplemental Fig. 14). Standard diet WT and iPLA2γ−/− liver contained 5.57 ± 0.18 and 7.78 ± 0.87 nmol of CL/mg of protein, respectively (p < 0.02), whereas HF diet WT and iPLA2γ−/− liver contained 3.95 ± 0.14 and 5.15 ± 0.18 nmol of CL/mg of protein, respectively (p < 0.02) (supplemental Fig 14C).
DISCUSSION
The results of this study demonstrate the surprising ability of the iPLA2γ−/− mouse to remain refractory to the development of obesity and insulin resistance during prolonged HF feeding. This phenotype could not be explained by either decreased dietary intake or malabsorption of ingested dietary fat. Instead, iPLA2γ−/− mice exhibited multiple cell type-specific alterations in mitochondrial function. First, respiration of mitochondria from skeletal muscle was uncoupled from ATP production that was due, at least in part, to pathologic alterations in CL metabolism resulting from iPLA2γ loss of function. Second, there was an unanticipated increase in epididymal adipocyte oxidative capacity using multiple substrates, which was due to an increase in adipocyte mitochondrial mass, increased levels of UCP3, and increased electron transport chain uncoupling. Third, the iPLA2γ−/− mouse demonstrated marked defects in glucose-stimulated insulin secretion that likely reflect the compromised state of mitochondrial function in pancreatic β cells. Collectively, these alterations protected the iPLA2γ−/− mouse from the downstream sequelae of HF feeding (i.e. obesity, insulin resistance, and hyperinsulinemia) that represent the biochemical progenitors of the metabolic syndrome. Thus, iPLA2γ plays essential upstream roles in integrating organismal responses to changes in substrate supply by mitochondrial uncoupling and cell type-specific alterations in transcriptional programs regulating mitochondrial function and cellular bioenergetics.
Direct examination of skeletal muscle mitochondria isolated from iPLA2γ−/− mice by high resolution respirometry identified a marked decrease in mitochondrial respiration that was accompanied by profound uncoupling. Similarly, analyses of adipocyte explants from iPLA2γ−/− mice demonstrated mitochondrial uncoupling and markedly increased respiration. Thus, mitochondria from the iPLA2γ−/− mouse possess a common bioenergetic defect (uncoupling) that is differentially manifest in each tissue through cell type-specific alterations in transcriptional regulation. For example, PPARγ transcriptional activation is known to result in robust mitochondrial biogenesis and mitochondrial remodeling (37). This study demonstrates that iPLA2γ loss of function results in increased PPARγ mRNA, and more importantly, the activation of its downstream response elements (ACC, FAS, aP2, adiponectin, and SCD1). Through this mechanism, increases in adipocyte mitochondrial mass are realized that compensate for the bioenergetic uncoupling manifest in adipocytes from the iPLA2γ−/− mouse. In sharp contrast, transcriptional regulation of oxidative programs in muscle are sharply down-regulated as demonstrated by the marked decreases in PPARα, PGC1α, CD36, and FOXO1. Thus, although the underlying biochemical mechanism, mitochondrial uncoupling, is the same in these two tissues, tissue-specific transcriptional regulation is a central mechanism contributing to the marked differences in adipocyte and skeletal muscle oxidative metabolism.
Multiple mechanisms exist through which mitochondrial uncoupling can occur, and respiration can be modulated. Prominent among these are alterations in the phospholipid composition of mitochondria and resultant changes in membrane molecular dynamics and surface charge density that modulate mitochondrial function (35, 36, 38). For example, symmetric cardiolipin molecular species provide an optimized membrane environment for supercomplex formation and efficient electron transport chain function (38, 39). Shotgun lipidomics identified large decreases in CL content in the skeletal muscle of iPLA2γ−/− mice that were accompanied by an altered molecular species distribution demonstrating the upstream mechanistic role of iPLA2γ in maintaining physiologic amounts of specific cardiolipin molecular species that optimize mitochondrial function. In addition, alterations in membrane physical properties modulate the kinetics of transmembrane ion channels and ion pumps that modify the pH and chemiosmotic gradients necessary for the efficient production of ATP. Furthermore, phospholipases generate a variety of lipid 2nd messengers that are known to play pivotal roles in mitochondrial bioenergetics. Most prominent among these is the release of fatty acids that are critical modulators of UCP function. The observed cell type-specific alterations in mitochondrial function suggest the possibility that iPLA2γ may provide important signaling molecules that can directly or indirectly modulate mitochondrial function in a cell type-specific fashion.
Previously, we demonstrated that differentiation of NIH 3T3-L1 cells into adipocytes was accompanied by increases in iPLA2γ message and protein and that siRNA directed against iPLA2γ could prevent their hormone-induced differentiation into fat cells (40). In this case, it is clear that the absence of increased adiposity in the epididymal fat pads was largely due to the absence of adipocyte hypertrophy. However, we cannot exclude potential decreases in adipocyte hyperplasia, especially in the abdomen where visible fat pads were nearly completely absent.
Many aspects of the physiologic regulation of adipocyte triglyceride metabolism were intact in iPLA2γ−/− mice as demonstrated by the robust increase in triglyceride hydrolysis by isoproterenol. In addition, qPCR demonstrated decreases in the mRNA levels of the adipocyte triglyceride lipases PNPLA2 and PNPLA3, which likely reflect an adaptive response to decreased triglyceride storage in triglyceride-depleted adipocytes in the iPLA2γ−/− mouse.
In early work, we demonstrated the importance of ATP in regulating calcium-independent phospholipase A2 activity thereby identifying an important role for this activity in cellular bioenergetics (41, 42). Detailed examination of pancreatic β cells identified the presence of iPLA2 activity that was robustly inhibited by the mechanism-based inhibitor (E)-6-(bromomethylene)-3-(1-naphthalenyl)-2H-tetrahydropyran-2-one (BEL). Moreover, we demonstrated that iPLA2 activity was necessary for the release of insulin from pancreatic β cells because inhibition of activity by BEL markedly attenuates insulin release that has now been confirmed through multiple genetic models (43–47). However, recent work has identified nine members of the iPLA2 family that are each inhibitable by BEL (48, 49) requiring investigation of the interplay of specific members of the iPLA2 family in glucose-stimulated insulin release (50, 51). Recent work using the genetic ablation of iPLA2β demonstrated a role of this enzyme in mediating glucose-induced insulin release after HF feeding or after repeated challenge with streptozotocin (47, 52). One of the most prominent characteristics of the iPLA2γ−/− mouse is its profound defect in glucose-stimulated insulin secretion.
The role of mitochondrial production of ATP in regulating glucose-induced insulin release has long been appreciated. In addition, the role of mitochondrial dysfunction as a key factor in mediating the development of type 2 diabetes has become increasingly recognized and is underscored by genetic alterations in many mitochondrial proteins that induce mitochondrial dysfunction accompanied by decreased glucose-stimulated insulin release (53). Although mitochondrial respiration in β cells cannot be assessed directly in the mouse, mitochondrial uncoupling is present in multiple tissues in the iPLA2γ−/− mouse. Thus, it seems likely that mitochondrial uncoupling in the β cell and the resultant decrease in glucose-stimulated ATP production is the likely mechanism for decreased insulin release in the iPLA2γ−/− mouse.
Although ablation of iPLA2γ results in multiple alterations of adipose and skeletal lipid composition, in contrast, alterations in the liver are much more modest and do not appear to be a major contributor to the observed phenotype. Specifically, no decreases in hepatic CL content or molecular species composition were observed with the standard diet or after HF feeding. Furthermore, no alterations in serum hepatic enzymes or ketone body production were detected. Collectively, these results suggest that the majority of the observed alterations in the phenotype of the iPLA2γ−/− mouse were not due to intrinsic alterations in hepatic function.
Many previous studies have stressed the importance of “cross-talk” between adipose and muscle tissue as an important key to understanding the etiology of obesity and the metabolic syndrome (54, 55). Although these studies have focused on the importance of increased mitochondrial fatty acid oxidation as the critical determinant in preventing ectopic lipid deposition and the concomitant development of insulin resistance (56–58), it is now clear that accumulation of triglycerides is not an obligatory precursor of insulin resistance or lipotoxicity. Instead, some recent studies have suggested that increased mitochondrial oxidation during HF feeding is responsible for activation of pathways that predispose to insulin resistance, including the activation of stress kinases by reactive oxygen species (59–61). The prevention of insulin resistance in the face of decreased skeletal muscle mitochondrial oxidation found in this study is consistent with the notion that overload of mitochondrial oxidation results in the production of toxic intermediates that contribute to insulin resistance.
While this manuscript was under review, Song et al. (62) also showed that the iPLA2γ−/− mouse that we previously generated and characterized (16) is resistant to diet-induced obesity and the development of high fat-induced hyperinsulinemia. The results of this study go on to identify the mechanism of resistance to diet-induced obesity as mitochondrial uncoupling and tissue-specific alterations in transcriptional regulation. More specifically, we demonstrate the activation of PPARγ and its downstream targets that lead to an increase in oxidative transcriptional programs in the adipocyte resulting in an increase in mitochondrial mass, increased fatty acid oxidation, and increased mitochondrial uncoupling. In sharp contrast, regulation of oxidative transcriptional programs in muscle was markedly down-regulated as demonstrated by the decrease in PPARα, PPARδ, PGC1α, and CD36. In addition, this study utilizes an enhanced shotgun lipidomics approach employing M + ½ isotopologues for accurate measurements of cardiolipin content and molecular species composition. The results of this study demonstrate a 40% decrease in skeletal muscle total cardiolipin content and a 50% decrease in symmetric tetra 18:2 cardiolipin molecular species that are essential in facilitating efficient coupling during oxidative phosphorylation.
In summary, the iPLA2γ−/− mouse is refractory to the development of obesity, insulin resistance, and hyperinsulinemia after HF feeding that results from mitochondrial uncoupling in conjunction with cell type-specific alterations in transcriptional oxidative programs. Through the use of high resolution respirometry and enhanced shotgun lipidomics, the central mechanism leading to mitochondrial uncoupling was identified as altered cardiolipin composition resulting from iPLA2γ loss of function. Thus, this study represents the first mechanistic demonstration of the obligatory upstream role of iPLA2γ in facilitating optimal electron transport chain function and coupling to ATP production in mitochondria.
Supplementary Material
Acknowledgment
The access to equipment for the DEXA and Oxymax analyses was provided by National Institutes of Health Grant P30 DK56341 from the NIDDK.
This work was supported, in whole or in part, by National Institutes of Health Grants 5PO1HL57278 and RO1HL41250. This work was also supported by Clinical Nutrition Research Unit Grant DK56351 and Scientist Development Grant 835140N from the American Heart Association (to X. S.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–14 and additional references.
- HF
- high fat
- iPLA2
- calcium-independent phospholipase A2
- iPLA2γ
- calcium-independent phospholipase A2γ
- BEL
- (E)-6-(bromomethylene)-3-(1-naphthalenyl)-2H-tetrahydropyran-2-one
- pAKT
- phospho-AKT
- p
- phospho-
- TAG
- triglyceride
- CL
- cardiolipin
- FCCP
- carbonyl cyanide p-trifluoromethoxyphenylhydrazone
- qPCR
- quantitative PCR
- DEXA
- dual-energy x-ray absorptiometry
- MDMS-SL
- multidimensional mass spectrometry-based Shotgun lipidomics
- ACC
- acetyl-CoA carboxylase
- FAS
- fatty-acid synthase
- PPARγ
- peroxisome proliferator-activated receptor γ.
REFERENCES
- 1.Moller D. E., Kaufman K. D. (2005) Annu. Rev. Med. 56, 45–62 [DOI] [PubMed] [Google Scholar]
- 2.Després J. P., Lemieux I. (2006) Nature 444, 881–887 [DOI] [PubMed] [Google Scholar]
- 3.Jo J., Gavrilova O., Pack S., Jou W., Mullen S., Sumner A. E., Cushman S. W., Periwal V. (2009) PLoS Comput. Biol. 5, e1000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huss J. M., Kelly D. P. (2005) J. Clin. Invest. 115, 547–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kinsey G. R., McHowat J., Beckett C. S., Schnellmann R. G. (2007) Am. J. Physiol. Renal Physiol. 292, F853–F860 [DOI] [PubMed] [Google Scholar]
- 6.Gadd M. E., Broekemeier K. M., Crouser E. D., Kumar J., Graff G., Pfeiffer D. R. (2006) J. Biol. Chem. 281, 6931–6939 [DOI] [PubMed] [Google Scholar]
- 7.Mancuso D. J., Jenkins C. M., Sims H. F., Cohen J. M., Yang J., Gross R. W. (2004) Eur. J. Biochem. 271, 4709–4724 [DOI] [PubMed] [Google Scholar]
- 8.Williams S. D., Gottlieb R. A. (2002) Biochem. J. 362, 23–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brustovetsky T., Antonsson B., Jemmerson R., Dubinsky J. M., Brustovetsky N. (2005) J. Neurochem. 94, 980–994 [DOI] [PubMed] [Google Scholar]
- 10.Seleznev K., Zhao C., Zhang X. H., Song K., Ma Z. A. (2006) J. Biol. Chem. 281, 22275–22288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yan W., Jenkins C. M., Han X., Mancuso D. J., Sims H. F., Yang K., Gross R. W. (2005) J. Biol. Chem. 280, 26669–26679 [DOI] [PubMed] [Google Scholar]
- 12.Lee C. H., Olson P., Hevener A., Mehl I., Chong L. W., Olefsky J. M., Gonzalez F. J., Ham J., Kang H., Peters J. M., Evans R. M. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 3444–3449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wanders R. J., Waterham H. R. (2006) Annu. Rev. Biochem. 75, 295–332 [DOI] [PubMed] [Google Scholar]
- 14.Fournier B., Smeitink J. A., Dorland L., Berger R., Saudubray J. M., Poll-The B. T. (1994) J. Inherited Metab. Dis. 17, 470–486 [DOI] [PubMed] [Google Scholar]
- 15.Mancuso D. J., Jenkins C. M., Gross R. W. (2000) J. Biol. Chem. 275, 9937–9945 [DOI] [PubMed] [Google Scholar]
- 16.Mancuso D. J., Sims H. F., Han X., Jenkins C. M., Guan S. P., Yang K., Moon S. H., Pietka T., Abumrad N. A., Schlesinger P. H., Gross R. W. (2007) J. Biol. Chem. 282, 34611–34622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Y., Zhou D., Abumrad N. A., Su X. (2010) Am. J. Physiol. Cell Physiol. 298, C921–C928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han X., Cheng H., Mancuso D. J., Gross R. W. (2004) Biochemistry 43, 15584–15594 [DOI] [PubMed] [Google Scholar]
- 19.Han X., Yang J., Yang K., Zhao Z., Abendschein D. R., Gross R. W. (2007) Biochemistry 46, 6417–6428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tchoukalova Y. D., Harteneck D. A., Karwoski R. A., Tarara J., Jensen M. D. (2003) J. Lipid Res. 44, 1795–1801 [DOI] [PubMed] [Google Scholar]
- 21.Nassir F., Wilson B., Han X., Gross R. W., Abumrad N. A. (2007) J. Biol. Chem. 282, 19493–19501 [DOI] [PubMed] [Google Scholar]
- 22.Hajri T., Han X. X., Bonen A., Abumrad N. A. (2002) J. Clin. Invest. 109, 1381–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hajri T., Hall A. M., Jensen D. R., Pietka T. A., Drover V. A., Tao H., Eckel R., Abumrad N. A. (2007) Diabetes 56, 1872–1880 [DOI] [PubMed] [Google Scholar]
- 24.Nahlé Z., Hsieh M., Pietka T., Coburn C. T., Grimaldi P. A., Zhang M. Q., Das D., Abumrad N. A. (2008) J. Biol. Chem. 283, 14317–14326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang K., Cheng H., Gross R. W., Han X. (2009) Anal. Chem. 81, 4356–4368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han X., Yang K., Gross R. W. (2008) Rapid Commun. Mass Spectrom. 22, 2115–2124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Han X., Gross R. W. (2005) Expert Rev. Proteomics 2, 253–264 [DOI] [PubMed] [Google Scholar]
- 28.Han X., Yang J., Cheng H., Ye H., Gross R. W. (2004) Anal. Biochem. 330, 317–331 [DOI] [PubMed] [Google Scholar]
- 29.Han X., Yang K., Yang J., Cheng H., Gross R. W. (2006) J. Lipid Res. 47, 864–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cannon B., Nedergaard J. (2001) Methods Mol. Biol. 155, 295–303 [DOI] [PubMed] [Google Scholar]
- 31.Sahlin K., Mogensen M., Bagger M., Fernström M., Pedersen P. K. (2007) Am. J. Physiol. Endocrinol. Metab. 292, E223–E230 [DOI] [PubMed] [Google Scholar]
- 32.Rasmussen U. F., Vielwerth S. E., Rasmussen H. N. (2004) Comp. Biochem. Physiol. A Mol. Integr. Physiol. 137, 435–446 [DOI] [PubMed] [Google Scholar]
- 33.Jüllig M., Hickey A. J., Chai C. C., Skea G. L., Middleditch M. J., Costa S., Choong S. Y., Philips A. R., Cooper G. J. (2008) Proteomics 8, 2556–2572 [DOI] [PubMed] [Google Scholar]
- 34.Chakravarthy M. V., Zhu Y., López M., Yin L., Wozniak D. F., Coleman T., Hu Z., Wolfgang M., Vidal-Puig A., Lane M. D., Semenkovich C. F. (2007) J. Clin. Invest. 117, 2539–2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang M., Mileykovskaya E., Dowhan W. (2002) J. Biol. Chem. 277, 43553–43556 [DOI] [PubMed] [Google Scholar]
- 36.Zhang M., Mileykovskaya E., Dowhan W. (2005) J. Biol. Chem. 280, 29403–29408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilson-Fritch L., Nicoloro S., Chouinard M., Lazar M. A., Chui P. C., Leszyk J., Straubhaar J., Czech M. P., Corvera S. (2004) J. Clin. Invest. 114, 1281–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Malhotra A., Edelman-Novemsky I., Xu Y., Plesken H., Ma J., Schlame M., Ren M. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 2337–2341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu Y., Zhang S., Malhotra A., Edelman-Novemsky I., Ma J., Kruppa A., Cernicica C., Blais S., Neubert T. A., Ren M., Schlame M. (2009) J. Biol. Chem. 284, 29230–29239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Su X., Mancuso D. J., Bickel P. E., Jenkins C. M., Gross R. W. (2004) J. Biol. Chem. 279, 21740–21748 [DOI] [PubMed] [Google Scholar]
- 41.Hazen S. L., Stuppy R. J., Gross R. W. (1990) J. Biol. Chem. 265, 10622–10630 [PubMed] [Google Scholar]
- 42.Hazen S. L., Gross R. W. (1991) J. Biol. Chem. 266, 14526–14534 [PubMed] [Google Scholar]
- 43.Gross R. W., Ramanadham S., Kruszka K. K., Han X., Turk J. (1993) Biochemistry 32, 327–336 [DOI] [PubMed] [Google Scholar]
- 44.Ramanadham S., Bohrer A., Mueller M., Jett P., Gross R. W., Turk J. (1993) Biochemistry 32, 5339–5351 [DOI] [PubMed] [Google Scholar]
- 45.Ramanadham S., Gross R. W., Han X., Turk J. (1993) Biochemistry 32, 337–346 [DOI] [PubMed] [Google Scholar]
- 46.Bao S., Bohrer A., Ramanadham S., Jin W., Zhang S., Turk J. (2006) J. Biol. Chem. 281, 187–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bao S., Jacobson D. A., Wohltmann M., Bohrer A., Jin W., Philipson L. H., Turk J. (2008) Am. J. Physiol. Endocrinol. Metab. 294, E217–E229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jenkins C. M., Han X., Mancuso D. J., Gross R. W. (2002) J. Biol. Chem. 277, 32807–32814 [DOI] [PubMed] [Google Scholar]
- 49.Jenkins C. M., Mancuso D. J., Yan W., Sims H. F., Gibson B., Gross R. W. (2004) J. Biol. Chem. 279, 48968–48975 [DOI] [PubMed] [Google Scholar]
- 50.Ramanadham S., Song H., Bao S., Hsu F. F., Zhang S., Ma Z., Jin C., Turk J. (2004) Diabetes 53, S179–S185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Song K., Zhang X., Zhao C., Ang N. T., Ma Z. A. (2005) Mol. Endocrinol. 19, 504–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bao S., Song H., Wohltmann M., Ramanadham S., Jin W., Bohrer A., Turk J. (2006) J. Biol. Chem. 281, 20958–20973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lu H., Koshkin V., Allister E. M., Gyulkhandanyan A. V., Wheeler M. B. (2010) Diabetes 59, 448–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Muoio D. M., Newgard C. B. (2006) Annu. Rev. Biochem. 75, 367–401 [DOI] [PubMed] [Google Scholar]
- 55.Argilés J. M., López-Soriano J., Almendro V., Busquets S., López-Soriano F. J. (2005) Med. Res. Rev. 25, 49–65 [DOI] [PubMed] [Google Scholar]
- 56.Lettner A., Roden M. (2008) Curr. Diab. Rep. 8, 185–191 [DOI] [PubMed] [Google Scholar]
- 57.Szendroedi J., Schmid A. I., Meyerspeer M., Cervin C., Kacerovsky M., Smekal G., Gräser-Lang S., Groop L., Roden M. (2009) Diabetes Care 32, 677–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Szendroedi J., Roden M. (2009) Curr. Opin. Lipidol. 20, 50–56 [DOI] [PubMed] [Google Scholar]
- 59.Nakamura S., Takamura T., Matsuzawa-Nagata N., Takayama H., Misu H., Noda H., Nabemoto S., Kurita S., Ota T., Ando H., Miyamoto K., Kaneko S. (2009) J. Biol. Chem. 284, 14809–14818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kennedy A., Overman A., Lapoint K., Hopkins R., West T., Chuang C. C., Martinez K., Bell D., McIntosh M. (2009) J. Lipid Res. 50, 225–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gupte A. A., Bomhoff G. L., Morris J. K., Gorres B. K., Geiger P. C. (2009) J. Appl. Physiol. 106, 1425–1434 [DOI] [PubMed] [Google Scholar]
- 62.Song H., Wohltmann M., Bao S., Ladenson J. H., Semenkovich C. F., Turk J. (2010) Am. J. Physiol. Endocrinol. Metab. 298, E1097–E1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.