

Abstract
The prion protein (PrPC) is a conserved glycosylphosphatidylinositol-anchored cell surface protein expressed by neurons and other cells. Stress-inducible protein 1 (STI1) binds PrPC extracellularly, and this activated signaling complex promotes neuronal differentiation and neuroprotection via the extracellular signal-regulated kinase 1 and 2 (ERK1/2) and cAMP-dependent protein kinase 1 (PKA) pathways. However, the mechanism by which the PrPC-STI1 interaction transduces extracellular signals to the intracellular environment is unknown. We found that in hippocampal neurons, STI1-PrPC engagement induces an increase in intracellular Ca2+ levels. This effect was not detected in PrPC-null neurons or wild-type neurons treated with an STI1 mutant unable to bind PrPC. Using a best candidate approach to test for potential channels involved in Ca2+ influx evoked by STI1-PrPC, we found that α-bungarotoxin, a specific inhibitor for α7 nicotinic acetylcholine receptor (α7nAChR), was able to block PrPC-STI1-mediated signaling, neuroprotection, and neuritogenesis. Importantly, when α7nAChR was transfected into HEK 293 cells, it formed a functional complex with PrPC and allowed reconstitution of signaling by PrPC-STI1 interaction. These results indicate that STI1 can interact with the PrPC·α7nAChR complex to promote signaling and provide a novel potential target for modulation of the effects of prion protein in neurodegenerative diseases.
Keywords: Heat Shock Protein, Neuron, Nicotinic Acetylcholine Receptors, Prions, Signal Transduction
Introduction
Prions are the infectious components of transmissible spongiform encephalopathies that can self-perpetuate by imprinting an anomalous conformation onto a glycosylphosphatidylinositol-anchored host protein known as the prion protein (PrPC).3 Conversion of PrPC to the protease-resistant prion is thought to be a major event in prion diseases (1). However, despite the wealth of knowledge on prions, the roles of PrPC on synaptic function and neuronal health are still not completely understood. Studies in yeast, mammalian cells, and mouse models support the hypothesis that PrPC plays a major role in neuroprotection and neuronal differentiation (for review, see Refs. 1–3).
In humans, the initial cognitive dysfunction observed in transmissible spongiform encephalopathies is remarkably similar to those observed in Alzheimer disease (AD). Therefore, synaptic failure is likely to play a major role in the cognitive alterations seen in these neurological disorders (4). For instance, in AD, the role for Aβ ligands in synaptic dysfunction has received considerable attention (5, 6). Interestingly, recent work provided evidence that PrPC functions as a receptor for Aβ and that this interaction mediates some of the effects of Aβ oligomers in synaptic plasticity (7) although these results are controversial at the moment (8, 9). Moreover, PrPC seems to regulate the β-secretase cleavage of amyloid precursor protein, thereby regulating the production of Aβ (10). In addition, α-secretase regulates the cleavage of PrPC, generating an N-terminal fragment with neuroprotective activity (11, 12).
PrPC also binds to transmembrane proteins such as the 67-kDa laminin receptor (13–15), neuronal cell adhesion molecule (16, 17), G protein-coupled serotonergic receptors (18), and low density lipoprotein receptor-related protein 1 (19, 20), which are able to promote intracellular signaling-mediated neuronal adhesion and differentiation as well as PrPC internalization. Remarkably, PrPC functions as a receptor or co-receptor for extracellular matrix proteins such as laminin (21, 22) and vitronectin (23), as well as the secreted co-chaperone stress-inducible protein 1 (STI1) (24). These data suggest that glycosylphosphatidylinositol-anchored PrPC is a potential scaffold receptor in a multiprotein, cell surface, signaling complex, that may be the basis for the multiple neuronal functions ascribed to PrPC (2, 3, 25).
We demonstrated previously that PrPC amino acids 113–128 constitute the STI1 binding site (24). Furthermore, PrPC-STI1 engagement rescues retinal and hippocampal neurons from staurosporine-induced programmed cell death through activation of protein kinase A (PKA). Additionally, PrPC-STI1 binding also induces the differentiation and protein synthesis in hippocampal neurons via extracellular signal-regulated kinase 1 and 2 (ERK1/2) and phosphoinositide 3-kinase (PI3K)–Akt-mTOR activation (26, 27). Trafficking of both PrPC and STI1 following binding at the membrane regulates the ERK1/2 pathway, but does not alter PrPC regulation of PKA signaling (28).
Although several signaling pathways triggered by PrPC have been identified, little is known regarding how intracellular signaling is activated by PrPC following its interaction with extracellular ligands. In this study we mapped the upstream signaling events triggered by PrPC-STI1 binding to identify the putative transmembrane protein responsible for connecting this extracellular complex to the intracellular milieu. Our data show that hippocampal neuronal signaling induced by PrPC-STI1 is dependent on calcium influx through the α7 nicotinic acetylcholine receptor (α7nAChR). These results provide a novel mechanism by which PrPC transduces extracellular signals, with implications for PrPC-mediated regulation of synaptic function and neuronal differentiation.
EXPERIMENTAL PROCEDURES
Reagents
Mouse recombinant STI1 (His6-STI1) and an STI1 deletion mutant lacking the PrPC binding site (STI1Δ230–245) were purified as described previously (24). The mitogen-activated protein kinase MAPK/ERK1/2 (p44/p42 MAPK) inhibitor 1,4-diamino-2,3-dicyano-1,4-bis (2-aminophenylthio) butadiene (U0126) was purchased from Promega. The PKA inhibitor KT5720, phosphodiesterase inhibitor isobutylmethylxanthine, and 4′-6-diamidino-2-phenylindole (DAPI) were purchased from Calbiochem. The PKA activator (forskolin) was purchased from LC Laboratories (Woburn, MA). The L-type calcium channel blocker (nifedipine), Ca2+-ATPase inhibitor (thapsigargin, THG), FLAG beads, FLAG peptide, and anti-FLAG antibodies were obtained from Sigma-Aldrich. The Q-type Ca2+ channel blocker (ω-conotoxin MCVII) was purchased from Latoxan (Valence, France). The store-operated calcium channel blocker (SKF-96365) and α7nAChR blocker (α-bungarotoxin, αBgt) were purchased from Tocris Biociences (Ellisville, MO). Fluo-3 AM, neurobasal medium, Opti-MEM, Hanks' buffered saline solution, and Lipofectamine were obtained from Invitrogen. Anti-phospho-ERK1/2 and anti-total ERK1/2 antibodies were purchased from Cell Signaling Technology (Beverly, MA), and peroxidase-coupled goat anti-rabbit secondary antibody and enhanced chemiluminescence (ECL) solution were obtained from GE Healthcare. Complete Protease Inhibitor Mixture was purchased from Roche Applied Science. The PKA assay system kit was purchased from Upstate Biotechnology. Anti-cleaved caspase-3, Alexa Fluor 488 anti-rabbit antibodies, and Alexa Fluor 647 αBgt were purchased from Molecular Probes. Immu-Mount was purchased from Thermo Scientific (Waltham, MA). The monoclonal antibodies 3F4 and 6H4 against PrPC were acquired from Abcam (Cambridge, MA) and Prionics (Schlieren, Switzerland).
Animals and Primary Neuronal Cultures
PrPC-null mice (Prnp0/0), descendants of the ZrchI line, were provided by Dr. Charles Weissmann (Scripps Florida). Wild-type animals were generated by crossing F1 descendants from 129/SV and C57BL/6J matings. All studies were conducted in accordance with National Institutes of Health guidelines for the care and use of animals and with animal protocols approved by the Institutional Animal Care and Use Committee.
Primary cultures of hippocampal neurons from embryonic day 17 (E17) wild-type (Prnp+/+) or PrPC-null mice (Prnp0/0) were obtained as previously described (26) and plated onto poly-l-lysine (5 μg/ml)-coated coverslips (35 mm) for 4 days.
Ca2+ Signaling and Data Analysis
Neurons and HEK 293 cells were loaded with 10 μm intracellular Ca2+ indicator Fluo-3 AM for 30 min at 37 °C in the presence of neurobasal medium or Opti-MEM supplemented with 2 mm CaCl2. Cells were washed three times with Hanks' buffered saline solution and resuspended in Krebs buffer (124 mm NaCl, 4 mm KCl, 25 mm Hepes, 1.2 mm MgSO4, 10 mm glucose) supplemented with 2 mm CaCl2. Ca2+-free experiments were performed in Krebs buffer without CaCl2 plus 1 mm EGTA. Cells were preincubated (30 min) in the presence of 25 μm SKF-96365, voltage-gated calcium channel (VGCC) inhibitors (1 μm ω-conotoxin MCVII plus 50 μm nifedipine) or 1 nm αBgt. Cells were treated with recombinant STI1 (1 μm for neurons and 2 μm for HEK 293), which mimics the effects of STI1 secreted from astrocytes (28, 29). HEK 293 cells were also pretreated with monoclonal antibodies against PrPC (6H4 and 3F4 both at 10 μg/ml) followed by recombinant STI1 (2 μm). In addition, recombinant STI1 deleted of the PrPC binding site, STI1Δ230–245 (2 μm) (26) was used in control experiments, and treatment with THG (1 μm) was performed to estimate calcium responses. Data acquisition was performed by confocal microscope using either a Bio-Rad Radiance 2100/Nikon (TE2000U) or a Zeiss LSM 510 (Zeiss, Toronto, ON) with excitation at 488 nm (argon laser) and emission collected with bandpass filter at 522–535 nm. The fluorescence was normalized as F1/F0 (F1, maximal fluorescence after drug addition and F0, basal fluorescence before drug addition). Software-based analysis (WCIF ImageJ (National Institutes of Health)) allowed quantification of fluorescence imaging in selected cells as a function of time. Experiments were carried out with at least three different cell cultures, and 40–50 cells were monitored in each experiment. Traces represent typical single-cell responses.
ERK1/2 Activity
Primary hippocampal neurons (1 × 106 cells) from Prnp+/+ mice were plated on dishes pretreated with poly-l-lysine and stimulated with STI1 (350 nm) for 1 min (26) in the presence or absence of 2 mm CaCl2. Some cells were also preincubated with 1 nm αBgt for 30 min prior to STI1 treatment. Cells were rinsed with ice-cold phosphate-buffered saline (PBS) and lysed in Laemmli buffer. Cell extracts were subjected to SDS-PAGE (10%), and proteins were transferred onto nitrocellulose membranes. The membranes were blocked (5% milk, 0.1% Tween 20 in Tris-buffered saline) for 1 h at room temperature, incubated with anti-phospho-ERK1/2 or anti-total ERK1/2 antibodies (1:2,000) overnight at 4 °C, followed by incubation with peroxidase-coupled, goat anti-rabbit secondary antibody (1:2,000) for 1 h at room temperature. Reactions were developed using ECL solution, and the bands obtained after x-ray film exposure to the membranes were analyzed by densitometric scanning and quantified using ImageJ software. Alternatively, in some experiments, a CCD-based system was used (Alpha Innotech). ERK1/2 band densities activity was quantified as a relative value representing the ratio between phospho-ERK1/2 and total ERK1/2 for each sample. Results represent five independent experiments for neurons and three separate experiments for HEK 293 cells.
PKA Activity
Primary hippocampal neurons (1 × 106 cells) were preincubated with 100 μm isobutylmethylxanthine for 1 h at 37 °C, followed by incubation with 1 μm STI1 in the presence or absence of extracellular CaCl2 (2 mm) or forskolin (10 μm) for 20 min at 37 °C as a positive PKA signaling control. Cells were washed with cold PBS and homogenized in ice-cold extraction buffer (150 mm NaCl, 20 mm MgCl2, 1% Triton X-100, and 25 mm Tris-HCl, pH 7.4), including Complete Protease Inhibitor mixture. Cellular debris was removed by centrifugation at 6,000 × g for 10 min. PKA activity was determined by [γ-32P]ATP incorporation to a PKA-specific substrate provided by the PKA assay system kit.
Neuritogenesis Assays
Primary hippocampal neurons (4 × 104) from wild-type (Prnp+/+) mice were treated with STI1 (350 nm) and incubated for 24 h at 37 °C. Neuritogenesis mediated by STI1 was evaluated after preincubation with 10 nm αBgt for 30 min. The cells were fixed with 4% paraformaldehyde and 0.12 m sucrose in PBS, pH 7.4, for 20 min at room temperature, washed three times with PBS, and stained with hematoxylin.
Morphometric analyses were performed using ImageJ software and the Neuron J plug in. The parameters analyzed were percentage of cells with neurites and percentage of neurons with neurites longer than 30 μm, which represents three or more times the average cell body. Approximately 200 cells were analyzed per sample.
Cell Death Assay
Primary hippocampal cultures (7 × 104 cells) were treated with STI1 (1.2 μm) for 1 h, followed by staurosporine (50 nm) treatment for 16 h as described previously (26). Alternatively, αBgt (10 nm) was added to the cultures 30 min prior to incubation with STI1. Cells were then fixed with 4% paraformaldehyde and 0.12 m sucrose in PBS, pH 7.4, for 20 min, and immunofluorescence reactions were performed to detect cleaved caspase-3. Briefly, cells were permeabilized and blocked with PBS/0.2% Triton X-100 plus 5% BSA for 1 h and subsequently incubated with anti-cleaved caspase-3 primary antibody (1:200) diluted in PBS/0.2% Triton X-100 plus 1% BSA for 2 h. After rinsing with PBS, cells were incubated with Alexa Fluor 488 anti-rabbit antibody and DAPI for 1 h. Immunolabeled cells were imaged with a BX61 Olympus Fluorescence microscope. Cell death induced by staurosporine was addressed by counting the percentage of cleaved caspase-3-positive cells. Results represent three independent experiments; at least 300 cells total were counted per condition.
DNA Constructs
The cDNA encoding the human α7nAChR subunit was cloned from a human universal QUICK-Clone cDNA library (Clontech) using the forward primer, 5′-CGACAGCCGAGACGTGGA-3′ and reverse primer, 5′-CCGATGGTACGGATGT GC-3′, designed to prime from the untranslated regions of the sequence (NCBI Reference Sequence: NM_000746). A forward primer, introducing a BamHI restriction site and minimal Kozak sequence (5′-GCCGGGATCCGCCACCATGCGCTGCTCGCCGGGA-3′) to the N terminus, and a reverse primer, introducing an XbaI restriction site, FLAG epitope tag sequence (DYKDDDDK) and stop codon (5′-CGGCTCTAGATTACTTGTCGTCGTCGTCCTTATAGTCCGCAAAGTTTTGGACACGGCC-3′) to the C terminus, were used to clone the FLAG-α7nAChR sequence into the pcDNA3.1 expression vector (Invitrogen).
The cDNA for human resistance to inhibitors of cholinesterase 3 homolog (Caenorhabditis elegans) (hRIC3; NCBI Reference Sequence: NM_024557.2) was purchased from OriGene (Rockville, MD). A forward primer, introducing a KpnI restriction site and minimal Kozak sequence (5′-GGCGGTACCGCCACCATGGCGTACTCCACAGTGCAGAGAGTC-3′) to the N terminus, and a reverse primer, introducing a NotI restriction site, HA epitope tag sequence (YPYDVPDYA) and stop codon (5′-GCGGCCGCCAACTCGCATAATCCCACATCATACGGATACTCTAAACCCTGGGGGTTACGCTTCCT-3′) to the C terminus, were used to clone the HA-hRIC3 sequence into the pcDNA5/FRT expression vector (Invitrogen).
HEK 293 Cell Transfection
Cells were transfected with plasmids expressing FLAG-α7nAChR with or without those expressing HA-hRIC 3 (1:1), using either a modified calcium phosphate method (30, 31) or Lipofectamine. To test transfection efficiency, cells transferred to coverslips were incubated with 500 nm Alexa Fluor 647-αBgt in Hanks' buffered saline solution containing 0.1% BSA for 1 h on ice. For confocal microscopy analyses, cells were washed with Hanks' buffered saline solution, fixed with PLP (0.2% periodate, 1.4% lysine, 2% paraformaldehyde), and coverslips were mounted onto glass microscope slides with Immu-Mount. Images are single z-sections captured by an LSM 510 Meta laser scanning microscope with excitation at 633 nm (HeNe laser) and a 650–710-nm bandpass emission filter.
Co-immunoprecipitation Assays
HEK 293 cells were transfected with plasmids expressing mouse PrPC bearing the 3F4 epitope (26) and FLAG-α7nAChR with or without HA-RIC 3 (1:1) cDNAs, as described above. Two days after transfection, 2–3 × 106 cells were lysed in 250 ml of 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, and 1% Triton X-100, plus protease inhibitors for 15 min on ice, followed by two sonication pulses. Protein extracts (800 μg) were incubated with 20 μl of FLAG beads for 16 h at 4 °C. Beads were washed three times with wash buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, pH 7.4, and proteins were eluted by incubating the beads with 15 μg of FLAG peptide (in 100 μl of 50 mm Tris-HCl, pH 7.4, and 150 mm NaCl) for 10 min at room temperature. One quarter of the eluate was resolved by 4–12% SDS-PAGE, followed by immunoblotting with anti-FLAG antibodies. The remaining eluate was resolved with a second 4–12% SDS-PAGE, followed by immunoblotting with anti-PrPC (3F4). These experiments were repeated four times.
Statistical Analysis
The statistical analyses were performed using GraphPad Prism 4. Results are represented as means ± S.E., and the total number of experiments is specified in each figure legend. Data were compared by one-way ANOVA and Newman-Keuls post test.
RESULTS
PrPC-STI1 Interaction Increases Intracellular Ca2+
We demonstrated previously that PrPC-STI1 engagement resulted in ERK1/2 phosphorylation and PKA activation, which in turn promoted neuritogenesis and neuroprotection, respectively (26). Ca2+ signaling is an early event upstream of both PKA and ERK1/2 activation (32, 33). In addition, PrPC has consistently been shown to modulate Ca2+ signaling (34–36). Therefore, to determine whether STI1 interaction with PrPC affects intracellular Ca2+ levels, we performed Ca2+ imaging experiments on cultured hippocampal neurons using Fluo-3 AM. STI1 treatment promoted intracellular Ca2+ increases in neurons derived from Prnp+/+ mice (Fig. 1, A and D), but no effect was observed in neurons from Prnp0/0 mice (Fig. 1, B and D). However, treatment with THG, a blocker of the endoplasmic reticulum Ca2+-ATPase, mobilized the release of intracellular Ca2+ stores (Fig. 1B), suggesting that these stores were available in Prnp0/0 neurons. Consistent with these observations, intracellular Ca2+ levels remained unchanged in hippocampal neurons treated with a mutant STI1 missing the PrPC binding site (STI1Δ230–245) (Fig. 1, C and D). Interestingly, when Ca2+ was removed from the extracellular medium, STI1 had no effect on intracellular Ca2+ levels (Fig. 1, A and D). These results suggest that PrPC-STI1 engagement can activate Ca2+ influx.
FIGURE 1.

STI1 interaction with PrPC promotes intracellular Ca2+ increase. A and B, Prnp+/+ (A) or Prnp0/0 (B) hippocampal neurons loaded with Fluo-3 AM were treated with STI1 (1 μm) in medium supplemented with (solid lines) or without (dashed line) Ca2+. THG-treated Prnp0/0 neurons show normal levels of intracellular Ca2+ stores. C, Prnp+/+ neurons were treated with an STI1 deletion mutant, STI1Δ230–245, lacking the PrPC binding site. D, relative intracellular Ca2+ levels in Prnp0/0 (white bars) and Prnp+/+ (black bars) neurons treated with STI1 or STI1Δ230–245 in the presence or absence of CaCl2 are indicated. The results represent the mean ± S.E. (error bars) of four independent experiments, and statistical significance was determined by one-way ANOVA and Newman-Keuls post test. *, p < 0.05 compared with controls.
Ca2+ Influx Induced by PrPC-STI1 Interaction Promotes ERK1/2 and PKA Activation
The effect of PrPC-STI1-mediated Ca2+ influx on ERK1/2 and PKA activation was tested in wild-type neuronal cultures. In the presence of extracellular Ca2+, STI1 treatment increased both PKA and ERK1/2 activation (Fig. 2). Forskolin, which activates adenylyl cyclase and increases intracellular levels of cAMP, was used as a positive control for PKA activation (Fig. 2A). Conversely, no effect was observed when cells were treated with STI1 in the absence of extracellular Ca2+ (Fig. 2). Thus, upstream Ca2+ signaling is required for PrPC-STI1-mediated ERK1/2 and PKA activation.
FIGURE 2.

PKA activation and ERK1/2 phosphorylation are dependent on PrPC-STI1-induced Ca2+ influx. PKA (A) and ERK1/2 (B) activation was induced by STI1 treatment in Prnp+/+ hippocampal neurons. Experiments were performed with or without CaCl2, as indicated. Forskolin-treated cells were used as positive controls for PKA activation. Relative levels of ERK1/2 activity represent the ratio between phosphorylated ERK1/2 (upper panel) and total ERK1/2 (lower panel) normalized to the untreated group. The results represent the mean ± S.E. (error bars) of four independent experiments, and statistical significance was determined by one-way ANOVA and Newman-Keuls post test. *, p < 0.05 compared with controls.
PrPC-STI1 Interaction Induces Ca2+ Influx through α7nAChR
The PrPC-STI1 interaction likely induces Ca2+ influx via modulation of an unidentified Ca2+ channel at the plasma membrane. Transmembrane Ca2+ channels include VGCCs and several ligand-gated channels. To determine the Ca2+ channel responsible for PrPC-STI1-mediated Ca2+ influx, we used a best candidate approach. To ascertain the role of VGCCs in this response, we used a VGCC inhibitor mixture (ω-conotoxin MCVIIC and nifedipine) targeting the majority of neuronal Ca2+ channels, including Cav2.1, Cav2.2, Cav1.1, Cav1.2, and Cav1.3. Hippocampal neurons still exhibited Ca2+ influx upon STI1 treatment in the presence of VGCC inhibitors (Fig. 3A), although Ca2+ influx due to KCl depolarization was blunted (data not shown).
FIGURE 3.

PrPC-STI1 interaction induces Ca2+ influx, PKA and ERK1/2 activation through α7nAChR. A and B, intracellular Ca2+ levels in Prnp+/+ hippocampal neurons were treated with STI1 (1 μm) in the presence (dashed line) or absence (solid line) of VGCC inhibitors (n = 3) (A) or in the presence (dashed line) or absence (solid line) of αBgt, a specific inhibitor of α7nAChR (n = 4) (B). C and D, PKA activity (n = 3) (C) and ERK1/2 (n = 3) phosphorylation (D) were measured in Prnp+/+ hippocampal neurons treated with STI1 in the presence of αBgt, as indicated. Forskolin was used as a positive control for PKA activation. Relative levels of ERK1/2 activity represent the ratio between phosphorylated ERK1/2 (upper panel) and total ERK1/2 (lower panel), normalized to the untreated group. The results were compared by one-way ANOVA and Newman-Keuls post test. *, p < 0.05 compared with controls.
To determine which ligand-gated channel might transduce PrPC-STI1-mediated Ca2+ influx, modulators of transient receptor potential channels and several neurotransmitter receptors (data not shown) were utilized. One candidate, the α7nAChR, has been shown to activate PKA and ERK1/2 (37) and to promote neuronal differentiation and survival (38–40). In the presence of the α7nAChR-specific inhibitor αBgt, the PrPC-STI1-mediated intracellular Ca2+ influx was abolished (Fig. 3B). Moreover, PKA activation (Fig. 3C) and ERK1/2 phosphorylation (Fig. 3D) were completely blocked in the presence of αBgt. These data show that STI1 binding to PrPC induces intracellular Ca2+ influx via modulation of α7nAChR, leading to ERK1/2 and PKA activation.
To verify whether α7nAChR can be modulated by PrPC-STI1, we used HEK 293 cells, which do not express endogenous α7nAChR, to reconstitute the expression of these receptors. HEK 293 cells were transfected with plasmid vectors encoding PrPC, α7nAChR, or α7nAChR and RIC3 a chaperone that is required for correct assembly of this receptor at the cell surface (41). The functional expression of α7nAChR at the cell surface was confirmed by binding of fluorescent αBgt only upon its co-expression with RIC3 (Fig. 4A).
FIGURE 4.

PrPC interacts with α7nAChR. A, HEK 293 cells were transfected with expression vector lacking insert (vector) or vectors encoding FLAG-tagged α7nAChR (α7) or FLAG-tagged α7nAChR and RIC3 (α7/RIC3). Cells were incubated with Alexa Fluor 647 αBgt and analyzed by confocal microscopy. Phase contrast images indicate the field of cells (top panels), and green fluorescence indicates Alexa Fluor 647 αBgt-labeled cells (bottom panels). B, HEK 293 cells were transfected with expression vector lacking insert (Vector) or expression vectors encoding PrPC either alone (PrPC) or co-transfected with FLAG-tagged α7nAChR and RIC3 (α7/RIC3). Immunoprecipitation (IP) was performed with anti-FLAG antibodies, and proteins from total lysates (Input) or eluates (IP-FLAG) were immunoblotted (IB) with anti-PrPC (IB-PrPC) or anti-FLAG (IB-FLAG) antibodies.
Co-immunoprecipitation experiments using anti-FLAG antibodies against FLAG-tagged α7nAChR, followed by immunoblotting using anti-3F4 antibodies toward 3F4-tagged mouse PrPC (28) or anti-FLAG antibodies, demonstrated that PrPC physically interacts with α7nAChR in these cells (Fig. 4B). To further test whether heterologous expression of α7nAChR was able to reconstitute PrPC-STI1-mediated Ca2+ signaling, we performed signaling experiments in HEK 293 cells. When these cells were transfected with empty vector (Fig. 5A), or with the vector encoding PrPC alone (Fig. 5B), STI1 treatment did not induce an increase in the level of intracellular Ca2+. Similar results were observed in cells transfected only with α7nAChR (Fig. 5C), likely due to the absence of RIC3. However, when HEK 293 cells were co-transfected with vectors encoding α7nAChR, RIC3, and PrPC, STI1 was able to increase intracellular Ca2+ (Fig. 5E). Interestingly, even in cells transfected only with α7nAChR/RIC3, STI1 was able to evoke a Ca2+ signal although this effect could be blocked by antibodies against PrPC (3F4 and 6H4) (Fig. 5D). In addition, STI1 deleted of the PrPC binding site (STI1Δ230–245) was unable to induce Ca2+ signaling (Fig. 5F). These results indicate that endogenous levels of PrPC expression in HEK 293 cells are sufficient to promote Ca2+ signaling when α7nAChR is present. These data are quantified in Fig. 5G.
FIGURE 5.

Expression of α7nAChR rescues PrPC-STI1-mediated Ca2+ influx in HEK 293 cells. A–F, HEK 293 cells were treated with 2 μm STI1 alone (A–C and E), STI1 in the presence of PrPC antibodies 6H4 or 3F4 (D), or 2 μm STI1 deleted of the PrPC binding site, STI1Δ230–245 (F). Cells were transfected with empty vector (n = 5) (A), PrPC (n = 4) (B), α7nAChR (n = 4) (C), α7nAChR and RIC3 (D) and treated with STI1 alone (n = 8) or in the presence of 6H4 (n = 4) or 3F4 (n = 3), PrPC, α7nAChR, and RIC3 (n = 4) (E) or α7nAChR and RIC3 (n = 5) (F). Cells were treated with THG, when indicated, to test the level of intracellular Ca2+ stores. G, relative levels of intracellular Ca2+ in cells transfected and treated as indicated are shown. The results represent the mean ± S.E. (error bars) of independent experiments compared by one-way ANOVA and Newman-Keuls post test. *, p < 0.01 compared with controls.
PrPC-STI1 Interaction Induces PKA and ERK1/2 Activation through α7nAChR
We also found that STI1 induced both PKA activation (Fig. 6A) and ERK1/2 phosphorylation (Fig. 6B) in HEK 293 cells transfected with both α7nAChR and RIC3. Conversely, activation of PKA or ERK1/2 was not observed upon STI1 treatment when these cells were transfected with empty vector or α7nAChR alone (Fig. 6 and data not shown).
FIGURE 6.

Expression of α7nAChR in HEK 293 cells rescues PrPC-STI1-induced PKA activity and ERK1/2 phosphorylation. PKA activity (A) and ERK1/2 phosphorylation (B) in HEK 293 cells transfected with empty vector or co-transfected with expression vectors for α7nAChR and RIC3. Cells were treated with forskolin, STI1, or choline chloride as indicated. Relative levels of ERK1/2 activity represent the ratio between phosphorylated ERK1/2 and total ERK1/2 (upper panel) in cells treated with STI1 and normalized to cells transfected with empty vector. The results represent the mean ± S.E. (error bars) of three independent experiments compared by one-way ANOVA and Newman-Keuls post test. *, p < 0.01 compared with controls.
PrPC-STI1 Interaction Induces Neuroprotection and Neuritogenesis through α7nAChR
To determine whether STI1-PrPC-mediated neuroprotection and neuritogenesis were also dependent on α7nAChR, we analyzed activated caspase-3 levels and neurite outgrowth in hippocampal neurons. In primary neuronal cultures, staurosporine-induced cell death was blocked by STI1 treatment, and this was reversed by preincubation with αBgt (Fig. 7A). In addition, the treatment of hippocampal neurons with αBgt blocked PrPC-STI1-mediated neuritogenesis, as measured by the percentage of cells with neurites (Fig. 7B) or the percentage of cells with neurite length >30 μm (Fig. 7C). Taken together, these data indicate that the PrPC-STI1 interaction modulates α7nAChR activity, thereby inducing Ca2+ influx and PKA and ERK1/2 activation and ultimately promoting neuronal protection and differentiation.
FIGURE 7.
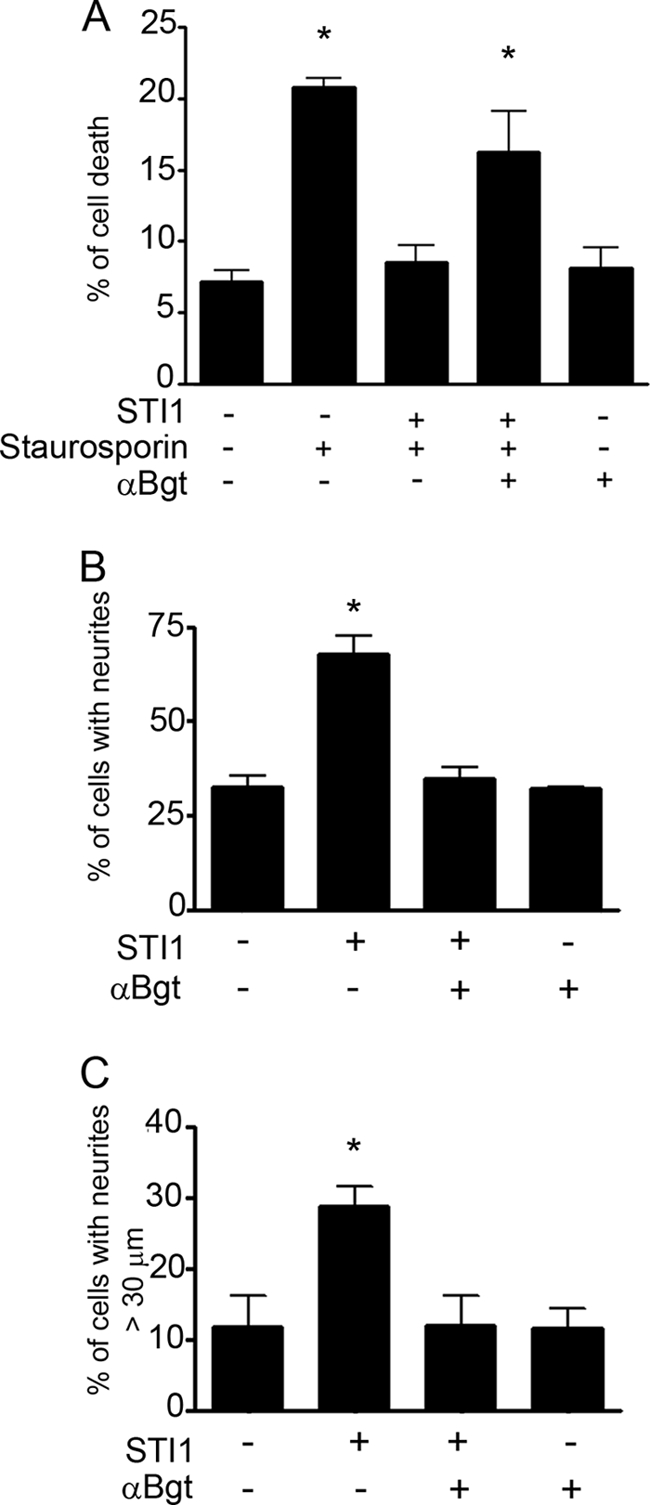
PrPC-STI1 interaction promotes neuroprotection and neuritogenesis through α7nAChR. A, Prnp+/+ hippocampal neurons were treated with STI1 (30 min) or STI1 and αBgt, followed by staurosporine, as indicated. Cells were immunolabeled with anti-activated caspase-3, which labels apoptotic cells. Data are represented as the percentage of activated caspase-3-positive cells. B and C, Prnp+/+ hippocampal neurons were treated with STI1 alone, or STI1 plus αBgt, as indicated, and neuritogenesis was measured as the percentage of cells with neurites or with neurites >30 μm, respectively. The results represent the mean ± S.E. (error bars) of three independent experiments, compared by one-way ANOVA and Newman-Keuls post test. *, p < 0.01 compared with controls.
DISCUSSION
In this study, we demonstrate that PrPC binding to STI1 increases Ca2+ influx in neurons through α7nAChR. These results provide new insight into the physiological role of PrPC as well as a novel mechanism by which PrPC transduces extracellular signals.
The PrPC-STI1 interaction has been shown to modulate neuronal differentiation and survival. Furthermore, these activities are dependent on activation of PKA, ERK1/2, and PI3K–mTOR via PrPC (26, 27). STI1 is secreted from astrocytes (29), and its activity is 100 times higher than recombinant STI1, probably due to a better folding or posttranslational modifications (28). The mechanisms associated with STI1 secretion are presently under investigation, but the protein is likely to use a nonconventional type of secretion due to the absence of a signal peptide (42). However, it is clear that secreted STI1 binds to PrPC, potentially acting as a neurotrophic-like factor (25). Interestingly, hippocampal infusion of STI1 or an STI1-derived peptide (STI1pep230–245), which mimics the PrPC binding site, increases PrPC-dependent memory consolidation. On the other hand, antibodies against either STI1 or its peptide 230–245 were able to impair memory formation (43). These results indicate that the STI1-PrPC interaction has physiological consequences in vivo.
PrPC appears to act as a receptor or co-receptor for a number of ligands, including transmembrane, aggregated and soluble proteins. Therefore, it is important to dissect the mechanisms by which glycosylphosphatidylinositol-anchored PrPC is able to transduce extracellular signals to the intracellular milieu. Previously, PrPC has been shown to interact with neuronal cell adhesion molecule to stimulate Fyn-kinase (17). Furthermore, PrPC has also been shown to associate with G protein-coupled serotonergic receptors, interfering with the intensities and/or dynamics of G protein activation by agonist-bound 5-HT receptors (18, 44). PrPC also acts as a receptor for Aβ1–42 oligomers to inhibit synaptic plasticity in the form of long term potentiation (7). In addition, PrPC interacts with the γ chain subunit of the extracellular matrix protein laminin (21). Our recent data demonstrate that laminin γ1 chain interaction with PrPC triggers neuronal signaling via group I metabotropic glutamate receptors, and regulates intracellular Ca2+ levels and neuritogenesis (62).
STI1 Activates α7nAChR and Ca2+-dependent Signaling Pathways
Our data strongly indicate that STI1 activation of Ca2+ influx is dependent on its engagement with PrPC, as neurons from PrPC-null mice do not respond to STI1. Moreover, in wild-type cultured hippocampal neurons, Ca2+ influx fails in the presence of STI1 lacking the PrPC binding site.
The inhibition of STI1-evoked Ca2+ influx, ERK1/2 phosphorylation, and PKA activation in αBgt-treated neurons is consistent with the fact that STI1 can modulate α7nAChR via PrPC. Moreover, the fact that STI1-mediated signaling is reconstituted in transfected HEK 293 cells supports the idea of a functional interaction between PrPC and α7nAChR. Indeed, we detected a physical interaction between PrPC and α7nAChR.
The mechanisms by which STI1 modulates α7nAChR activity through its interaction with PrPC are unknown. For instance, it is unclear whether STI1 and PrPC act together to activate α7nAChR, or whether they modulate α7nAChR sensitivity to agonists. It should be noted that choline, a proposed α7nAChR agonist (45), is normally found in the extracellular milieu, albeit at a concentration too low to activate α7nAChR (45). Hence, it is possible that STI1-PrPC engagement modulates the response of α7nAChR to extracellular choline. Further experiments using electrophysiology techniques will be necessary to refine possible mechanisms.
Remarkably, the effects of STI1 on neuritogenesis and protection against staurosporine-induced neuronal cell death were also completely blocked by αBgt. Extensive studies have shown that α7nAChR regulates many brain functions (45). These receptors are formed by homomeric assembly of five subunits and are preferentially permeable to Ca2+ (45). Furthermore, α7nAChR activation has been shown to be neuroprotective (46–48) and to regulate learning and memory (for review, see Ref. 49) in an ERK1/2- and PKA-dependent manner (50).
The α7nAChR has also been implicated in AD (51–54), as expression of these receptors is either decreased or increased in AD brains (55, 56) and animal models of AD (57). Furthermore, there is evidence that α7nAChR interacts with Aβ1–42 oligomers (58, 59). Moreover, recent experiments involving α7nAChR knock-out mice crossed with transgenic mice expressing mutated amyloid precursor protein suggest that the synaptic toxicity observed in the latter may be due in part to α7nAChR (60). On the other hand, experiments using a separate transgenic mouse model for AD indicate that these receptors may be protective at early ages. Interestingly, drugs that prevent binding of Aβ1–42 to α7nAChR seem to be beneficial in a model of AD (58). Hence, it seems that STI1 binding to PrPC can hijack one of the key signaling pathways related to AD and learning and memory. Therefore, it is possible that STI1 modulation of a complex containing PrPC and α7nAChR may play an important role in AD.
Little is known regarding the role of nAChRs in prion diseases. Recent data demonstrated that PrPC is co-localized with the β4 subunit of nAChR in the brain and gastrointestinal tract. However, infection experiments using the Rocky Mountain Laboratory prion strain in β4nAChR knock-out mice demonstrated that these animals presented with the same disease incubation period when compared with controls (61).
In summary, we show that STI1 interaction with PrPC can modulate Ca2+ influx via α7nAChR, thereby promoting neuronal survival and differentiation (Fig. 8). Future experiments will need to define the mechanisms involved in PrPC modulation of α7nAChR as well as the pathophysiological roles of this new signaling complex in prion diseases and AD.
FIGURE 8.

Schematic model of signaling events mediated by PrPC-STI1 interaction. PrPC-STI1 interaction modulates α7nAChR, leading to Ca2+ influx and PKA and ERK1/2 activation. PKA and ERK1/2 activation promotes neuroprotection and neuritogenesis, respectively.
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo Grant 03-13189-2 and Programa Institutos Nacionais de Ciência e Tecnologia, do Conselho Nacional de Desenvolvimento Científico e Tecnológico (to V. R. M.). This work was also supported by grants from PrioNet-Canada (to M. A. M. P.) and Canadian Institutes for Health Research (to R. J. R. and M. A. M. P.) and fellowships from Fundação de Amparo à Pesquisa do Estado de São Paulo (to F. H. B., C. P. A., T. G. S., and N. G. T. Q.) and the Department of Foreign Affairs and International Trade-Canada (to F. H. B.).
- PrPC
- prion protein
- α7nAChR
- α7 nicotinic acetylcholine receptor
- αBgt
- α-bungarotoxin
- AD
- Alzheimer disease
- STI1
- stress-inducible protein 1
- THG
- thapsigargin
- VGCC
- voltage-gated calcium channel.
REFERENCES
- 1.Westergard L., Christensen H. M., Harris D. A. (2007) Biochim. Biophys. Acta 1772, 629–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Linden R., Martins V. R., Prado M. A., Cammarota M., Izquierdo I., Brentani R. R. (2008) Physiol. Rev. 88, 673–728 [DOI] [PubMed] [Google Scholar]
- 3.Linden R., Martins V. R, Prado M. A. (2009) Prion protein. UCSD-Nature Molecule Pages, 29July2009, doi:10.1038/mp.a003935.01 [Google Scholar]
- 4.Nimmrich V., Ebert U. (2009) Rev. Neurosci. 20, 1–12 [DOI] [PubMed] [Google Scholar]
- 5.Walsh D. M., Selkoe D. J. (2007) J. Neurochem. 101, 1172–1184 [DOI] [PubMed] [Google Scholar]
- 6.Klein W. L. (2002) Neurochem. Int. 41, 345–352 [DOI] [PubMed] [Google Scholar]
- 7.Laurén J., Gimbel D. A., Nygaard H. B., Gilbert J. W., Strittmatter S. M. (2009) Nature 457, 1128–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Calella A. M., Farinelli M., Nuvolone M., Mirante O., Moos R., Falsig J., Mansuy I. M., Aguzzi A. (2010) EMBO Mol. Med. 2, 306–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kessels H. W., Nguyen L. N., Nabavi S., Malinow R. (2010) Nature 466, E3–E4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parkin E. T., Watt N. T., Hussain I., Eckman E. A., Eckman C. B., Manson J. C., Baybutt H. N., Turner A. J., Hooper N. M. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 11062–11067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cissé M. A., Sunyach C., Lefranc-Jullien S., Postina R., Vincent B., Checler F. (2005) J. Biol. Chem. 280, 40624–40631 [DOI] [PubMed] [Google Scholar]
- 12.Guillot-Sestier M. V., Sunyach C., Druon C., Scarzello S., Checler F. (2009) J. Biol. Chem. 284, 35973–35986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rieger R., Edenhofer F., Lasmézas C. I., Weiss S. (1997) Nat. Med. 3, 1383–1388 [DOI] [PubMed] [Google Scholar]
- 14.Gauczynski S., Peyrin J. M., Haïk S., Leucht C., Hundt C., Rieger R., Krasemann S., Deslys J. P., Dormont D., Lasmézas C. I., Weiss S. (2001) EMBO J. 20, 5863–5875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hundt C., Peyrin J. M., Haïk S., Gauczynski S., Leucht C., Rieger R., Riley M. L., Deslys J. P., Dormont D., Lasmézas C. I., Weiss S. (2001) EMBO J. 20, 5876–5886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmitt-Ulms G., Legname G., Baldwin M. A., Ball H. L., Bradon N., Bosque P. J., Crossin K. L., Edelman G. M., DeArmond S. J., Cohen F. E., Prusiner S. B. (2001) J. Mol. Biol. 314, 1209–1225 [DOI] [PubMed] [Google Scholar]
- 17.Santuccione A., Sytnyk V., Leshchyns'ka I., Schachner M. (2005) J. Cell Biol. 169, 341–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mouillet-Richard S., Pietri M., Schneider B., Vidal C., Mutel V., Launay J. M., Kellermann O. (2005) J. Biol. Chem. 280, 4592–4601 [DOI] [PubMed] [Google Scholar]
- 19.Taylor D. R., Hooper N. M. (2007) Biochem. J. 402, 17–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parkyn C. J., Vermeulen E. G., Mootoosamy R. C., Sunyach C., Jacobsen C., Oxvig C., Moestrup S., Liu Q., Bu G., Jen A., Morris R. J. (2008) J. Cell Sci. 121, 773–783 [DOI] [PubMed] [Google Scholar]
- 21.Graner E., Mercadante A. F., Zanata S. M., Forlenza O. V., Cabral A. L., Veiga S. S., Juliano M. A., Roesler R., Walz R., Minetti A., Izquierdo I., Martins V. R., Brentani R. R. (2000) Brain Res. Mol. Brain Res. 76, 85–92 [DOI] [PubMed] [Google Scholar]
- 22.Graner E., Mercadante A. F., Zanata S. M., Martins V. R., Jay D. G., Brentani R. R. (2000) FEBS Lett. 482, 257–260 [DOI] [PubMed] [Google Scholar]
- 23.Hajj G. N., Lopes M. H., Mercadante A. F., Veiga S. S., da Silveira R. B., Santos T. G., Ribeiro K. C., Juliano M. A., Jacchieri S. G., Zanata S. M., Martins V. R. (2007) J. Cell Sci. 120, 1915–1926 [DOI] [PubMed] [Google Scholar]
- 24.Zanata S. M., Lopes M. H., Mercadante A. F., Hajj G. N., Chiarini L. B., Nomizo R., Freitas A. R., Cabral A. L., Lee K. S., Juliano M. A., de Oliveira E., Jachieri S. G., Burlingame A., Huang L., Linden R., Brentani R. R., Martins V. R. (2002) EMBO J. 21, 3307–3316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martins V. R., Beraldo F. H., Hajj G. N., Lopes M. H., Lee K. S., Prado M. M., Linden R. (2010) Curr. Issues Mol. Biol. 12, 63–86 [PubMed] [Google Scholar]
- 26.Lopes M. H., Hajj G. N., Muras A. G., Mancini G. L., Castro R. M., Ribeiro K. C., Brentani R. R., Linden R., Martins V. R. (2005) J. Neurosci. 25, 11330–11339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roffé M., Beraldo F. H., Bester R., Nunziante M., Bach C., Mancini G., Gilch S., Vorberg I., Castilho B. A., Martins V. R., Hajj G. N. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 13147–13152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caetano F. A., Lopes M. H., Hajj G. N., Machado C. F., Pinto A. C., Magalhães A. C., Vieira M. P., Américo T. A., Massensini A. R., Priola S. A., Vorberg I., Gomez M. V., Linden R., Prado V. F., Martins V. R., Prado M. A. (2008) J. Neurosci. 28, 6691–6702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lima F. R., Arantes C. P., Muras A. G., Nomizo R., Brentani R. R., Martins V. R. (2007) J. Neurochem. 103, 2164–2176 [DOI] [PubMed] [Google Scholar]
- 30.Cullen B. R. (1987) Methods Enzymol. 152, 684–704 [DOI] [PubMed] [Google Scholar]
- 31.Dale L. B., Bhattacharya M., Anborgh P. H., Murdoch B., Bhatia M., Nakanishi S., Ferguson S. S. (2000) J. Biol. Chem. 275, 38213–38220 [DOI] [PubMed] [Google Scholar]
- 32.Cooper D. M., Mons N., Karpen J. W. (1995) Nature 374, 421–424 [DOI] [PubMed] [Google Scholar]
- 33.Fierro A. F., Wurth G. A., Zweifach A. (2004) J. Biol. Chem. 279, 25646–25652 [DOI] [PubMed] [Google Scholar]
- 34.Brini M., Miuzzo M., Pierobon N., Negro A., Sorgato M. C. (2005) Mol. Biol. Cell 16, 2799–2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Korte S., Vassallo N., Kramer M. L., Kretzschmar H. A., Herms J. (2003) J. Neurochem. 87, 1037–1042 [DOI] [PubMed] [Google Scholar]
- 36.Whatley S. A., Powell J. F., Politopoulou G., Campbell I. C., Brammer M. J., Percy N. S. (1995) Neuroreport 6, 2333–2337 [DOI] [PubMed] [Google Scholar]
- 37.Bitner R. S., Bunnelle W. H., Anderson D. J., Briggs C. A., Buccafusco J., Curzon P., Decker M. W., Frost J. M., Gronlien J. H., Gubbins E., Li J., Malysz J., Markosyan S., Marsh K., Meyer M. D., Nikkel A. L., Radek R. J., Robb H. M., Timmermann D., Sullivan J. P., Gopalakrishnan M. (2007) J. Neurosci. 27, 10578–10587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Broide R. S., Leslie F. M. (1999) Mol. Neurobiol. 20, 1–16 [DOI] [PubMed] [Google Scholar]
- 39.Shimohama S., Greenwald D. L., Shafron D. H., Akaika A., Maeda T., Kaneko S., Kimura J., Simpkins C. E., Day A. L., Meyer E. M. (1998) Brain Res. 779, 359–363 [DOI] [PubMed] [Google Scholar]
- 40.Jonnala R. R., Buccafusco J. J. (2001) J. Neurosci. Res. 66, 565–572 [DOI] [PubMed] [Google Scholar]
- 41.Williams M. E., Burton B., Urrutia A., Shcherbatko A., Chavez-Noriega L. E., Cohen C. J., Aiyar J. (2005) J. Biol. Chem. 280, 1257–1263 [DOI] [PubMed] [Google Scholar]
- 42.Keller M., Rüegg A., Werner S., Beer H. D. (2008) Cell 132, 818–831 [DOI] [PubMed] [Google Scholar]
- 43.Coitinho A. S., Lopes M. H., Hajj G. N., Rossato J. I., Freitas A. R., Castro C. C., Cammarota M., Brentani R. R., Izquierdo I., Martins V. R. (2007) Neurobiol. Dis. 26, 282–290 [DOI] [PubMed] [Google Scholar]
- 44.Mouillet-Richard S., Schneider B., Pradines E., Pietri M., Ermonval M., Grassi J., Richards J. G., Mutel V., Launay J. M., Kellermann O. (2007) Ann. N.Y. Acad. Sci. 1096, 106–119 [DOI] [PubMed] [Google Scholar]
- 45.Albuquerque E. X., Pereira E. F., Alkondon M., Rogers S. W. (2009) Physiol. Rev. 89, 73–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dajas-Bailador F., Wonnacott S. (2004) Trends Pharmacol. Sci. 25, 317–324 [DOI] [PubMed] [Google Scholar]
- 47.Akaike A., Takada-Takatori Y., Kume T., Izumi Y. (2010) J. Mol. Neurosci. 40, 211–216 [DOI] [PubMed] [Google Scholar]
- 48.Dajas-Bailador F. A., Lima P. A., Wonnacott S. (2000) Neuropharmacology 39, 2799–2807 [DOI] [PubMed] [Google Scholar]
- 49.Thomsen M. S., Hansen H. H., Timmerman D. B., Mikkelsen J. D. (2010) Curr. Pharm. Des 16, 323–343 [DOI] [PubMed] [Google Scholar]
- 50.Dajas-Bailador F. A., Soliakov L., Wonnacott S. (2002) J. Neurochem. 80, 520–530 [DOI] [PubMed] [Google Scholar]
- 51.Hernandez C. M., Kayed R., Zheng H., Sweatt J. D., Dineley K. T. (2010) J. Neurosci. 30, 2442–2453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ren K., King M. A., Liu J., Siemann J., Altman M., Meyers C., Hughes J. A., Meyer E. M. (2007) Neuroscience 148, 230–237 [DOI] [PubMed] [Google Scholar]
- 53.Kihara T., Shimohama S., Sawada H., Honda K., Nakamizo T., Shibasaki H., Kume T., Akaike A. (2001) J. Biol. Chem. 276, 13541–13546 [DOI] [PubMed] [Google Scholar]
- 54.Buckingham S. D., Jones A. K., Brown L. A., Sattelle D. B. (2009) Pharmacol. Rev. 61, 39–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jones I. W., Westmacott A., Chan E., Jones R. W., Dineley K., O'Neill M. J., Wonnacott S. (2006) J. Mol. Neurosci. 30, 83–84 [DOI] [PubMed] [Google Scholar]
- 56.Teaktong T., Graham A. J., Court JA, Perry R. H., Jaros E., Johnson M., Hall R., Perry E. K. (2004) J. Neurol. Sci. 225, 39–49 [DOI] [PubMed] [Google Scholar]
- 57.Dineley K. T., Xia X., Bui D., Sweatt J. D., Zheng H. (2002) J. Biol. Chem. 277, 22768–22780 [DOI] [PubMed] [Google Scholar]
- 58.Wang H. Y., Stucky A., Liu J., Shen C., Trocme-Thibierge C., Morain P. (2009) J. Neurosci. 29, 10961–10973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang H. Y., Lee D. H., D'Andrea M. R., Peterson P. A., Shank R. P., Reitz A. B. (2000) J. Biol. Chem. 275, 5626–5632 [DOI] [PubMed] [Google Scholar]
- 60.Dziewczapolski G., Glogowski C. M., Masliah E., Heinemann S. F. (2009) J. Neurosci. 29, 8805–8815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Petrakis S., Irinopoulou T., Panagiotidis C. H., Engelstein R., Lindstrom J., Orr-Urtreger A., Gabizon R., Grigoriadis N., Sklaviadis T. (2008) Eur. J. Neurosci. 27, 612–620 [DOI] [PubMed] [Google Scholar]
- 62.Beraldo F. H., Arantes C. P., Santos T. G., Machado C. F., Roffe M., Hajj G. N., Lee K. S., Magalhães A. C., Caetano F. A., Mancini G. L., Lopes M. H., Americo T. A., Magdesian M. H., Ferguson S. S., Linden R., Prado M. A., Martins V. R. (2010) FASEB J., in press [Google Scholar]