

Abstract
Malignant gliomas (MG) are highly infiltrative tumors that consistently recur despite aggressive treatment. Brain fatty acid-binding protein (FABP7), which binds docosahexaenoic acid (DHA) and arachidonic acid (AA), localizes to sites of tumor infiltration and is associated with a poor prognosis in MG. Manipulation of FABP7 expression in MG cell lines affects cell migration, suggesting a role for FABP7 in tumor infiltration and recurrence. Here, we show that DHA inhibits and AA stimulates migration in an FABP7-dependent manner in U87 MG cells. We demonstrate that DHA binds to and sequesters FABP7 to the nucleus, resulting in decreased cell migration. This anti-migratory effect is partially dependent on peroxisome proliferator-activated receptor γ, a DHA-activated transcription factor. Conversely, AA-bound FABP7 stimulates cell migration by activating cyclooxygenase-2 and reducing peroxisome proliferator-activated receptor γ levels. Our data provide mechanistic insight as to why FABP7 is associated with a poor prognosis in MG and suggest that relative levels of DHA and AA in the tumor environment can make a profound impact on tumor growth properties. We propose that FABP7 and its fatty acid ligands may be key therapeutic targets for controlling the dissemination of MG cells within the brain.
Keywords: Fatty Acid, Fatty Acid-binding Protein, Lipid, Lipid-binding Protein, Tumor, Cell Migration, Malignant Glioma
Introduction
Anaplastic astrocytoma (grade III astrocytoma) and glioblastoma multiforme (grade IV astrocytoma), collectively called malignant gliomas (MG),2 are the most common cancers of the central nervous system (CNS). The prognosis for these cancers is dismal, with median survival times of 30 months for grade III astrocytoma and less than 1 year for grade IV astrocytoma (1). Despite aggressive treatment involving surgical resection, radiation therapy, and adjuvant chemotherapy using nitrosourea-based compounds and temozolamide (2), these highly infiltrative tumors consistently recur in the brain at sites that can be either proximal or distal to the original tumor mass. Thus, improving patient survival will likely depend on developing new therapies that target infiltrative cells while minimizing damage to normal brain tissue.
Brain fatty acid-binding protein (FABP7; BLBP) is expressed in astrocytoma tumor biopsies as well as in a subset of MG cell lines (3), and FABP7 is up-regulated in brain tumor tissue compared with normal adult brain (4, 5). FABP7 expression has been associated with decreased survival times and/or tumor progression in patients with grade IV astrocytoma (4, 6), melanoma, basal-type breast cancer, and renal cell carcinoma (7–9). Introduction of FABP7 into FABP7-negative MG cells confers a pro-migratory/pro-invasive phenotype to the cells, whereas knockdown of FABP7 in MG cells that naturally express FABP7 results in reduced migration/invasion (4, 10). These combined data suggest that FABP7 may enhance the infiltrative and/or invasive properties of tumor cells, thereby contributing to tumor recurrence and decreased patient survival.
FABP7 is a member of the FABP family of lipid chaperones involved in the uptake and intracellular trafficking of fatty acids. In vitro ligand binding studies have shown that the polyunsaturated fatty acid (PUFA) ω-3-docosahexaenoic acid (DHA; 22:6) is the preferred ligand of FABP7. FABP7 also binds ω-6-arachidonic acid (AA; 20:4), albeit with an ∼4-fold lower affinity (11). DHA and AA have been shown to have opposite effects on tumor growth, with DHA inhibiting growth and AA promoting growth (12).
FABPs, found in both the nucleus and cytoplasm, are believed to play a role in gene regulation by activating peroxisome proliferator-activated receptors (PPARs), nuclear receptors that function as transcription factors. Specifically, liver FABP (FABP1) has been shown to bind and activate PPARα and PPARγ (13). Adipocyte FABP (FABP4) and keratinocyte FABP (FABP5) bind PPARγ and PPARβ, respectively (14, 15), whereas FABP7 interacts with PPARγ (16). It has been postulated that nuclear FABPs can deliver their fatty acid ligands to PPARs, thereby regulating PPAR transcriptional activity. Increased expression of PPARα is associated with a worse prognosis in MG (17), whereas PPARγ is generally associated with growth arrest and apoptosis in these tumors (18, 19).
Although relationships between specific fatty acids and FABPs, FABPs and cancer, and fatty acids and cancer have been described previously, there has been no concerted effort to investigate the interdependence of fatty acids and FABPs on tumorigenic properties. In a previous study, Wang et al. (20) demonstrated ∼50% growth inhibition of breast cancer cells stably transfected with an FABP7 expression construct in the presence of DHA. Furthermore, treatment of FABP5-expressing PC12 cells with DHA increased neurite extension (21). These data highlight the importance of FABPs in determining tumor cell response to fatty acids. Here, we examine the effect of DHA and AA on FABP7-mediated cell migration in MG. We describe an inhibitory role for DHA in FABP7-mediated cell migration and a permissive role for AA in FABP7-mediated cell migration. Intriguingly, DHA-mediated inhibition of cell migration functions through PPARγ and requires nuclear localization of FABP7, whereas stimulation of cell migration by FABP7 and AA is dependent on activation of cyclooxygenase 2 (COX-2) and prostaglandin E2 (PGE2) production. We propose a model whereby relative levels of DHA and AA, and the fatty acid-dependent subcellular distribution of FABP7, determine the migratory potential of MG cells. The importance of our findings resonates 2-fold as they provide a molecular mechanism for FABP7-induced MG cell migration and point to the potential use of DHA as an anti-infiltrative therapeutic agent in the treatment of MG.
EXPERIMENTAL PROCEDURES
Cell Lines and Transfectants
The origin and FABP7 status of MG cell lines U87, U251, U373, and M049, as well as the generation of stable U87 clonal populations transfected with either empty pREP4 vector (FABP7(−)) or a pREP4-FABP7 expression construct (FABP7(+)), have been described previously (10, 22, 23). Transient transfection of U87 cells was by calcium phosphate-DNA precipitation, with 40–50% transfection efficiency. Transiently transfected cells were analyzed 48 h after removal of the DNA. Unless otherwise stated, cells were cultured in Dulbecco's modified essential medium (DMEM) supplemented with 10% FCS, 100 μg/ml streptomycin, and 100 units/ml penicillin.
Western Blot Analysis
Whole cell lysates were prepared by incubating cells on ice for 20 min in 0.5 m Tris-HCl, pH 7.5, 0.15 m NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 1 mm NaF, 0.1% SDS, 1× protease inhibitor mixture (Roche Applied Sci), 1 mm PMSF, 2 mm DTT. Nuclear cell extracts were prepared by lysing cells at 4 °C in 10 mm HEPES, pH 7.9, 10 mm KCl, 0.1 mm EDTA, 0.4% Nonidet P-40, 1 mm DTT, 0.5 mm PMSF. Nuclei were collected by centrifugation (15,000 × g at 4 °C for 3 min) and lysed by vigorous shaking in 20 mm HEPES, pH 7.9, 0.4 m NaCl, 1 mm EDTA, 10% glycerol, 1 mm DTT, 0.5 mm PMSF. Nuclear lysates were centrifuged at 14,000 × g for 5 min at 4 °C, and the supernatants were collected. Lysates were electrophoresed in a 13.5% SDS-polyacrylamide gel followed by electroblotting to nitrocellulose membranes. Membranes were immunostained with rabbit anti-FABP7 (3), mouse anti-HA (Santa Cruz Biotechnology), mouse anti-actin (Sigma-Aldrich), rabbit anti-FABP5 (HyCult Biotechnology), goat anti-COX-2 (sc-1747; Santa Cruz Biotechnology), mouse anti-PPARα (Chemicon), mouse anti-PPARβ (Chemicon), or mouse anti-PPARγ (Chemicon) antibodies. Primary antibodies were detected with horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch) using the ECL detection system (GE Healthcare).
Cell Migration
Twenty thousand cells were plated in DMEM (without FCS) in the top chamber of HTS multiwell inserts with a porous 8 μm PET membrane separating the top and bottom chambers (Transwell; BD Biosciences). Cells were incubated for 6 h in a CO2 incubator to allow cell migration to the bottom chamber containing chemoattractant (DMEM plus 10% FCS). Cells that migrated to the bottom chamber were fixed and stained with 2% crystal violet in 20% methanol. The cells were photographed using a 2.5× lens and four frames combined to reconstruct the entire well. Cells were then counted using Metamorph version 7.7 imaging software.
Immunofluorescence Microscopy
Cells were fixed in 1% paraformaldehyde for 10 min and then permeabilized in PBS + 0.5% Triton X-100 for 5 min. Affinity-purified rabbit anti-FABP7 antibody was used at a 1:200 dilution, followed by secondary Alexa 488-conjugated goat anti-rabbit secondary antibody (Cedarlane Laboratories). All images were collected on a Zeiss LSM 510 confocal microscope with a 40×/1.3 oil immersion lens.
Delipidation of FABP7 and Fatty-acid Binding Assay
Lipidex® 1000 (Sigma) was used to delipidate recombinant FABP7 and carry out the fatty acid binding assays. Lipidex 1000 removes unbound and/or protein-bound fatty acids from aqueous solutions in a temperature-dependent manner. For delipidation, recombinant GST-tagged FABP7 was incubated in 1.0-ml amber glass vials along with 50% w/v Lipidex in 10 mm potassium phosphate buffer, pH 7.4, for 10 min at 37 °C. The Lipidex/protein mixture was then spun down for 2 min at 4,000 × g, and the incubation repeated twice using fresh Lipidex-conjugated beads. For fatty acid binding assays, delipidated FABP7 was incubated in 10 mm potassium phosphate buffer, pH 7.4, with varying concentrations of [1-14C]DHA (Moravek Biochemicals) and incubated at 37 °C for 10 min. Unbound fatty acids were removed from the solution by adding 50 μl of an ice-cold Lipidex/buffer suspension (1:1, w/v) followed by a 10-min incubation at 4 °C. The mixture was then centrifuged for 5 min at 4,000 × g, and fatty acid binding was calculated from the amount of radioactivity present in the supernatant as measured by a scintillation counter. Graphs were generated and analyzed using GraphPad Prism software version 5.
Knockdown of FABP7, PPARγ, and PPARβ by siRNA
Cells were transfected with 10 nm of the following Stealth siRNAs (Invitrogen) using LipofectamineTM RNAiMAX (Invitrogen): FABP7 (5′-CAAACCAACGGUAAUUAUCAGUCAA-3′ (NM_001446_stealth_405)); PPARγ (5′-CCAGUGGUUGCAGAUUACAAGUAUG-3′ (NM_13872_stealth_425)); PPARγ (5′-AGGGAGUUUCUAAAGAGCCUGCGAA-3′ (NM_13772_stealth_1295)); PPARβ (5′-CCACUACGGUGUUCAUGCAUGUGAG-3′ (NM_006238_stealth_561)); PPARβ (5′-UCAGUGAUAUCAUUGAGCCUAAGUU-3′ (NM_006238_stealth_1280)); scrambled siRNA (StealthTM RNAi Negative Control Low and Medium GC Duplex). The Transwell migration assay was carried out 72 h post-transfection using 20,000 cells/well. The remaining cells were used for the preparation of cell lysates and used for Western blot analysis to verify knockdown.
Enzyme-linked Immunosorbent Assay (ELISA)
PGE2 levels were determined by ELISA using the protocol supplied by the manufacturer (prostaglandin E2 Biotrak Enzyme Immunoassay System, GE Healthcare). Cells were seeded in triplicate on 96-well plates at 50,000 cells/well and cultured for 24 h. Plates were read at 450 nm using a microplate reader (FLUOstar OPTIMA, BMG LABTECH).
Preparation of Fatty Acids and COX-2 Inhibitor NS398
Fatty acids (Sigma) were dissolved in ethanol and then complexed to fatty acid-free BSA (Roche Applied Science) over a steady stream of nitrogen gas. The final concentration of ethanol in our experiments was 0.1%. NS398 (Sigma) was dissolved in DMSO (Sigma) to generate a 200 mm (1000×) stock solution. The final concentration of DMSO in the medium was 0.1%.
Site-directed Mutagenesis
The 400-bp human FABP7 cDNA encoding the entire open reading frame was inserted into the pcDNA3.1 vector at the EcoRV site. To generate the nuclear localization signal (NLS) mutant, site-directed mutagenesis of FABP7 at K21A (AAG→GCG), R30A (AGG→GCG), and Q31A (CAG→GCG) was carried out by sequential PCR (24) using partially complementary primers carrying the appropriate point mutations. These primers were used in conjunction with pcDNA3.1 vector primers to generate DNA fragments corresponding to full-length FABP7. To generate the nonfatty acid-binding mutant, the same technique was applied, using primers carrying the following point mutations: F104A (TTT→AGA), R126A (CGC→GCC), and Y128A (TAT→GCT). These primers, which contained an XhoI recognition site, were used along with primers to the pcDNA3.1 vector sequence spanning the BamHI site to allow directional cloning into pcDNA3.1. The FABP7 cDNA inserts were sequenced to ensure that they were error-free. Expression of mutant FABP7 was confirmed by transfection into U87MG and Western blot analysis.
RESULTS
FABP7-induced MG Cell Migration Is Fatty Acid Ligand-dependent
Previous work has shown that stable transfection of an FABP7 expression construct into FABP7(−) U87MG cells results in increased motility and migration (10). In vitro ligand binding studies indicate that FABP7 binds both DHA and AA, with equilibrium dissociation constants (Kd) of 53.4 and 207 nm, respectively (11). We therefore used the Transwell assay to examine the effect of DHA and AA on the migration of FABP7(−) and FABP7(+) U87 cells. Cells were cultured in growth medium (DMEM plus 10% FCS) supplemented with 60 μm BSA-complexed DHA, AA, palmitic acid (PA; 16:0), or BSA alone for 24 h. As demonstrated in Fig. 1A, none of the fatty acids tested had an effect on FABP7(−) U87 cell migration, with an average of ∼250 cells migrating through the upper chamber of Transwell inserts. As expected, FABP7-expressing U87 cells cultured in 10% FCS supplemented with BSA showed increased migration compared with FABP7(−) U87 cells (1553 versus 294 cells/well). Growth in PA had no effect on FABP7(+) U87 cell migration; however, growth in DHA resulted in a 5-fold decrease in cell migration, with an average of 279 cells migrating to the bottom chamber compared with 1553 cells in the BSA control (p < 0.01) (Fig. 1A). In contrast, FABP7(+) U87 cells cultured in AA showed a small but significant increase in cell migration, with 1812 cells/well migrating to the bottom chamber compared with 1553 in the BSA control (p < 0.01). These results indicate a strong link between FABP7-mediated cell migration and the presence of DHA or AA.
FIGURE 1.

Cell migration of U87-FABP7(−) and U87-FABP7(+) cells in the presence of fatty acids. A, cells cultured in DMEM plus 10% FCS (growth medium) were treated with 60 μm BSA, 60 μm DHA, 60 μm AA, or 60 μm PA for 24 h. B, cells were serum-starved for 24 h and then treated with either BSA or the indicated fatty acids for 24 h. C, cells were cultured in growth medium and treated with different ratios of DHA and AA as follows: 1 = 30 μm; 2, = 60 μm; 3 = 90 μm; 4 = 120 μm. A–C, cells were trypsinized, and 20,000 cells (in DMEM) were plated in the upper chambers of Transwell inserts. The bottom chambers contained DMEM plus 10% FCS. After 6 h, the cells migrating through the porous membrane were fixed, stained, and counted using Metamorph imaging software version 7.7. Statistical significance was determined using the unpaired t test. Error bars represent standard deviation.
Studying clonal populations of U87 cells that differ in whether they have been transfected with empty vector or an FABP7 expression construct represents a highly controlled experimental situation. To broaden the scope of our study, we examined the effect of AA and DHA on three MG cell lines that naturally express FABP7: U251, U373, and M049 (Fig. 2A). U251 and U373 cultured in the presence of BSA had migratory activities that were similar to that of FABP7(+) U87 cells (Fig. 2A). Although growth of U251 and U373 cells in AA did not significantly increase cell migration, U251 and U373 cells cultured in DHA showed the same decrease in migration observed for FABP7(+) U87 cells. In contrast, M049 cells showed significantly reduced migration compared with the other FABP7-expressing MG cell lines. Similar to FABP7(−) U87 cells, neither DHA nor AA had an effect on M049 cell migration. These results indicate that factors other than FABP7 can affect MG cell migration. Of note, FABP5 is highly expressed in M049 compared with U251 and U373 (Fig. 2A).
FIGURE 2.

Cell migration of U251, U373, and M049 cells in the presence of fatty acids. A, FABP7(+) U251, U373, and M049 MG cells were cultured for 24 h in DMEM + 10% FCS containing 60 μm BSA, 60 μm DHA, or 60 μm AA. B, U251 cells transfected with control (scrambled) siRNA and FABP7 siRNA were cultured for 24 h in DMEM + 10% FCS containing 60 μm BSA, 60 μm DHA, or 60 μm AA. A and B, 20,000 cells (in DMEM) were plated in the upper chambers of Transwell inserts. The bottom chambers contained DMEM plus 10% FCS. After 6 h, the cells migrating through the porous membrane were fixed, stained, and counted using Metamorph imaging software version 7.7. Statistical significance was determined using the unpaired t test (*, p < 0.01). Error bars represent standard deviation. For Western blot analysis, proteins (25 μg/lane) were separated in a 13.5% SDS-polyacrylamide gel, transferred to nitrocellulose membranes, and immunostained with rabbit anti-FABP7 antibody, rabbit anti-FABP5 antibody, and mouse anti-actin antibody, followed by HRP-conjugated secondary antibodies.
We then used siRNA to reduce FABP7 levels in U251 cells. Knockdown of FABP7 resulted in a significant decrease in cell migration, with 641 FABP7(↓) U251 cells migrating to the bottom chamber in comparison with 1490 cells for U251 control (p < 0.01) (Fig. 2B). Growth in the presence of DHA or AA had little effect on the migration of FABP7(↓) U251 cells, in keeping with the results obtained with FABP7(−) U87 cells. Because there was general, if not complete, agreement between the results obtained with U87, U251, and U373, we pursued our analyses with the U87-FABP7(−) and U87-FABP7(+) clonal populations.
Fetal calf serum contains >20 different fatty acids, including DHA and AA at concentrations of 4 and 11.5 μm, respectively. To study the effects of DHA and AA on migration in the absence of FCS, U87-FABP7(−) and U87-FABP7(+) MG cells were cultured in DMEM without FCS for 24 h and then treated with DHA, AA, PA, or BSA in the absence of FCS for 24 h (i.e. 48-h total serum starvation). Although there was a 30% decrease in the number of serum-starved BSA-treated U87-FABP7(+) cells that migrated to the bottom chamber compared with cells cultured in 10% FCS (Fig. 1B), the overall trend was similar to that observed for cells grown in the presence of FCS, with DHA inhibiting and AA stimulating cell migration. There was an average of 1202 cells/well for the BSA control compared with 199 cells/well for DHA-treated cells and 1446 cells/well for AA-treated cells. Again, PA treatment did not affect cell migration, and none of the fatty acids affected the migration of U87-FABP7(−) cells. These results indicate that DHA and AA can regulate FABP7-mediated U87 cell migration.
Ratio of DHA:AA Determines the Migration of FABP7(+) U87 MG Cells
A number of studies have shown that ω-3 fatty acids have anti-tumorigenic properties, inhibiting tumor growth and preventing vascularization and cell migration. In contrast, ω-6 fatty acids have pro-tumorigenic properties. It has been postulated that the ratio of ω-3 to ω-6 fatty acids is key to the pathogenesis of many diseases, including cancer (25). To address whether the ratio of DHA:AA, as opposed to individual concentrations of DHA or AA, is driving the effects observed in Fig. 1, A and B, we treated U87-FABP7(+) MG cells with varying concentrations of both DHA and AA, and we measured their migration using the Transwell assay (Fig. 1C).
The migration of U87-FABP7(+) cells cultured in growth medium supplemented with 60 μm each of DHA and AA was inhibited by 75%, with an average of 371 cells/well migrating to the bottom chamber compared with 1513 cells/well in the untreated control (Fig. 1C). Cell migration was also inhibited by 74% when the concentration of DHA was halved (30 μm DHA; 60 μm AA). However, when levels of AA exceeded those of DHA by 3-fold or more, the inhibitory effects of DHA on cell migration were negated, with 1596 cells/well migrating to the bottom chamber when the DHA:AA ratio was 1:3 (30 μm DHA; 90 μm AA), and 1646 cells/well migrating to the bottom chamber with a DHA:AA ratio of 1:4 (30 μm DHA; 120 μm AA). These results are expected in light of in vitro data indicating that DHA has a four times greater binding affinity for FABP7 than AA (11). Increasing the concentration of DHA, from 60 to 120 μm, in the presence of 30 μm AA, did not result in further inhibition of cell migration (Fig. 1C). PA at concentrations ranging from 30 to 120 μm had no effect on cell migration (Fig. 3). Based on cellular morphology and growth rate, high concentrations of fatty acids had no adverse effect on the survival of FABP7(+) U87 cells. These combined data suggest that different pathways are activated when FABP7 is bound to DHA or AA, with DHA inhibiting and AA stimulating cell migration.
FIGURE 3.
Cell migration of U87-FABP7(+) cells treated with varying fatty acid ratios. U87-FABP7(+) cells were grown in DMEM plus 10% FCS and treated with varying fatty acid ratios for 24 h: 1 = 30 μm, 2 = 60 μm, 3 = 90 μm, 4 = 120 μm. Cell migration was measured using the Transwell assay. Twenty thousand cells/well were plated and incubated for 6 h, and the cells migrating through the porous membrane were fixed, stained, and counted using Metamorph imaging software. Statistical significance was determined using the unpaired t test. Error bars represent standard deviation.
Subcellular Localization of FABP7 Is Dependent on Binding to PUFAs
FABPs play an important role in the transport of their fatty acid ligands to various subcellular compartments, including endoplasmic reticulum, mitochondria, and nucleus. FABP7 is found in both the cytoplasm and nucleus of MG cells, with some tumors preferentially expressing FABP7 in either the nucleus or cytoplasm (4, 6). To investigate whether the subcellular localization of FABP7 might be dependent on availability of specific fatty acid ligands, we examined the subcellular distribution of FABP7 in U87-FABP7(+) and U251, in the presence and absence of DHA and AA. When cells were cultured in medium depleted of fatty acids (i.e. serum-starved for 24 h), FABP7 was primarily found in the cytoplasm (Fig. 4, A and D). Addition of 60 μm DHA to DMEM resulted in FABP7 localization to the nucleus (Fig. 4, B and E), suggesting that DHA-bound FABP7 is sequestered to the nucleus. In contrast, a relatively uniform distribution of FABP7 was observed throughout the cell in the presence of 60 μm AA (Fig. 4, C and F). Alterations in the subcellular distribution of FABP7 were not accompanied by any changes in the levels of FABP7 protein (Fig. 5). The dynamic shuttling of FABP7 from the cytoplasm to the nucleus in the presence of DHA (and to a lesser extent AA) suggests a role for FABP7 in delivering its fatty acid ligands to the nucleus.
FIGURE 4.

PUFA-dependent subcellular localization of FABP7 in U87-FABP7(+) and U251MG cell lines. U87-FABP7(+) and U251MG cells expressing endogenous FABP7 were serum-starved for 24 h (A and D) and then were either treated with 60 μm DHA in the absence of FCS for an additional 24 h (B and E) or treated with 60 μm AA in the absence of FCS for 24 h (C and F). The subcellular localization of FABP7 in U87-FABP7(+) and U251MG was analyzed by immunofluorescence using anti-FABP7 antibody followed by Alexa 488-conjugated secondary antibody. The DNA was counterstained with DAPI.
FIGURE 5.

Western blot analysis of FABP7 in U87-FABP7(+) stable transfectants treated with 60 μm BSA, AA, DHA, or PA. Whole cell lysates (25 μg/lane) were prepared from U87-FABP7(+) and electrophoresed in a 13.5% SDS-polyacrylamide gel. Proteins were transferred to nitrocellulose membranes and sequentially immunostained with rabbit anti-FABP7 antibody and mouse anti-actin antibody, followed by HRP-conjugated secondary antibodies. C, cytoplasmic; N, nuclear.
FABP7-induced Cell Migration Is Dependent on a Direct FABP7-Fatty Acid Ligand Interaction
Based on x-ray crystallography, Balendiran et al. (11) predicted that the amino acid Phe-104 is essential for the formation of π-π interactions between FABP7 and DHA, whereas the guanidinium group of Arg-126 and the hydroxyl group of Tyr-128 were predicted to bind directly to the carboxylate moiety of DHA. We therefore mutated the FABP7 Phe-104/Arg-126/Tyr-128 residues to Ala-104/Ala-126/Ala-128 (FABP7FAB) to gain further insight into the importance of fatty acid binding to FABP7-mediated migration in MG cells. FABP7FAB was cloned into the pcDNA3.1 expression vector and transfected into U87-FABP7(−) MG cells. Western blot analysis demonstrated the presence of FABP7FAB in the cytoplasm with little mutant protein in the nucleus (Fig. 6). The abundance of FABP7FAB in the cytoplasm indicates that any structural changes resulting from the site-directed mutagenesis had little or no effect on protein stability. Subcellular localization experiments verified that FABP7FAB mostly resides in the cytoplasm, in contrast to wild-type FABP7 (FABP7WT), which is abundant in the nucleus (Fig. 7, A and B). These findings are in keeping with our previous results indicating that serum starvation prevents FABP7 from accumulating in the nucleus.
FIGURE 6.
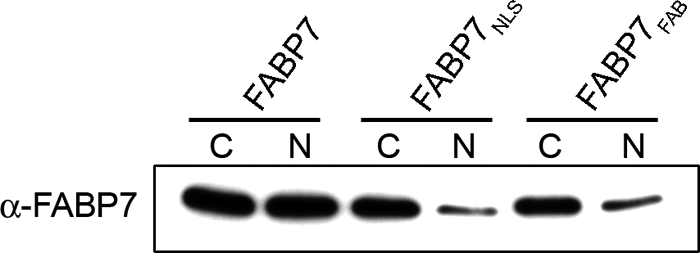
FABP7 levels in U87MG cells transfected with wild-type and mutant FABP7 expression constructs. U87MG cells were transfected with pcDNA3.1-FABP7WT, -FABP7NLS, and -FABP7FAB expression constructs. Cytoplasmic (C) and nuclear (N) fractions were prepared, and 25 μg of protein was loaded in each lane. Proteins were electrophoresed in a 13.5% SDS-polyacrylamide gel, transferred to nitrocellulose membranes, and immunostained with anti-FABP7 antibody. The signal was detected using ECL reagent (GE Healthcare.
FIGURE 7.

Fatty acid binding is required for migration. Subcellular localization of FABP7 in U87MG cells transiently transfected with pcDNA3.1-FABP7WT (A) or pcDNA3.1-FABP7FAB (B) expression constructs. Transfection efficiency was ∼40%. Immunofluorescence was carried out 48 h after transfection using anti-FABP7 antibody followed by Alexa 488-conjugated secondary antibody. DNA was counterstained with DAPI. C, migration of U87MG cells transfected with empty vector, pcDNA3.1-FABP7WT, and pcDNA3.1-FABP7FAB was measured using the Transwell assay as described for Fig. 1. Statistical significance was determined using the unpaired t test (n = 3; *, p < 0.05). Error bars represent standard deviation.
Next, we assessed the binding of fatty acids to recombinant FABP7FAB using Lipidex and radiolabeled DHA. As shown in Fig. 8, there was no detectable binding of FABP7FAB to [1-14C]DHA. These results confirm the predictions made from the three-dimensional model of the FABP7-ligand complex and indicate that the three mutated residues are essential for fatty acid binding to FABP7.
FIGURE 8.
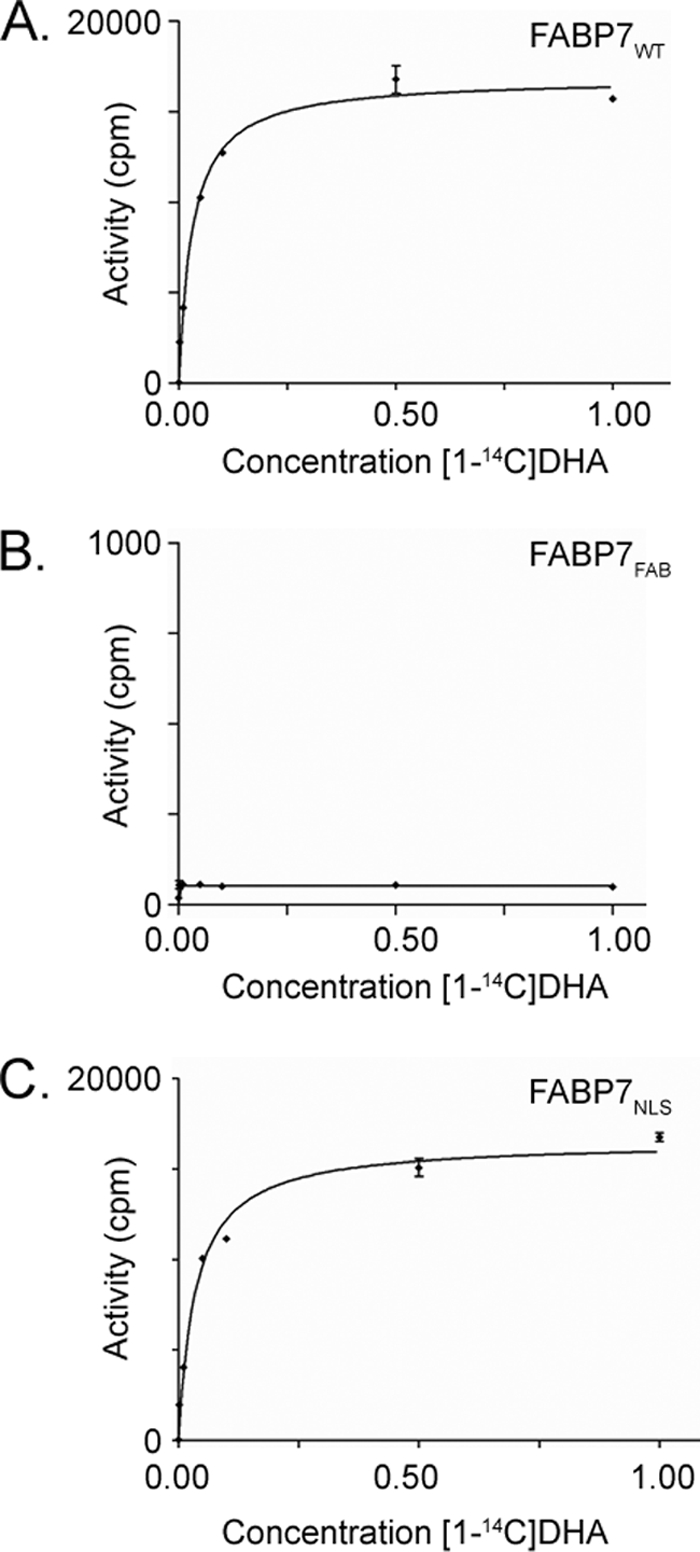
Binding of DHA to FABP7WT, FABP7FAB, and FABP7NLS proteins. Reactions were carried out in the presence of different concentrations of [1-14C]DHA ranging from 0.001 to 5.0 μm (0.001, 0.01, 0.05, 0.1, 0.5, 1, and 5 μm) using 5 μg of recombinant FABP7WT (A), FABP7FAB (B), and FABP7NLS (C). Data points are the mean ± S.D. (n = 3).
To directly test the effect of fatty acid binding on FABP7-mediated MG cell migration, U87 cells were transiently transfected with FABP7WT or FABP7FAB expression constructs, and migration assays were conducted 48 h post-transfection. As shown in Fig. 7C, transient transfection of FABP7WT resulted in a significant increase in migration compared with control transfectants, with an average of 736 cells and 400 cells migrating to the bottom chamber, respectively (p < 0.05). Cells transiently transfected with the FABP7FAB expression construct showed no increase in cell migration compared with control cells with an average of 329 cells migrating to the lower chamber. These results indicate that FABP7-mediated migration is dependent on FABP7 binding to its fatty acid ligands.
FABP7-induced Migration Is Independent of Nuclear Localization
In light of the inhibitory effects on migration observed in the presence of DHA, and the nuclear localization of FABP7 in the presence of DHA, we reasoned that the increased migration observed in FABP7-expressing U87 cells might be mediated through AA and cytoplasmic FABP7. To address this hypothesis, we generated a mutant FABP7 that could no longer localize to the nucleus. The NLS of FABP4 consists of three basic residues situated in the N-terminal helix-loop-helix region (26). The sequence similarity and structural homology of FABP4 to FABP7 raise the possibility that nuclear import of the latter may also be mediated through an N-terminal NLS, consisting of conserved residues Lys-21, Arg-30, and Gln-31. An FABP7 mutant with all three residues mutated to alanines (FABP7NLS) was generated and cloned into pcDNA3.1. FABP7NLS and FABP7WT were transfected into U87 cells and protein distribution patterns examined. Overall levels of FABP7NLS and FABPWT were similar in transfected cells, although very little FABP7NLS was detected in the nuclear fraction (Fig. 6). FABP7NLS was clearly excluded from the nucleus based on immunofluorescence analysis (Fig. 9A).
FIGURE 9.

FABP7-induced cell migration is independent of nuclear localization. A, U87MG cells were transfected with a pcDNA3.1-FABP7NLS expression construct. FABP7NLS localization was analyzed 48 h after transfection by immunofluorescence using anti-FABP7 antibody followed by Alexa 488-conjugated secondary antibody. DNA was counterstained with DAPI to illustrate the location of FABP7 relative to the nucleus. B, cell migration of U87-FABP7WT and U87-FABP7NLS cells treated with BSA, 60 μm AA, or 60 μm DHA was measured using the Transwell assay as described for Fig. 1. Statistical significance was determined using the unpaired t test (n = 6; *, p < 0.05). Error bars represent standard deviation.
As the subcellular localization pattern of the NLS mutant was similar to that of the FABP7FAB mutant, we examined whether the ability of FABP7NLS to bind to fatty acid ligands was compromised. Based on Fig. 8C, there was no impairment in fatty acid binding to FABP7NLS. The similarity of the FABP7NLS and FABP7WT binding curves indicates that the NLS mutations did not interfere with fatty acid binding.
We next examined the effect of the FABP7NLS mutant on cell migration. U87 cells were transiently transfected with FABP7WT or FABP7NLS expression constructs and cultured in the presence of 10% FCS supplemented with 60 μm BSA (control), DHA, or AA. Expression of either wild-type or NLS mutant construct resulted in a significant increase in migration (average of 736 or 687 cells/well) compared with U87 cells transfected with empty vector (average of 400 cells/well) (Fig. 9B). However, in contrast to FABP7WT-transfected cells, DHA had no significant effect on the migration of FABP7NLS-transfected cells. A small but significant effect was seen on migration when FABP7NLS-transfected cells were cultured in the presence of AA, with an average of 802 cells/well (p < 0.05). These results indicate that FABP7 localization to the nucleus is not required for FABP7-mediated cell migration; however, inhibition of migration by DHA is dependent on localization of FABP7 to the nucleus.
Inhibition of COX-2 Blocks FABP7-induced Cell Migration
COX-2 is up-regulated in a variety of cancers, including malignant gliomas (27). Importantly, the conversion of AA to prostaglandins by COX-2 has been linked to an increase in cell migration (28). PGE2 is a primary metabolite of AA and has been shown to function as a mediator of cell migration (29). COX-2 protein levels are increased ∼5-fold in U87-FABP7(+) compared with U87-FABP7(−) cells (Fig. 10A). This increase in COX-2 levels is accompanied by a concomitant >6 times increase in PGE2 production (Fig. 10B). To determine whether COX-2 might be involved in the increase in cell migration observed in FABP7-expressing U87 cells, cells cultured in 60 μm AA were treated with the COX-2 inhibitor NS398 and tested for cell migration using the Transwell assay. A significant decrease (46%; p < 0.01) in the number of cells migrating to the bottom chamber was observed in the presence of the COX-2 inhibitor (Fig. 10C). In comparison, there were 5-fold fewer migrating cells when the medium was supplemented with DHA. These results suggest opposing pathways governing cell migration in FABP7-expressing MG cells, with AA/COX-2 increasing migration through cytoplasmic FABP7 and DHA decreasing migration through nuclear FABP7.
FIGURE 10.

COX-2 involvement in FABP7-induced cell migration. A, Western blot analysis of whole cell lysates (25 μg/lane) prepared from U87 cells stably transfected with FABP7 or empty vector. Lysates were electrophoresed in a 13.5% SDS-polyacrylamide gel, and the proteins were transferred to nitrocellulose membranes. Membranes were sequentially immunostained with goat anti-COX-2 antibody and mouse anti-actin antibody. B, ELISA was used to measure PGE2 levels in U87-FABP7(−) versus U87-FABP(+) cell populations. The data were obtained from two independent experiments measured in triplicate. No error bars are shown for U87-FABP7(+) as all the wells were saturated for PGE2. C, U87-FABP7(+) stable transfectants were treated 60 μm AA, 60 μm DHA, and/or 200 μm NS398, as indicated. Cell migration was measured using the Transwell assay as described previously. Statistical significance was determined using the unpaired t test (*, p < 0.01). Error bars represent standard deviation.
Nuclear Receptors PPARβ and PPARγ Play a Role in FABP7-induced Cell Migration
As FABPs can interact with and activate members of the PPAR family in a fatty acid-dependent manner, we examined the expression of PPARs in U87-FABP7(−) and U87-FABP7(+) cultures. As shown in Fig. 11A, FABP7 expression in U87 cells was accompanied by an increase in PPARβ and a decrease in PPARγ levels. PPARα was not affected by FABP7 expression. To determine whether PPARβ and/or PPARγ might be involved in FABP7-induced cell migration, we used siRNAs to reduce PPARβ and PPARγ protein levels in U87-FABP7(+) cells (Fig. 11B). All three populations of transfected cells (scrambled siRNA control, PPARβ siRNA, and PPARγ siRNA) were cultured in growth medium supplemented with BSA, AA, or DHA for 24 h, and cell migration was examined using the Transwell assay. A significant decrease in migration was observed in cells transfected with PPARβ siRNA compared with cells transfected with scrambled siRNA (1065 versus 1712 cells/well for BSA control; 206 versus 365 cells/well for DHA-treated cells; 843 versus 2035 for AA-treated cells) (Fig. 11C). No such effect was seen in cells transfected with PPARγ siRNA (1712 versus 1729 cells/well, respectively for BSA control). Intriguingly, there was a partial reversal of DHA inhibition upon PPARγ knockdown (791 cells/well for DHA-treated PPARγ knockdown compared with 365 cells/well in DHA-treated control). Knockdown of PPARγ in the presence of AA had little effect on cell migration (2317 cells/well in control cells treated with AA compared with 2035 cells/well in PPARγ knockdown cells treated with AA). These combined data suggest that the effect of AA on cell migration is not mediated through either PPARβ or PPARγ but that the inhibitory effect of DHA on cell migration is mediated through PPARγ.
FIGURE 11.

Role of PPARs in FABP7/FA-mediated cell migration. A, Western blot analysis of PPARα, PPARβ, and PPARγ in whole cell lysates (25 μg/lane) prepared from U87-FABP7(−) and U87-FABP7(+) cell populations. Lysates were electrophoresed in a 13.5% SDS-polyacrylamide gel. B, Western blot of U87-FABP7(+) cells transfected with scrambled siRNA (control), siRNAs targeting PPARβ, or PPARγ. C, 24 h after transfection, cells cultured in DMEM plus 10% FCS were treated with 60 μm BSA, 60 μm DHA, or 60 μm AA for an additional 24 h and subjected to the Transwell migration assay. The data were obtained from two independent experiments carried out in triplicate.
DISCUSSION
The hallmark of MG cells is their ability to infiltrate surrounding brain tissue, thus escaping therapies aimed at eradicating the main tumor mass. Importantly, MG is rarely metastatic, suggesting that controlling local disease may be key to the successful treatment of these tumors. FABP7, a radial glia/neural progenitor cell marker, is often expressed in MG where it localizes to regions associated with tumor infiltration such as the subpial surface and surrounding blood vessels (10). FABP7 expression is associated with decreased patient survival in grade IV astrocytomas (6, 30). Manipulation of FABP7 levels in MG cell lines demonstrates a direct link between FABP7 expression and cell migration, with increased FABP7 levels associated with increased migration (10). Here, we provide evidence of a central role for FABP7 in regulating the growth properties of MG cells, particularly as related to cell migration.
FABP7 binds the long chain PUFAs DHA (Kd 53 nm) and AA (Kd 207 nm) (11). Treatment of cells with DHA and AA generally leads to opposite outcomes, with DHA having anti-tumorigenic properties and AA having pro-tumorigenic properties (12). Our results support a role for FABP7 at the apex of two competing fatty acid-dependent signaling pathways, one pro-migratory, the other anti-migratory. In particular, our data indicate the following: (i) FABP7-mediated cell migration is dependent on availability of AA and activation of COX-2 and PGE2 production, and (ii) FABP7 bound to DHA has anti-migratory effects through activation of the nuclear receptor PPARγ. Importantly, these pro- and anti-migratory effects are governed by the ratio of DHA to AA rather than their empirical levels in the cell.
Transfection of FABP7 into U87MG, a cell line that normally does not express FABP7, is accompanied by increased cell migration in regular growth medium (3:1 ratio of AA:DHA), as well as increased COX-2 expression. A significant proportion of FABP7 is expected to be bound to AA under these conditions, suggesting a functional link between FABP7 bound to AA and COX-2. In support of this idea, cyclooxygenases, along with lipooxygenases, convert AA into eicosanoids, biologically active signaling molecules that include PGE2 (31, 32). Eicosanoids stimulate cell proliferation and promote cell survival and migration by activating factors such as VEGF and EGF receptor (33). Thus, one can postulate a feedback loop whereby increased FABP7/AA gives rise to increased PGE2 production (through increased COX-2 expression) which in turn maintains elevated levels of COX-2, and drives the pro-migratory phenotype (Fig. 12).
FIGURE 12.

Model of cytoplasmic and nuclear roles for FABP7/fatty acids in cell migration. Cytosolic FABP7 can bind to either DHA or AA depending on the relative abundance and availability of these two fatty acids. Binding of FABP7 to AA induces cell migration through transfer of AA to the COX-2 pathway, whereby AA is metabolized into pro-migratory signaling molecules such as PGE2. Binding of FABP7 to DHA results in translocation of FABP7 to the nucleus where DHA is transferred to PPARγ, resulting in down-regulation of pro-migratory genes.
COX-2 is overexpressed in many different types of cancers, including breast, prostate, pancreas, skin, and lung and is found at higher levels in grades III and IV astrocytomas than in low grade astrocytomas and normal brain (27). Others have reported that treatment of U251 (FABP7+) and U87 (FABP7−) with the COX-2 inhibitor, NS398, results in reduced migration in both cell lines (34). Inhibition of cell migration in U87 was shown to be an indirect effect, caused by increased apoptosis and decreased cell proliferation. It may therefore be useful to measure FABP7 expression to predict the effect of AA and COX-2 on MG growth properties.
It is well established that DHA suppresses tumorigenesis in vivo (35, 36) and cancer cell proliferation in vitro (37, 38); however, the mechanism by which DHA exerts its tumor suppressing activity remains controversial, with competing theories proposing that DHA can indirectly or directly modulate gene expression (39). Indirect effects may be a consequence of DHA competing with AA for modification by COX, leading to the production of metabolically inactive compounds such as Δ17-6-keto-PGF and thromboxane B3 (40). However, the role of COX proteins in the conversion of DHA into secondary compounds remains to be proven. In contrast, direct effects of DHA have been postulated to be a consequence of binding and activating transcription factors (e.g. PPARs and RXRs) in the nucleus (41).
Our results show that DHA is a strong inhibitor of AA/COX-2/PGE2-mediated cell migration and that this anti-migratory effect is dependent not only on FABP7 but on localization of FABP7 to the nucleus. Furthermore, our data demonstrate that binding of DHA to FABP7 allows accumulation of FABP7 in the nucleus, indicating that DHA binding may alter the configuration of FABP7 in such as way as to promote its translocation to the nucleus. This possibility is supported by x-ray crystallography data indicating that binding of FABP4 to nucleus-homing ligands stabilizes structures that expose a nuclear localization signal (42).
Protein interaction studies by fluorescence resonance energy transfer and co-immunoprecipitation indicate that FABPs and fatty acids can cooperate in the activation of transcription factors in the nucleus (13–15). There is a growing body of evidence indicating that ligand-dependent activation of PPARs can inhibit tumor growth. For example, activation of PPARγ in MG using synthetic ligands can induce apoptosis and inhibit growth, migration, and invasion in animal models (43–46).
DHA has been reported to bind PPARγ but not PPARα or PPARβ (47, 48). Using siRNAs targeting either PPARβ or PPARγ, we demonstrate that PPARγ is important for DHA-dependent inhibition of migration in U87-FABP7(+) cells. We postulate that FABP7 activates PPARγ in the nucleus by DHA-ligand transfer, thereby inhibiting cell migration through alteration in the transcription of PPARγ target genes (Fig. 12). In light of studies demonstrating that activated PPARγ down-regulates COX-2 expression (49), we propose that COX-2 may be a direct target of PPARγ. In keeping with our observation that PPARγ levels are decreased and COX-2 levels are increased in U87-FABP7(+) versus U87-FABP7(−) cells, others have reported that PPARγ is down-regulated in response to prostaglandins produced by COX-2 (50). These data indicate complex feedback loops regulating the FABP7/PUFA/COX-2/PPARγ pathways. To add additional levels of complexity, our data suggest a relationship between PPARβ and FABP7, and PPARs have been directly implicated in the transcription of at least four FABPs (FABP1–4) (51–53).
Our experiments indicate that the migratory response of FABP7-expressing U87 cells depends on the ratio of DHA:AA, suggesting cross-talk between the DHA- and AA-related signaling/gene regulation pathways through FABP7. Such molecular cross-talk is not surprising because FABP7-expressing cells are unlikely to be exposed to only AA or DHA in their natural environment. During brain development, when cell migration is important and FABP7-expressing radial glial cells are abundant, levels of AA exceed that of DHA (54). The DHA:AA ratio gradually increases upon brain maturation, along with a reduced need for cell migration and down-regulation of FABP7 (54). A number of reports indicate that the DHA:AA ratio is considerably lower in MG compared with adult brain tissue (55, 56). Thus, FABP7 may be more likely to be bound to AA than DHA in MG tumors compared with brain tissue. Combined with the increased levels of FABP7 in MG and the association between FABP7 and MG cell migration, our data support a key role for FABP7 in controlling the infiltrative/migratory properties of MG tumors. However, it is clear that factors other than FABP7 can influence cell migration and response to AA and DHA, as evidenced by results obtained with MG cell lines that naturally express FABP7. In this regard, it is noteworthy that FABP5, which is highly expressed in the nonmigratory FABP7(+) M049 cell line, binds poorly to both DHA and AA (21) and interacts with PPARβ, in contrast to FABP7, which binds strongly to DHA and interacts with PPARγ.
Many phase II clinical trials involving MG have been carried out over the past years. Some of these clinical trials, including one involving both COX-2 inhibitors and PPARγ agonists (57), have shown moderate effects suggesting that the treatment regimen might be suited to only a subset of patients. As DHA/AA, COX-2, and PPARγ appear to be linked through FABP7, FABP7 may be a key factor in controlling which signaling pathways are activated or repressed in MG. We propose that early use of DHA as an adjuvant therapy in those tumors that express FABP7 may alter the migration potential and infiltrative properties of MG cells.
Acknowledgments
We thank Judy Lin, Stanley Poon, and Darryl Glubrecht for their expert technical assistance with cloning and protein preparations.
This work was supported by a grant from the Alberta Cancer Research Institute (to R. G.) and studentships from the Alberta Cancer Foundation (to R. M.) and the Canadian Institutes of Health Research (to M. J. B.).
- MG
- malignant glioma
- FABP
- fatty acid-binding protein
- DHA
- docosahexaenoic acid
- AA
- arachidonic acid
- PPAR
- peroxisome proliferator-activated receptor
- PUFA
- polyunsaturated fatty acid
- PA
- palmitic acid
- PG
- prostaglandin
- NLS
- nuclear localization signal.
REFERENCES
- 1.Lin C. L., Lieu A. S., Lee K. S., Yang Y. H., Kuo T. H., Hung M. H., Loh J. K., Yen C. P., Chang C. Z., Howng S. L., Hwang S. L. (2003) Surg. Neurol. 60, 402–406 [DOI] [PubMed] [Google Scholar]
- 2.Mason W. P., Maestro R. D., Eisenstat D., Forsyth P., Fulton D., Laperrière N., Macdonald D., Perry J., Thiessen B. (2007) Curr. Oncol. 14, 110–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Godbout R., Bisgrove D. A., Shkolny D., Day R. S., 3rd (1998) Oncogene 16, 1955–1962 [DOI] [PubMed] [Google Scholar]
- 4.Liang Y., Diehn M., Watson N., Bollen A. W., Aldape K. D., Nicholas M. K., Lamborn K. R., Berger M. S., Botstein D., Brown P. O., Israel M. A. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 5814–5819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tso C. L., Shintaku P., Chen J., Liu Q., Liu J., Chen Z., Yoshimoto K., Mischel P. S., Cloughesy T. F., Liau L. M., Nelson S. F. (2006) Mol. Cancer Res. 4, 607–619 [DOI] [PubMed] [Google Scholar]
- 6.Kaloshi G., Mokhtari K., Carpentier C., Taillibert S., Lejeune J., Marie Y., Delattre J. Y., Godbout R., Sanson M. (2007) J. Neurooncol. 84, 245–248 [DOI] [PubMed] [Google Scholar]
- 7.Zhang H., Rakha E. A., Ball G. R., Spiteri I., Aleskandarany M., Paish E. C., Powe D. G., Macmillan R. D., Caldas C., Ellis I. O., Green A. R. (2010) Breast Cancer Res. Treat. 121, 41–51 [DOI] [PubMed] [Google Scholar]
- 8.Seliger B., Lichtenfels R., Atkins D., Bukur J., Halder T., Kersten M., Harder A., Ackermann A., Malenica B., Brenner W., Zobawa M., Lottspeich F. (2005) Proteomics 5, 2631–2640 [DOI] [PubMed] [Google Scholar]
- 9.Slipicevic A., Jørgensen K., Skrede M., Rosnes A. K., Trøen G., Davidson B., Flørenes V. A. (2008) BMC Cancer 8, 276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mita R., Coles J. E., Glubrecht D. D., Sung R., Sun X., Godbout R. (2007) Neoplasia 9, 734–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balendiran G. K., Schnutgen F., Scapin G., Borchers T., Xhong N., Lim K., Godbout R., Spener F., Sacchettini J. C. (2000) J. Biol. Chem. 275, 27045–27054 [DOI] [PubMed] [Google Scholar]
- 12.Larsson S. C., Kumlin M., Ingelman-Sundberg M., Wolk A. (2004) Am. J. Clin. Nutr. 79, 935–945 [DOI] [PubMed] [Google Scholar]
- 13.Wolfrum C., Borrmann C. M., Borchers T., Spener F. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 2323–2328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tan N. S., Shaw N. S., Vinckenbosch N., Liu P., Yasmin R., Desvergne B., Wahli W., Noy N. (2002) Mol. Cell. Biol. 22, 5114–5127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Helledie T., Jørgensen C., Antonius M., Krogsdam A. M., Kratchmarova I., Kristiansen K., Mandrup S. (2002) Mol. Cell. Biochem. 239, 157–164 [PubMed] [Google Scholar]
- 16.Adida A., Spener F. (2006) Biochim. Biophys. Acta 1761, 172–181 [DOI] [PubMed] [Google Scholar]
- 17.Benedetti E., Galzio R., Laurenti G., D'Angelo B., Melchiorre E., Cifone M. G., Fanelli F., Muzi P., Coletti G., Alecci M., Sotgiu A., Cerù M. P., Cimini A. (2010) Int. J. Immunopathol. Pharmacol. 23, 235–246 [DOI] [PubMed] [Google Scholar]
- 18.Papi A., Tatenhorst L., Terwel D., Hermes M., Kummer M. P., Orlandi M., Heneka M. T. (2009) J. Neurochem. 109, 1779–1790 [DOI] [PubMed] [Google Scholar]
- 19.Chearwae W., Bright J. J. (2008) Br. J. Cancer 99, 2044–2053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang M., Liu Y. E., Ni J., Aygun B., Goldberg I. D., Shi Y. E. (2000) Cancer Res. 60, 6482–6487 [PubMed] [Google Scholar]
- 21.Liu J. W., Almaguel F. G., Bu L., De Leon D. D., De Leon M. (2008) J. Neurochem. 106, 2015–2029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bisgrove D. A., Monckton E. A., Packer M., Godbout R. (2000) J. Biol. Chem. 275, 30668–30676 [DOI] [PubMed] [Google Scholar]
- 23.Brun M., Coles J. E., Monckton E. A., Glubrecht D. D., Bisgrove D., Godbout R. (2009) J. Mol. Biol. 391, 282–300 [DOI] [PubMed] [Google Scholar]
- 24.Cormack B., Castaño I. (2002) Methods Enzymol. 350, 199–218 [DOI] [PubMed] [Google Scholar]
- 25.Simopoulos A. P. (2009) World Rev. Nutr. Diet. 99, 1–16 [DOI] [PubMed] [Google Scholar]
- 26.Ayers S. D., Nedrow K. L., Gillilan R. E., Noy N. (2007) Biochemistry 46, 6744–6752 [DOI] [PubMed] [Google Scholar]
- 27.Perdiki M., Korkolopoulou P., Thymara I., Agrogiannis G., Piperi C., Boviatsis E., Kotsiakis X., Angelidakis D., Diamantopoulou K., Thomas-Tsagli E., Patsouris E. (2007) Mol. Cell. Biochem. 295, 75–83 [DOI] [PubMed] [Google Scholar]
- 28.Giese A., Hagel C., Kim E. L., Zapf S., Djawaheri J., Berens M. E., Westphal M. (1999) Neuro. Oncol. 1, 3–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buchanan F. G., Wang D., Bargiacchi F., DuBois R. N. (2003) J. Biol. Chem. 278, 35451–35457 [DOI] [PubMed] [Google Scholar]
- 30.Liang Y., Bollen A. W., Aldape K. D., Gupta N. (2006) BMC Cancer 6, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Funk C. D. (2001) Science 294, 1871–1875 [DOI] [PubMed] [Google Scholar]
- 32.Soberman R. J., Christmas P. (2003) J. Clin. Invest. 111, 1107–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu T. (2006) Cancer Treat. Rev. 32, 28–44 [DOI] [PubMed] [Google Scholar]
- 34.Joki T., Heese O., Nikas D. C., Bello L., Zhang J., Kraeft S. K., Seyfried N. T., Abe T., Chen L. B., Carroll R. S., Black P. M. (2000) Cancer Res. 60, 4926–4931 [PubMed] [Google Scholar]
- 35.Noguchi M., Minami M., Yagasaki R., Kinoshita K., Earashi M., Kitagawa H., Taniya T., Miyazaki I. (1997) Br. J. Cancer 75, 348–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rose D. P., Connolly J. M., Coleman M. (1996) Clin. Cancer Res. 2, 1751–1756 [PubMed] [Google Scholar]
- 37.Chajès V., Sattler W., Stranzl A., Kostner G. M. (1995) Breast Cancer Res. Treat. 34, 199–212 [DOI] [PubMed] [Google Scholar]
- 38.Connolly J. M., Gilhooly E. M., Rose D. P. (1999) Nutr. Cancer 35, 44–49 [DOI] [PubMed] [Google Scholar]
- 39.Biondo P. D., Brindley D. N., Sawyer M. B., Field C. J. (2008) J. Nutr. Biochem. 19, 787–796 [DOI] [PubMed] [Google Scholar]
- 40.Rose D. P., Connolly J. M. (1999) Pharmacol. Ther. 83, 217–244 [DOI] [PubMed] [Google Scholar]
- 41.Crawford M. A., Golfetto I., Ghebremeskel K., Min Y., Moodley T., Poston L., Phylactos A., Cunnane S., Schmidt W. (2003) Lipids 38, 303–315 [DOI] [PubMed] [Google Scholar]
- 42.Gillilan R. E., Ayers S. D., Noy N. (2007) J. Mol. Biol. 372, 1246–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zang C., Wächter M., Liu H., Posch M. G., Fenner M. H., Stadelmann C., von Deimling A., Possinger K., Black K. L., Koeffler H. P., Elstner E. (2003) J. Neurooncol. 65, 107–118 [DOI] [PubMed] [Google Scholar]
- 44.Coras R., Hölsken A., Seufert S., Hauke J., Eyüpoglu I. Y., Reichel M., Tränkle C., Siebzehnrübl F. A., Buslei R., Blümcke I., Hahnen E. (2007) Mol. Cancer Ther. 6, 1745–1754 [DOI] [PubMed] [Google Scholar]
- 45.Grommes C., Landreth G. E., Sastre M., Beck M., Feinstein D. L., Jacobs A. H., Schlegel U., Heneka M. T. (2006) Mol. Pharmacol. 70, 1524–1533 [DOI] [PubMed] [Google Scholar]
- 46.Spagnolo A., Glick R. P., Lin H., Cohen E. P., Feinstein D. L., Lichtor T. (2007) J. Neurosurg. 106, 299–305 [DOI] [PubMed] [Google Scholar]
- 47.Itoh T., Fairall L., Amin K., Inaba Y., Szanto A., Balint B. L., Nagy L., Yamamoto K., Schwabe J. W. (2008) Nat. Struct. Mol. Biol. 15, 924–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Banga A., Unal R., Tripathi P., Pokrovskaya I., Owens R. J., Kern P. A., Ranganathan G. (2009) Am. J. Physiol. Endocrinol. Metab. 296, E480–E489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bren-Mattison Y., Meyer A. M., Van Putten V., Li H., Kuhn K., Stearman R., Weiser-Evans M., Winn R. A., Heasley L. E., Nemenoff R. A. (2008) Mol. Pharmacol. 73, 709–717 [DOI] [PubMed] [Google Scholar]
- 50.Yan H., Kermouni A., Abdel-Hafez M., Lau D. C. (2003) J. Lipid Res. 44, 424–429 [DOI] [PubMed] [Google Scholar]
- 51.Poirier H., Niot I., Monnot M. C., Braissant O., Meunier-Durmort C., Costet P., Pineau T., Wahli W., Willson T. M., Besnard P. (2001) Biochem. J. 355, 481–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fujishiro K., Fukui Y., Sato O., Kawabe K., Seto K., Motojima K. (2002) Mol. Cell. Biochem. 239, 165–172 [PubMed] [Google Scholar]
- 53.Motojima K. (2000) Int. J. Biochem. Cell Biol. 32, 1085–1092 [DOI] [PubMed] [Google Scholar]
- 54.Liu R. Z., Mita R., Beaulieu M., Gao Z., Godbout R. (2010) Int. J. Dev. Biol. 54, 1229–1239 [DOI] [PubMed] [Google Scholar]
- 55.Martin D. D., Robbins M. E., Spector A. A., Wen B. C., Hussey D. H. (1996) Lipids 31, 1283–1288 [DOI] [PubMed] [Google Scholar]
- 56.Marszałek R., Pisklak M., Jankowski W., £ukaszkiewicz J., Horsztyñski D., Wawer I. (2010) Acta Pol. Pharm. 67, 129–136 [PubMed] [Google Scholar]
- 57.Hau P., Kunz-Schughart L., Bogdahn U., Baumgart U., Hirschmann B., Weimann E., Muhleisen H., Ruemmele P., Steinbrecher A., Reichle A. (2007) Oncology 73, 21–25 [DOI] [PubMed] [Google Scholar]