

Abstract
Hyperpolarization-activated cAMP-regulated (HCN) channels play important physiological roles in both cardiovascular and central nervous systems. Among the four HCN isoforms, HCN2 and HCN4 show high expression levels in the human heart, with HCN4 being the major cardiac isoform. The previously published crystal structure of the mouse HCN2 (mHCN2) C-terminal fragment, including the C-linker and the cyclic-nucleotide binding domain (CNBD), has provided many insights into cAMP-dependent gating in HCN channels. However, structures of other mammalian HCN channel isoforms have been lacking. Here we used a combination of approaches including structural biology, biochemistry, and electrophysiology to study cAMP-dependent gating in HCN4 channel. First we solved the crystal structure of the C-terminal fragment of human HCN4 (hHCN4) channel at 2.4 Å. Overall we observed a high similarity between mHCN2 and hHCN4 crystal structures. Functional comparison between two isoforms revealed that compared with mHCN2, the hHCN4 protein exhibited marked different contributions to channel function, such as a ∼3-fold reduction in the response to cAMP. Guided by structural differences in the loop region between β4 and β5 strands, we identified residues that could partially account for the differences in response to cAMP between mHCN2 and hHCN4 proteins. Moreover, upon cAMP binding, the hHCN4 C-terminal protein exerts a much prolonged effect in channel deactivation that could have significant physiological contributions.
Keywords: Cyclic AMP (cAMP), Ion Channels, Ligand Binding Protein, Protein Structure, X-ray Crystallography, Cyclic-nucleotide Binding Domain, Human HCN4 Channel, Mouse HCN2 Channel
Introduction
Ion channels form the molecular basis for cell membrane excitability (1). Other than under the direct influence of membrane potential changes, ion channels also respond to chemical cues acting from either the extracellular or intracellular side. Cyclic nucleotides, including cAMP and cGMP (together hereafter referred to as cNMP), are important intracellular second messengers. They bind directly to and regulate not only protein kinases like PKA but also transmembrane ion channels. In the superfamily of voltage-gated potassium channels, CNG, HCN, and EAG channels all contain a sequence fragment homologous to other cNMP-binding proteins downstream from the last transmembrane domain (S6) on the intracellular side (2–5). Indeed, both cAMP and cGMP directly bind to and open CNG and HCN channels. CNG channels function mainly in the olfactory and visual sensory systems; they open upon direct cNMP binding but respond weakly to membrane potential changes. On the other hand, HCN channels are mainly voltage-gated and open upon membrane hyperpolarization (6). The opening of HCN channels can be dramatically facilitated upon cNMP binding. cNMP binding shifts the voltage-dependent channel activation curve toward more positive potentials, increasing the macroscopic current amplitude and making the channel activate faster and deactivate slower.
Each functional HCN channel is composed of four subunits, and each subunit contains a transmembrane domain, similar to other voltage-gated K+ channels, and a C-terminal cyclic nucleotide binding domain (CNBD).3 Between the last transmembrane domain S6 and CNBD, there is a 90-amino acid sequence called the C-linker. Previous extensive biophysical studies have provided valuable insights into cAMP-dependent binding and gating for mammalian HCN channels (7–9). In 2003, the structure of the mouse HCN2 C-terminal fragment was published (10). This crystal structure contained the C-linker and CNBD, with either cAMP or cGMP bound to the binding pocket. Each CNBD has a very similar fold as other cNMP-binding proteins, containing eight β-strands (1–8) flanked by four α-helixes (A-C and P). The negatively charged cyclic-phosphate group in cNMP interacts with a highly conserved positively charged arginine residue (Arg591), which has been shown to be critical for cNMP binding in both CNG and HCN channels (11–13). The C-linker region is composed of 6 α-helixes (A′-F′). Through intricate inter-subunit interactions, especially interactions in the C-linker region, four subunits form a tetrameric structure with 4-fold symmetry. Stemmed from the crystal structure of the mouse HCN2 protein, other biophysical studies further enhanced our understanding of ligand binding and gating in HCN channels (12, 14–16).
The mammalian HCN channel family contains four homologous members, HCN1, HCN2, HCN3, and HCN4. Among them, HCN2 and HCN4 are the dominant forms expressed in heart. In the human heart, HCN2 shows significant expression in various regions, especially in ventricle (17, 18). The expression level of HCN4 is much higher, especially in the sinoatrial node, where impulses initiate and propagate throughout the heart. Cardiac automaticity is accomplished through a complicated coordination among different types of ion channels and transporters and involves not only HCN channels but also various calcium, potassium, sodium channels, and sodium-calcium exchangers, etc. (19). Multiple lines of evidence support the important roles played by HCN channels in cardiac pacemaking. For example, several clinically approved compounds used for slowing heart rate, such as ivabradine, are rather specific HCN channel blockers (20). Genetic evidence from transgenic mice, and more importantly, human patients, directly associate HCN channel genes with cardiac automaticity (21, 22). Currently, it is widely accepted that the HCN channel is one of the major players involved in cardiac physiology; however, the exact role it plays during heart pacemaking and cardiac automaticity is still an open question.
To date, five different mutations in the HCN4 channel have been reported in human patients showing sick sinus syndromes. Interestingly, three of these mutations are single-point mutations located in the C-terminal fragment containing the C-linker and CNBD domain (22). One of these mutations leads to deletion of the C terminus including CNBD (573X) (23), whereas the other two are single amino acid missense mutations, leading to either reduced channel expression (D553N) or compromised channel function (S672R) (24, 25). Corresponding to the loss-of-function phenotype of these mutant HCN4 channels, patients carrying these mutations show symptoms of cardiac arrhythmias or bradycardia (slower heart rate). Thus, these observations have further underscored the critical roles played by HCN channels and the regulation by cAMP in cardiac physiology.
Even though all four HCN channel isoforms share high sequence identity, they respond differently to increases in intracellular cAMP concentration. Indeed, when we replaced the CNBD and C-linker domain of HCN2 with the corresponding ones from HCN4, we observed a marked difference in cAMP-dependent regulation of channel gating from HCN2 controls. We proceeded to crystallize the human HCN4 C-linker and CNBD fragment and solved its structure in complex with cAMP to 2.4-Å resolution. Furthermore, we identified structural differences between hHCN4 and mHCN2 proteins and carried out further studies to investigate the different responses to cAMP between two HCN isoforms.
EXPERIMENTAL PROCEDURES
Functional Expression in Xenopus Oocytes and Electrophysiological Characterization
To make a chimera between hHCN4 and mHCN2, we first amplified by PCR the C-linker and CNBD region from hHCN4. Then we cut and inserted this DNA fragment (Asp521-Asn739) into the pGH-mHCN2 vector through restriction sites of pflmI and BsmI. Plasmids encoding HCN channels in the pGH vector were linearized overnight by digestion with SphI and further purified by phenol-chloroform extraction. mMessage machine (Ambion) was used for in vitro cRNA synthesis. 40–50 ng of cRNA was injected into each oocyte at stage IV. After incubating the injected oocyte at 17 °C for 3–5 days, we recorded macroscopic HCN channel currents from membrane patches using the inside-out patch clamp recording configuration. We used the following pipette/bath solution during experiments (in mm): KCl, 107; NaCl, 5; MgCl2, 1; EGTA, 1; HEPES, 10, pH 7.4, adjusted by KOH. We used EPC9 (a generous gift from Dr. Siegelbaum) for data acquisition, Pulsefit 8.0 for data analysis, and OriginPro 8.0 for graph plotting.
DNA Cloning, Protein Expression, and Purification
A DNA fragment corresponding to residues 521–724 of the human HCN4 channel (hereafter referred to as hHCN4C) was cloned into pSMT3 vector through BamHI and XhoI sites. The resulting plasmid, pSMT3-hHCN4C, was transformed into Escherichia coli BL21(DE3) Gold (Novagen) cells where hHCN4C was expressed as a Smt3 fusion protein with a N-terminal His tag. This fusion protein was expressed with the autoinduction protocol according to Dr. William Studier (26).
After induction, cells were harvested by centrifugation, resuspended in ice-cold 2× PBS, and then lysed with sonication. The following purification steps were carried out at 4 °C. The Smt3-hHCN4C fusion protein was first purified on a HisTrap column (buffer A, 2× PBS + 300 mm NaCl; and buffer B, 2× PBS + 400 mm imidazole). After cleaving the Smt3 tag with Ulp1 protease, the protein sample was dialyzed overnight in a buffer containing 20 mm Tris-HCl, pH 7.5, 300 mm NaCl, and 1 mm DTT. The Smt3 tag was separated from hHCN4C using a HiTrap Q column because Smt3 bound to the Q column, whereas hHCN4C did not. Next we adjusted the pH of the flow- through after a HiTrap Q column to 6.0 with 1 m MES, pH 5.5, and loaded it onto a HiTrap S column. The bound hHCN4C was eluted with a linear NaCl gradient (buffer A, 30 mm MES, pH 6.0; and buffer B, 30 mm MES, pH 6.0, + 1 m NaCl), and two well separated peaks, S1 and S2, were observed. Each peak was concentrated separately and further purified with size-exclusion chromatography on a HiLoad 16/60 Superdex 75 column (buffer used: 20 mm MES, pH 6.0, + 200 mm NaCl). Finally, the purified protein was concentrated to >10 mg/ml in buffer containing 5 mm MES-NaOH, pH 6.0, and 50 mm NaCl. The pSMT3 vector-Ulp1 protease system was a generous gift from Dr. Chris Lima. All columns used during protein purification were from GE Healthcare.
Sedimentation Equilibrium
Concentrated protein samples were separated into two pools and dialyzed against buffers containing (in mm) 20 HEPES, 300 NaCl, and 1 DTT, pH 7.2, either with or without 5 mm cAMP. We used an XL-1 analytical ultracentrifuge (Beckman/Coulter) equipped with Rayleith interference optics. Protein samples were loaded at concentrations of 4, 2, or 1 mg/ml and spun sequentially at 12,000, 17,000, and 25,000 rpm at 4 °C. We monitored the progress of equilibrium by taking scans every hour and analyzed the readings using WinMatch. We used WinReed to process the final scans and then globally fit the traces by WinNonLin. We used a three-species model containing monomer, dimer, and tetramer. After obtaining σ from sedimentation equilibrium experiments, we calculated the molecular weight using SEDNTERP software, using the following equation,
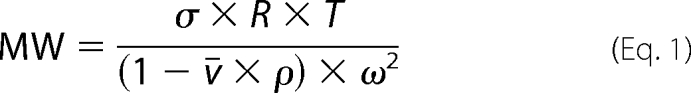 |
which uses the following parameters: ¯ν, partial specific volume; ρ, solvent density, ω, rotor speed; σ, reduced molecular weight, R, gas constant (8.314 × 107 erg/mol/K), T, temperature.
cAMP Binding Assays: Isothermal Titration Calorimetry (ITC) and Fluorescence Anisotropy (FA)
We did cAMP binding assays on purified HCN C-terminal protein samples using two equilibrium methods, ITC and FA. For ITC experiments, we sequentially injected 1.25 mm cAMP into the sample cell containing the protein sample at a concentration of 75 μm at a fixed time interval (iTC200 Microcalorimeter by Microcal). Then we obtained the Kd value by fitting the curve of heat exchange versus protein:cAMP ratio. For FA experiments, we mixed different concentrations of protein samples with a fluorescent cAMP analog (8-Fluo-cAMP from Biolog.de) and used BEACON 2000 to measure the fluorescence anisotropy. We obtained the Kd value from the binding curve of FA versus protein concentration.
Crystallization, Data Collection, and Model Building
Crystallization screens were set up at both room temperature and 4 °C using the hanging drop vapor diffusion method. The concentrated protein was mixed with the reservoir solution at a 1:1 ratio. hHCN4C crystals were grown at 4 °C using 12% ethanol, sodium citrate, pH 5.5, and 200 mm Li2SO4. Prior to flash freezing in liquid nitrogen, these crystals were cryoprotected by soaking in 12% ethanol, sodium citrate, pH 5.5, 200 mm Li2SO4, and 15% MPD.
Two native data sets were collected from a single frozen crystal at the X4C beamline of the National Synchrotron Light Source (NSLS) at the Brookhaven National Laboratory. One native data set diffracted to 2.4 Å was collected at a detector distance of 175 mm. Due to overloaded reflections, a second data set, a low resolution data set, was collected from the same crystal at a detector distance of 300 mm. These two data sets were indexed, merged, and processed using the HKL2000 (27). This crystal belongs to the I422 space group.
Using a previously published mouse HCN2 structure (Protein Data Bank code 1Q43) as search model, we did molecular replacement in PHASER to obtain the phase information (28). Further manual model building was carried out with COOT (29). Initial refinement was performed with CNS using iterative cycles of simulated annealing (30), and in the final stages of the refinement, we used REFMAC (31). The final refined model has overall good stereochemistry with only one Ramachandran outlier, Ala645, on a loop. We used VMD (32) and PYMOL (The PyMOL Molecular Graphics System, Schrödinger, LLC.) for structure presentation.
RESULTS
Functional Differences between the mHCN2 and hHCN4 C-terminal Fragments
Prompted by the differential distribution of HCN2 and HCN4 isoforms in the heart and the different responses to cAMP, we carried out electrophysiological experiments to analyze the function of the human HCN4 C-terminal fragment and compare it to the corresponding part from mouse HCN2, which had been crystallized and well characterized (10). We chose Xenopus oocyte as the expression system and used the inside-out patch-clamp recording configuration to quantify the responses to cAMP. Therefore, we exposed the intracellular side (CNBD) to the bath solution to facilitate the exchange of different concentrations of cAMP. This is advantageous over the whole cell recording configuration on mammalian cells, in which the cAMP concentration is determined by both the pipette solution as well as the intracellular signaling pathways and thus cannot be easily manipulated.
However, unlike the mouse HCN2 channel, the full-length human HCN4 channel does not express well in Xenopus oocytes. Therefore, we used the full-length mHCN2 channel as a reference and replaced its C-linker + CNBD fragment with the corresponding region from hHCN4, aiming to directly compare the cAMP-dependent gating machineries between the two isoforms. We named this chimeric channel mHCN2-h4 (Fig. 1A). Inside-out patches from oocytes expressing this construct showed a robust expression of HCN currents of typical characteristics (Fig. 1, B and C). Applying cAMP to the intracellular side significantly facilitated channel gating for both mHCN2 and mHCN2-h4. Based on the amplitude of tail currents recorded at −40 mV, we generated voltage-dependent channel activation curves and then fit the tail current amplitudes with the Boltzmann equation to obtain the V½ value (Fig. 1, D and E). With relatively high concentrations of cAMP applied (10 μm), we measured the maximal shift in V½ as 16.8 ± 0.7 mV (n = 6) for mHCN2 and 14.9 ± 1.3 mV (n = 6) for mHCN2-h4 (Fig. 1F).
FIGURE 1.

Construction and characterization of the mHCN2-h4 chimeric channel. A, construction of the mHCN2-h4 channel by swapping the C-linker and CNBD region from human HCN4 (red) to mouse HCN2 channel (blue). B, macroscopic currents of mouse HCN2 in response to a series of hyperpolarizing voltage steps in 10-mV intervals. Voltage steps used are shown in the top. Left, control without cAMP; right, 10 μm cAMP applied to the intracellular side. C, macroscopic currents of mHCN2-h4. Voltage steps used are shown in the top. Left, control without cAMP; right, 10 μm cAMP. D, voltage-dependent channel activation curve for mHCN2 channel based on the tail currents measured from the recordings shown in B. Open circle, control no cAMP; filled circle, 10 μm cAMP applied to the intracellular side. E, voltage-dependent channel activation curve for mHCN2-h4 channel based on the recordings shown in C. Open circle, control no cAMP; filled circle, 10 μm cAMP. F, dose-response curves showing the shift in the voltage-dependent channel activation curve (ΔV½) as a function of cAMP concentration (black, mHCN2; red, mHCN2-h4).
We further compared the mHCN2 and mHCN2-h4 proteins by applying different concentrations of cAMP and measuring the corresponding ΔV½ (the shift in V½) and then obtained the dose-response curve. Even though we observed a similar maximal shift with saturating concentrations of cAMP, there was a significant difference in K½ or EC50, the apparent affinity or half-maximal effective concentration, between the two channels (Fig. 1F). For mHCN2, the K½ for cAMP to shift the voltage-dependent channel activation curve was 0.08 ± 0.01 μm with a Hill coefficient of 1.25 ± 0.16. However, when the C-linker and CNBD were replaced by corresponding regions from the hHCN4 protein (mHCN2-h4), the K½ was increased by 3-fold to 0.24 ± 0.02 μm with the Hill coefficient of 1.4 ± 0.13. The K½ value measured from this mHCN2-h4 chimera should be close to that of full-length hHCN4 channel, as it was previously shown that the response to cAMP was largely determined by the C terminus, especially the C-linker and CNBD (33). Moreover, the mHCN2 and hHCN4 channel isoforms share identical sequences in the S4–S5 linker of the transmembrane domain as well as in the first α-helix of the C-linker, all of which have been shown to be important for coupling between ligand binding and channel gating (9, 34–37).
Protein Purification and Characterization of hHCN4 C-terminal Fragment
The above results (Fig. 1) established a difference in the response to cAMP responsiveness between mHCN2 and hHCN4 channels, which is most likely attributed to the C-terminal fragment. Thus, we next pursued structural studies of the hHCN4 C-terminal fragment to discern the structural determinants of its differential cAMP sensitivity compared with the corresponding mHCN2 region.
We expressed the hHCN4 C-terminal region containing residues 521 to 724 (hHCN4C) in E. coli. This region includes the C-linker region and the CNBD (Fig. 2). We purified this protein through 3 major steps using a HisTrap affinity column, ion-exchange HiTrap S column, and the size exclusion Superdex 75 column. Interestingly, during elution of the HiTrap S column, we observed two well separated peaks and named them S1 and S2 (Fig. 3A). Both peaks ran at the same position on the SDS-PAGE gel, indicating that they are two populations of the hHCN4C protein (Fig. 3B). We collected S1 and S2 separately and further purified each on size exclusion columns.
FIGURE 2.

Primary sequence alignment in the C-linker and CNBD region from representative HCN channels. Protein primary sequences for the C-linker and CNBD region from the mouse HCN1–3 channels and human HCN4 channel were aligned by ClustalW (45). Secondary structures are labeled on the top of the sequence. Red bar, α-helix; blue arrow, β-strand.
FIGURE 3.

Purification and biochemical characterization of the hHCN4C protein. A, elution profile of the hHCN4C protein from the ion-exchange HiTrap S column. Blue trace, UV absorbance (left y axis); red trace, the percentage of buffer B (1 m NaCl, right y axis). The two UV absorbance peaks are labeled S1 and S2, respectively. B, SDS-PAGE of purified hHCN4 S1 and S2 fractions. 1, 2, and 4 μg of S1 and S2 were loaded on the gel, and stained with Coomassie Blue. C, elution profiles of S1 (red) and S2 (black) fractions on the Superdex 200 10/30 size exclusion column. Dashed lines, control without cAMP; solid lines, 5 μm cAMP.
Next, we analyzed the differences between S1 and S2 fractions using size exclusion chromatography and analytical ultracentrifugation sedimentation equilibrium assays. On the size exclusion Superdex 200 column, the elution volumes of S1 and S2 were 16.03 and 15.50 ml, respectively (Fig. 3C, dashed lines; supplemental Fig. S1). This indicates that S1 had a lower apparent molecular weight than S2 and thus probably had a lower oligomerization state than S2. Interestingly, when cAMP was added to the running buffer, both S1 and S2 were both eluted out at similar volumes, about 15 ml (Fig. 3C, solid lines). These results are consistent with previous observations that cAMP binding promotes the formation of tetramers in HCN channels (10, 38). Corresponding to the differential behavior by size exclusion chromatography, S1 and S2 showed distinct tendencies for oligomerization as revealed by analytical ultracentrifugation sedimentation experiments. For the S1 fraction, we observed a significant difference after adding cAMP. After fitting the data, the calculated molecular mass without cAMP for S1 was 28,908 Da, which is close to the expected molecular mass of a monomer (24,105 Da). However, the calculated molecular mass in the presence of cAMP was 55,627 Da, suggesting that the presence of cAMP shifted the equilibrium toward a higher order oligomeric state. In contrast, for the S2 fraction, adding cAMP did not lead to any obvious changes in the analytical ultracentrifugation results. The calculated molecular masses for S2 were 54,778 (with cAMP) or 57,778 Da (without cAMP), respectively, and both values were close to the S1 value in the presence of cAMP. Given these results, we hypothesized that the S1 fraction was most likely the cAMP-free form, whereas the S2 fraction was the cAMP-bound form (bound with endogenous cAMP from E. coli), even with extensive dialysis before loading onto the HiTrap S column. Our hypothesis was confirmed by the following crystallographic studies.
Crystal Structure of the hHCN4 C-terminal Fragment and Alignment with the Corresponding mHCN2 Structure
We set up crystallization screens for the purified S1 or S2 fractions without adding cAMP, and only obtained crystals for the S2 fraction. However, when we added cAMP to the crystallization drops, we obtained similar crystals for both the S1 and S2 fractions. After optimizing the crystallization and cryoprotection conditions, we collected a native data set at 2.4-Å resolution at the National Synchrotron Light Source (NSLS), the Brookhaven National laboratory. The crystals were in an I422 space group with a = 69.264 Å and c = 191.616 Å, with one monomer in each asymmetric unit. Because the sequence identity between hHCN4 and mHCN2 in this region is as high as 94.1%, we used molecular replacement to obtain the phases and then solved the structure.
After refinement at 2.4-Å resolution to a Rwork of 23.7% and Rfree of 27.5%, the final model contained residues 521 to 717 with no break in the middle (Table 1). The last 7 residues at the extreme C terminus were too disordered to model. The overall model has good stereochemistry with only one Ramachandran outlier on a loop. As expected, we did observe well defined electron density in the cAMP binding pocket, and more importantly, cAMP could be fitted well in this density (Fig. 4D). Therefore, we concluded that the S2 fraction did contain cAMP in the binding pocket. Because we did not add cAMP during the purification process, hHCN4C in the S2 fraction must have bound to endogenous cAMP molecules from E. coli during expression.
TABLE 1.
Diffraction data and refinement
Values in parentheses indicate the corresponding statistics in the highest resolution shell.
| Crystal parameters | |
| Space group | I422 |
| a (Å) | 69.264 |
| c (Å) | 191.616 |
| Zaa/solvent content (%) | 1/48.43 |
| Data collection | |
| Wavelength (Å) | 0.97923 |
| Bragg spacings (Å) | 50-2.4 (2.44-2.4) |
| Total reflections | 734,711 |
| Unique reflections | 9,567 |
| Rmerge (%)b | 5.3 (27.7) |
| Average I/σ | 91.2 (5.2) |
| Completeness (%) | 94.0 (95.9) |
| Redundancy | 18.8 (8.4) |
| Refinement | |
| Bragg spacings (Å) | 50-2.4 |
| Rwork (%)c | 23.7 |
| Rfree (%)c | 27.5 |
| Averge B-factor (Å2) | 79.1 |
| Root mean square deviation: bond/angle ideality (Å/°) | 0.009/1.082 |
| Total atoms | 1,612 |
| Protein residues/cAMP/H2O | 197/1/29 |
| Ramachandran analysisd | |
| Most favored | 87.4% |
| Additional allowed | 12.1% |
| Generously allowed | 0.0% |
| Disallowed | 0.6% |
a Za, number of molecules per asymmetric unit.
b Rsym = (Σ|Ih − 〈Ih〉|)/ΣIh, where 〈Ih〉 is the average intensity over symmetry equivalent.
c Rwork = Σ‖Fo|−|Fc|/Σ|Fo|. Rfree is equivalent to Rwork, but calculated for a randomly chosen 5% of reflection, which were omitted from the refinement process.
d Analysis was performed with PROCHECK from the CCP4 suite.
FIGURE 4.

Crystal structure of the human HCN4 C terminus and alignment with the mouse HCN2 structure. A, ribbon diagram of the hHCN4C structure. The regions of C-linker and CNBD are labeled. The cAMP molecule has sticks for bonds. B, superposition of hHCN4C with mHCN2C based on C-α atoms. hHCN4C is red and mHCN2C is blue. The cAMP molecule has sticks for bonds. C, ribbon diagram of the hHCN4C tetramer. A tetrameric assembly was built based on crystallographic symmetry. Each subunit is shown in a different color, and the tetramer is viewed parallel to the 4-fold axis, presumably from the cell membrane into the intracellular side. D, electron density of the cAMP molecule in the hHCN4C structure. The model-phased (2|Fo| − |Fc|) electron density map of cAMP was drawn as yellow three-dimensional baskets contoured at σ level 1.0 after refinement at 2.4-Å resolution. The cAMP molecule has sticks for bonds.
The overall structure of hHCN4C showed high similarity in its folding pattern to the previously published mouse HCN2 protein (Fig. 4A). When aligned, the root mean square deviation between the Cα atoms of these two structures was 0.791 Å (Fig. 4B). Like the mHCN2 structure, the C-linker contained 6 α-helices (A′ to F′) and the CNDB contained eight β-strands (1–8) flanked by four α-helixes (A-C and P). The cAMP was observed to stay in anti-conformation and coordinated in a similar way in the binding pocket of hHCN4. Based on crystallographic symmetry, we were also able to build a tetramer with similar intersubunit contacts (Fig. 4C).
Structure-function Analysis of the Different Responses to cAMP between mHCN2 and mHCN2-h4 Channels
Even though the overall structures of hHCN4C and mHCN2C were similar to each other, they did display some obvious differences. In the loop region between strands β4 and β5, the end of the loop was shifted away by about 3.0 Å in the hHCN4C structure (Fig. 5A). Interestingly, these two proteins shared identical residues in this loop region. From adjacent β4 and β5 strands, the amino acid differences are in β5, Thr650 and Ala653 in hHCN4 (Fig. 5B). A careful comparison of two crystal structures revealed that in the mHCN2 structure, the long hydrophobic side chain of Met572 interacts with the adenosine ring of cAMP at the entrance of the cAMP binding pocket. However, in the hHCN4 structure, the side chain of Thr650 was too short to form any contact with cAMP. Corresponding to this difference, the cAMP molecule shifts slightly into the cAMP binding pocket in the hHCN4 structure by about 0.5 Å. Could this residue be responsible for the structural and functional differences between these two proteins? Next we mutated Met572 in mHCN2 to threonine to test whether we could make the cAMP response of mHCN2 like that of the mHCN4-h4. Indeed, sensitivity to cAMP in the mHCN2/M572T mutant channel was reduced to 0.38 ± 0.07 μm, which was close to that of mHCN2-h4 (Fig. 5, C and D). However, the converse mutation in the mHCN2-h4 channel, T650M, did not lead to an increase in sensitivity to cAMP as expected (0.51 ± 0.09 μm, Fig. 5D, right, magenta). Then we went one step further to check the contribution from another residue in β5, Ala653, by mutating both residues (Fig. 5D, right). This construct, mHCN2-h4/T650M + A653S (green), did show an improved response to cAMP as measured by K½ (0.28 ± 0.05 μm) but was still different from mHCN2 (0.08 ± 0.01 μm).
FIGURE 5.

Point mutation of M572T in mHCN2 diminishes the difference in response to cAMP from hHCN4. A, left, structure alignment of mHCN2C (blue) and hHCN4C (red) monomers. The β4 loop-β5 region is highlighted with a green circle. Right, a zoomed view over the loop region between β4 and β5. B, primary sequence alignment of the β4 loop-β5 region for mHCN2 and hHCN4. The β4 and β5 strands are highlighted in red. The differences in primary sequence are indicated by arrows. C, representative recordings of mHCN2/M572T (left, control; right, 10 μm cAMP). D, dose-response curves showing the shift in the voltage-dependent channel activation curve (ΔV½) versus cAMP concentration for WT mHCN2 (black), mHCN2-h4 (red), mHCN2/M572T (blue), mHCN2-h4/T650M (magenta), and mHCN2-h4/T650M + A653S (green).
To further investigate the response to cAMP, we specifically focused on cAMP binding by measuring binding affinity using the purified HCN channel C-terminal proteins. We applied two solution-based equilibrium methods: ITC and FA (Fig. 6). Surprisingly, ITC results revealed ∼3-fold tighter binding of cAMP to the hHCN4 protein (0.83 ± 0.04 μm) than the mHCN2 protein (2.58 ± 0.44 μm). This result is in contrast with functional assays showing that the hHCN4 protein introduced a weaker response to cAMP as measured by the value of K½. Furthermore, ITC experiments showed that the M572T mutation in the mHCN2 protein leads to a slight increase in cAMP binding; conversely, the T650M mutation in the hHCN4 protein results in a decrease in cAMP binding. To further check the binding results, we measured cAMP binding by FA, using a fluorescent cAMP analog, 8-Fluo-cAMP. FA results showed that the Kd values for all four protein samples were all around 0.3 μm, which was in the micromolar range similar to ITC results, but the difference was apparent. To correlate ITC and FA studies, we measured binding of 8-Fluo-cAMP to the wild type (WT) mHCN2 protein by ITC and obtained the Kd value of 3.75 ± 0.84 μm (supplemental Fig. S2), which was close to the Kd of cAMP (2.58 ± 0.44 μm). Thus, we concluded that the discrepancy in binding affinities by ITC and FA was due to an intrinsic difference between these two popular binding assays. The similar Kd values obtained for four different proteins by FA is probably related to the fact that the extra chemical group (fluorescein), attached to the purine ring in the cAMP molecule, compromises the differential recognition of the ligand.
FIGURE 6.

Biochemical binding assays on purified HCN C-terminal proteins. A, original ITC data showing the binding of cAMP to WT hHCN4 protein. Top, the rate of heat exchange is plotted as a function of time. Each spike represents injection of cAMP into the sample cell. Bottom, the plot of heat exchange as a function of protein to the cAMP ratio. B, FA results showing the binding of 8-Fluo-cAMP to WT hHCN4 (top) and T650M mutant hHCN4 (bottom) proteins. C, summary of the Kd value obtained from ITC or FA experiments on different protein samples. *, results for the WT mHCN2 protein are listed for comparison purpose.
Other than shifting the voltage-dependent channel activation to more depolarized potentials, cAMP binding also facilitates channel gating by affecting channel opening and closing kinetics. For both mHCN2 and mHCN2-h4 channels, upon cAMP application, we observed a significant acceleration in channel opening kinetics and a slowdown in channel closing kinetics (Fig. 7, A and B). We used a simple single-exponential function to fit channel activation and deactivation kinetics and examined the effect of cAMP. In the absence of cAMP, both mHCN2 and mHCN2-h4 opened quite slowly. τopen measured at −160 mV was 1.08 ± 0.08 s (n = 21) for mHCN2 or 0.79 ± 0.06 s (n = 14) for mHCN2-h4. Both channels closed similarly in the absence of cAMP (Fig. 7, A and B, top). With a saturating concentration of cAMP (10 μm) applied, both channels opened much faster and the time constants measured at −160 mV were 0.33 ± 0.03 s (n = 11) for mHCN2 and 0.36 ± 0.06 s (n = 6) for mHCN2-h4. At voltage steps more negative than −150 mV, the channel activation was largely voltage-insensitive (Fig. 7C, solid lines). This observation correlates with previous reports that at these extreme negative voltages the limiting factor for channel opening is voltage-insensitive and cAMP binding to the mHCN2 channel speeds up the voltage-independent channel opening (39). Interestingly, we observed a change in the channel deactivation time constant, especially when cAMP was applied. The closing rate τclose+cAMP measured at −40 mV with 10 μm cAMP applied was 0.17 ± 0.01 s (n = 7) for mHCN2 but 0.29 ± 0.02 s (n = 9) for mHCN2-h4 (Fig. 7D), which indicates that hHCN4C made the channel deactivate much slower. Thus, our results showed that cAMP binding makes these two channels open with similar fast rates but dramatically slows down the closing rate for the mHCN2-h4 channel.
FIGURE 7.

Effects of cAMP binding on channel opening and closing kinetics in mHCN2 and mHCN2-h4 channels. A, the mHCN2 channel was activated by a voltage step from −40 to −140 mV. Then channel deactivation kinetics were measured at different holding potentials ranging from +40 to −50 mV. Voltage protocol is shown on the top. Recording traces are shown in the middle, control without cAMP; bottom, 10 μm cAMP. B, channel deactivation in the mHCN2-h4 channel. Voltage protocol is shown at the top. Middle, control without cAMP; bottom, 10 μm cAMP. C, voltage-dependent channel activation kinetics. A series of hyperpolarizing voltage steps as used in Fig. 1, B and C, were used to activate mHCN2 (black) and mHCN2-h4 (red) to difference levels. Open circle and the dashed line represent the conditions without cAMP, whereas closed circle and solid line represent the conditions with cAMP. D, voltage-dependent channel deactivation kinetics. The voltage protocol and representative current traces are shown in A and B for mHCN2 (black) and mHCN2-h4 (red), respectively. Open circle and dashed line represent the conditions without cAMP, whereas closed circle and solid line represent the conditions with cAMP.
DISCUSSION
In this article, we established that the hHCN4 C-terminal fragment confers differential cAMP sensitivity to the mouse HCN2 channel. At the same time, we determined the crystal structure of the human HCN4 C-terminal fragment, which is similar to an equivalent structure from the mouse HCN2 channel. Furthermore, we characterized the contribution to ligand binding and gating by this hHCN4 protein and compared it with the corresponding part from the well characterized mHCN2 channel. Following a structural difference in a loop region, we identified residues that at least partially contributed to the differential responses to cAMP. Furthermore, our results showed that the hHCN4 C-terminal fragment facilitated the function of the channel by significantly slowing down the deactivation rate.
Similar to the previously published mHCN2 structure, this hHCN4C structure contained a 4-fold symmetry along the center of the molecule. Extensive intersubunit interactions, especially the interactions between the C-linkers from neighboring subunits, mediate the assembly of the four subunits. This new structure of hHCN4 further underscores the high structural conservation for this region. Given the drastically different responses to cAMP by channels ranging from invertebrate HCN channels, such as the homolog SPIH channel, to mutant mHCN2 channels, it is interesting to see that all these structures show high similarity and the maximal root mean square deviation of C-α atoms is less than 1 Å (16, 40, 41). This structural conservation is possibly due to an intrinsically stable fold, which can tolerate dramatic perturbations such as the absence of cAMP from the binding pocket or point mutations. Thus, it remains a very intriguing question regarding the structural changes that occur upon cAMP binding during the cAMP-dependent gating process.
So far there have been multiple reports on the biophysical properties of HCN4 channels but with some discrepancies (42). One study reported that the maximal shift in V½ with a saturating concentration of cAMP is ∼10 mV and the K½ value is ∼1.5 μm (25). Other studies have shown maximal shifts in the 20–24 mV range without mentioning the value of K½ (43, 44). Here our results from the mHCN2-h4 chimera showed that the maximal shift in ΔV½ was about ∼16 mV and the K½ about ∼0.3 μm. Because both mHCN2 and hHCN4 contain identical sequences in the S4–S5 linker as well as in the first α-helix in the C-linker region, our studies on the chimera of mHCN2-h4 should largely reflect the contributions from the hHCN4 C-terminal fragment.
Based on alignment of the mHCN2 and hHCN4 structures, we observed a structural difference in the loop region between the β4 and β5. Following this lead, we focused on the sequence in this region and identified a residue in β5 adjacent to this loop, Met572 in mHCN2 or Thr650 in hHCN4. Mutating this residue from Met to Thr in mHCN2 largely diminished the difference in K½ between two channels. This suggests that Met572 in the mHCN2 channel is important for strengthening the response to cAMP. Because the purine ring of the cAMP molecule is largely hydrophobic, a residue like methionine containing a hydrophobic side chain is probably more preferred than a polar residue like threonine. However, the converse mutation, from Thr to Met in hHCN4, did not lead to an expected increase in response to cAMP. We went on and tested another neighboring residue, Ala653 in hHCN4, but only obtained a slight increase in the response to cAMP when we mutated Ala653 to Ser. Thus, we concluded that compared with mHCN2, the reduced response to cAMP in the hHCN4 channel is most likely due to a distributed mechanism. How about other HCN channel subunits? It is noticeable that HCN1 and -2 share the same residue, methionine, but HCN3 and -4 share the other residue, threonine, at this position. It has been shown that the responses to cAMP are very similar between HCN1 (K½, 0.06 μm) and HCN2 (0.10 μm) (33). In contrast, HCN4 has a reduced response to cAMP and HCN3 has an almost diminished or even negative response to cAMP (46, 47). Therefore, it is intriguing to further investigate the structure-function relationship for this position as well as the adjacent structural elements across different HCN isoforms.
For the ligand-gated ion channels, it is known that the value of EC50 or K½ contains information from both ligand binding and gating efficacies, which are difficult to separate (48). We tried to tease out these two issues by separately studying cAMP binding using purified proteins. Indeed, ITC results showed that cAMP binding to the hHCN4 protein is around 3-fold tighter than the mHCN2 protein, which is in contrast with characterization of the functional channels showing that hHCN4 has a reduced sensitivity to cAMP as measured by the value of K½. Thus, our study provides a further case supporting that ligand binding and gating efficacy are two separate issues. Moreover, the fact that the M572T mutation reduces the response to cAMP (K½) but actually increases the binding affinity in mHCN2 suggests that this residue might play a more important role in the coupling between cAMP binding to channel opening rather than cAMP binding.
Regarding channel kinetics, there is a significant difference between the HCN2 and HCN4 channels, HCN4 activates and deactivates much slower that HCN2 (49). For channel activation, it has been shown that elements in the transmembrane domain, including the first transmembrane domain, the second transmembrane domain, and the loop between, are responsible for this difference (50). However, it is unclear which region in the HCN4 channel is responsible for the slow deactivation kinetics. Our results show that under a saturating concentration of cAMP, similar to the full-length hHCN4 channel, mHCN2-h4 deactivates about twice slower than mHCN2, indicating that the hHCN4C fragment plays a major role in this interesting channel behavior. This observation suggests that when the CNBD of hHCN4 is occupied with cAMP, the channel not only opens faster but also closes slower after the hyperpolarization membrane potential change is withdrawn. Thus, cAMP binding to the HCN4 channel might lead to larger net currents going through and thus greater contributions to the diastolic depolarization in pacemaking cells. However, the impact of the difference between the two major isoforms of the HCN channel in heart physiology remains to be further clarified. In summary, given the fact that HCN4 is the major HCN isoform expressed in the heart and carries all the mutations found in cardiac patients, our structural and functional studies of the hHCN4 C-terminal fragment will be very useful in studying HCN channel biophysics as well as pathophysiological mechanisms for the HCN channel-related cardiac disorders.
Supplementary Material
Acknowledgments
We thank Dr. Siegelbaum and Dr. Biel for providing the cDNAs for mouse HCN2 and human HCN4 clones, the staff of Beamline X4A/C at the National Synchrotron Light Source (NSLS) for assisting in diffraction data collection, and Dr. Faik Musayev for help with structure refinement. We also thank Dr. Logothetis and Dr. Siegelbaum for carefully reading the manuscript and providing insightful comments.
This work was supported by startup funds from the Virginia Commonwealth University School of Medicine (to L. Z. and Q. L.) and a New Scholar Award in Aging from the Ellison Medical Foundation (to Q. L.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
The atomic coordinates and structure factors (code 3OTF) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- CNBD
- cyclic-nucleotide binding domain
- ITC
- isothermal titration calorimetry
- FA
- fluorescence anisotropy.
REFERENCES
- 1.Hille B. (2001) Ion Channels of Excitable Membranes, 3rd Ed., Sinauer, Sunderland, MA [Google Scholar]
- 2.Jan L. Y., Jan Y. N. (1990) Nature 345, 672. [DOI] [PubMed] [Google Scholar]
- 3.Zagotta W. N., Siegelbaum S. A. (1996) Annu. Rev. Neurosci. 19, 235–263 [DOI] [PubMed] [Google Scholar]
- 4.Biel M., Wahl-Schott C., Michalakis S., Zong X. (2009) Physiol. Rev. 89, 847–885 [DOI] [PubMed] [Google Scholar]
- 5.Ganetzky B., Robertson G. A., Wilson G. F., Trudeau M. C., Titus S. A. (1999) Ann. N.Y. Acad. Sci. 868, 356–369 [DOI] [PubMed] [Google Scholar]
- 6.Bell D. C., Yao H., Saenger R. C., Riley J. H., Siegelbaum S. A. (2004) J. Gen. Physiol. 123, 5–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ulens C., Siegelbaum S. A. (2003) Neuron 40, 959–970 [DOI] [PubMed] [Google Scholar]
- 8.Wang J., Chen S., Nolan M. F., Siegelbaum S. A. (2002) Neuron 36, 451–461 [DOI] [PubMed] [Google Scholar]
- 9.Wainger B. J., DeGennaro M., Santoro B., Siegelbaum S. A., Tibbs G. R. (2001) Nature 411, 805–810 [DOI] [PubMed] [Google Scholar]
- 10.Zagotta W. N., Olivier N. B., Black K. D., Young E. C., Olson R., Gouaux E. (2003) Nature 425, 200–205 [DOI] [PubMed] [Google Scholar]
- 11.Tibbs G. R., Liu D. T., Leypold B. G., Siegelbaum S. A. (1998) J. Biol. Chem. 273, 4497–4505 [DOI] [PubMed] [Google Scholar]
- 12.Zhou L., Siegelbaum S. A. (2007) Structure 15, 655–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen S., Wang J., Siegelbaum S. A. (2001) J. Gen. Physiol. 117, 491–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou L., Siegelbaum S. A. (2008) Biophys. J. 94, L90–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Craven K. B., Zagotta W. N. (2004) J Gen. Physiol. 124, 663–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flynn G. E., Black K. D., Islas L. D., Sankaran B., Zagotta W. N. (2007) Structure 15, 671–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaborit N., Le Bouter S., Szuts V., Varro A., Escande D., Nattel S., Demolombe S. (2007) J. Physiol. 582, 675–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stillitano F., Lonardo G., Zicha S., Varro A., Cerbai E., Mugelli A., Nattel S. (2008) J. Mol. Cell Cardiol. 45, 289–299 [DOI] [PubMed] [Google Scholar]
- 19.Mangoni M. E., Nargeot J. (2008) Physiol. Rev. 88, 919–982 [DOI] [PubMed] [Google Scholar]
- 20.DiFrancesco D. (2005) Curr. Med. Res. Opin. 21, 1115–1122 [DOI] [PubMed] [Google Scholar]
- 21.Ludwig A., Herrmann S., Hoesl E., Stieber J. (2008) Prog. Biophys. Mol. Biol. 98, 179–185 [DOI] [PubMed] [Google Scholar]
- 22.Nof E., Antzelevitch C., Glikson M. (2010) Pacing Clin. Electrophysiol. 33, 100–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schulze-Bahr E., Neu A., Friederich P., Kaupp U. B., Breithardt G., Pongs O., Isbrandt D. (2003) J. Clin. Invest. 111, 1537–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ueda K., Nakamura K., Hayashi T., Inagaki N., Takahashi M., Arimura T., Morita H., Higashiuesato Y., Hirano Y., Yasunami M., Takishita S., Yamashina A., Ohe T., Sunamori M., Hiraoka M., Kimura A. (2004) J. Biol. Chem. 279, 27194–27198 [DOI] [PubMed] [Google Scholar]
- 25.Milanesi R., Baruscotti M., Gnecchi-Ruscone T., DiFrancesco D. (2006) N. Engl. J. Med. 354, 151–157 [DOI] [PubMed] [Google Scholar]
- 26.Studier F. W. (2005) Protein Expr. Purif. 41, 207–234 [DOI] [PubMed] [Google Scholar]
- 27.Otwinowski Z., Minor W. (1997) Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 28.McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Emsley P., Cowtan K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 30.Brunger A. T. (2007) Nature Protocols 2, 2728–2733 [DOI] [PubMed] [Google Scholar]
- 31.Bailey S. (1994) Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 32.Humphrey W., Dalke A., Schulten K. (1996) J. Mol. Graph. 14, 33–38, 27–38 [DOI] [PubMed] [Google Scholar]
- 33.Wang J., Chen S., Siegelbaum S. A. (2001) J. Gen. Physiol. 118, 237–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnson J. P., Jr., Zagotta W. N. (2001) Nature 412, 917–921 [DOI] [PubMed] [Google Scholar]
- 35.Sanguinetti M. C., Xu Q. P. (1999) J. Physiol. 514, 667–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Decher N., Chen J., Sanguinetti M. C. (2004) J. Biol. Chem. 279, 13859–13865 [DOI] [PubMed] [Google Scholar]
- 37.Chen J., Mitcheson J. S., Tristani-Firouzi M., Lin M., Sanguinetti M. C. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 11277–11282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou L., Olivier N. B., Yao H., Young E. C., Siegelbaum S. A. (2004) Neuron 44, 823–834 [DOI] [PubMed] [Google Scholar]
- 39.Chen S., Wang J., Zhou L., George M. S., Siegelbaum S. A. (2007) J. Gen. Physiol. 129, 175–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Craven K. B., Olivier N. B., Zagotta W. N. (2008) J. Biol. Chem. 283, 14728–14738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Taraska J. W., Puljung M. C., Olivier N. B., Flynn G. E., Zagotta W. N. (2009) Nat. Methods 6, 532–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Verkerk A. O., van Ginneken A. C., Wilders R. (2009) Int. J. Cardiol. 132, 318–336 [DOI] [PubMed] [Google Scholar]
- 43.Zolles G., Klöcker N., Wenzel D., Weisser-Thomas J., Fleischmann B. K., Roeper J., Fakler B. (2006) Neuron 52, 1027–1036 [DOI] [PubMed] [Google Scholar]
- 44.Harzheim D., Pfeiffer K. H., Fabritz L., Kremmer E., Buch T., Waisman A., Kirchhof P., Kaupp U. B., Seifert R. (2008) EMBO J. 27, 692–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thompson J. D., Gibson T. J., Higgins D. G. (2002) Curr. Protoc. Bioinformatics 2.3.1–2.3.22, John Wiley and Sons, Inc., New York: [DOI] [PubMed] [Google Scholar]
- 46.Stieber J., Stöckl G., Herrmann S., Hassfurth B., Hofmann F. (2005) J. Biol. Chem. 280, 34635–34643 [DOI] [PubMed] [Google Scholar]
- 47.Mistrík P., Mader R., Michalakis S., Weidinger M., Pfeifer A., Biel M. (2005) J. Biol. Chem. 280, 27056–27061 [DOI] [PubMed] [Google Scholar]
- 48.Colquhoun D. (1998) Br. J. Pharmacol. 125, 924–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ludwig A., Zong X., Stieber J., Hullin R., Hofmann F., Biel M. (1999) EMBO J. 18, 2323–2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stieber J., Thomer A., Much B., Schneider A., Biel M., Hofmann F. (2003) J. Biol. Chem. 278, 33672–33680 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.