

Abstract
Both trimellitic anhydride (TMA), a small molecular weight chemical, and ovalbumin (OVA), a reference protein allergen, cause asthma with eosinophilia. To test the hypothesis that different allergens elicit symptoms of asthma via different effector pathways, gene expression was compared in lungs of Balb/c mice sensitized with either TMA or OVA, followed by intratracheal challenge with TMA conjugated to mouse serum albumin (TMA-MSA) or OVA, respectively. Sensitized animals challenged with mouse serum albumin (MSA) alone were controls. Seventy-two hours after challenge, lung eosinophil peroxidase indicated that both allergens caused the same significant change in eosinophilia. Total RNA was isolated from lung lobes of 6–8 animals in each of four treatment groups and hybridized to Affymetrix U74Av2 GeneChips. False discovery rates (q-values) were calculated from an overall F test to identify candidate genes with differences in expression for the four groups. Using a q-value cutoff of 0.1, 853 probe sets had significantly different expression across the four treatment groups. Of these 853 probe sets, 376 genes had an Experimental/Control ratio of greater than 1.2 or less than 1/1.2 for either OVA- or TMA-treated animals, and 249 of the 376 genes were uniquely up- or down-regulated for OVA or TMA (i.e., differentially expressed with the allergen). qRT-PCR analysis of selected transcripts confirmed the gene expression analysis. Increases in both arginase transcript and enzyme activity were significantly greater in OVA-induced asthma compared to TMA-induced asthma. These data suggest that pathways of arginine metabolism and the importance of nitric oxide may differ in OVA- and TMA-induced asthma.
Keywords: rodent, eosinophils, allergy, lung, nitric oxide
Symptoms of occupational asthma include reversible airway obstruction, airway hyperresponsiveness, mucus secretion, airway remodeling, and inflammatory cell infiltration consisting particularly of eosinophils. Up to 200 different substances encountered in the environment and/or the workplace are implicated in asthmatic responses (Van Kampen et al., 2000), including trimellitic anhydride (TMA) and ovalbumin (OVA). TMA is an organic acid anhydride that is produced in billion-pound quantities per year and used in the paint and plastics industries (Zeiss et al., 1990). The allergic immunotoxic response to this low-molecular-weight chemical has been extensively studied, and TMA is a known cause of occupational asthma in humans (Zeiss et al., 1977). TMA elicits an immune response by acting as a hapten, combining with host proteins to produce chemical protein conjugates that are immunogenic. OVA is a protein used extensively as a reference allergen in immunology and a known cause of occupational asthma (Boeniger et al., 2001).
In general, it is assumed that mechanisms and mediators of the effector phase of asthma are common to all allergens, including low-molecular-weight occupational allergens such as TMA. Zimmermann et al. (2003) used two different antigens, OVA and Aspergillus, to elicit the asthmatic phenotype in the mouse and found that expression of 291 genes in the lung changed in common for both allergens. They described these as “signature genes” for allergic airway inflammation and have pursued their role in the effector phase of asthma. However, 236 and 205 genes were unique to sensitization and challenge with Aspergillus and OVA, respectively, suggesting that mechanisms may differ with the allergen. Zimmermann et al. (2003) attributed these differences in gene expression between the two allergens to differences in exposure regimens (i.e., acute exposure to OVA and chronic exposure to Aspergillus). Clearly some differences in mechanisms of asthma may be related to differences in exposure conditions to the allergen. For example, in mouse models of asthma using OVA as the allergen, differences in the pathophysiology of the response have been attributed in part to variation in the dose and intensity of antigen administration, routes of sensitization and challenge, as well as the inbred strain of animal used in the mouse asthma models (Humbles et al., 2002; Ma et al., 2002; Regal, 2004). Other evidence in guinea pig asthma models suggests that the role of the complement system and arachidonate metabolites may differ depending on whether the inciting allergen is OVA or TMA (Arakawa et al., 1993; Fraser et al., 1995; Regal and Fraser, 1996). Also, in humans, variability of the asthma phenotype suggests that different mechanisms may result in asthma symptoms of different severity and character (Bel, 2004).
We hypothesized that different allergens would evoke unique patterns of gene expression suggestive of unique effector mechanisms leading to the asthma phenotype. To test this hypothesis, gene expression was examined in the effector phase in lungs from Balb/c mice sensitized and challenged with two different allergens, TMA and OVA, evaluating eosinophilia as the asthma endpoint. To insure that expression pattern differences reflected mechanistic differences due to the allergen and not the exposure regimen (Humbles et al., 2002; Ma et al., 2002; Regal, 2004; Zimmermann et al., 2003), identical acute sensitization and challenge regimens were used. Doses of TMA and OVA were chosen which evoked similar changes in eosinophil infiltration into the mouse lung. OVA-induced changes in gene expression in the present study confirmed increases in many genes reported previously, including arginase (Daheshia et al., 2002; Scheerens et al., 2002; Zhu et al., 2004; Zimmermann et al., 2003; Zuhdi Alimam et al., 2000). In addition, TMA sensitization and challenge revealed a unique gene expression pattern compared to OVA. Further investigation focused on differences in arginase and changes in expression of genes involved in arginine metabolism and nitric oxide production. These data suggest unique effector mechanisms involving arginase exist for OVA- and TMA-induced asthma, and optimal therapy may need to target different pathways for asthma induced by different allergens.
Materials and Methods
Experimental procedures for assessment of cellular infiltration into the lung
Female BALB/c mice (BALB/cAnNHsd) were purchased from Harlan (Portage, MI) and were 6 to 11 weeks of age (average body weight 18.1 ± 0.2 g) at time of sensitization. Mice were fed Purina Rodent Chow and water ad libitum and maintained on a 12-h light-dark cycle. All animal studies were approved by the University of Minnesota Institutional Animal Care and Use Committee and were carried out in accordance with the Guide for the Care and Use of Laboratory animals as adopted by the U.S. National Institutes of Health. Intratracheal instillation was performed by aspiration as previously described (Regal et al., 2001) using mice anesthetized intramuscularly with 1 mg ketamine and 0.2 mg xylazine. TMA was conjugated to mouse serum albumin (TMA-MSA) as described previously in our studies using guinea pig serum albumin (Fraser et al., 1995). MSA was obtained from Sigma Chemical Co. (St. Louis, MO). The degree of substitution was 18–21 mol TMA per mole MSA.
Animals were sensitized with OVA (0.3%) and TMA (3%) as previously described (Regal et al., 2001). Briefly, animals received OVA and TMA suspensions in corn oil intradermally on days 1 and 3, and intratracheally administered OVA or TMA-MSA (30 μg) on day 12. Animals were challenged on day 19 with 400 μg OVA, TMA-MSA, or MSA intratracheally to evoke the asthma phenotype. Seventy-two hours after intratracheal challenge, animals were anesthetized with pentobarbital and bled by cardiac puncture or retroorbital bleed. Lungs were removed for RNA isolation and analysis of eosinophil peroxidase (Regal et al., 2001) as a measure of lung eosinophilia. Lung lobes for RNA isolation were flash frozen in liquid nitrogen or added to RNAlater (Ambion). Lobes for eosinophil peroxidase (EPO) were processed as previously described (Regal et al., 2001). In a subset of these animals, a lung lobe was also removed for measurement of myeloperoxidase as an indicator of lung neutrophilia. In a separate group of animals (n= 19), after anesthesia with pentobarbital, lungs were lavaged with 2 × 0.9 ml PBS, and bronchoalveolar lavage (BAL) cells were identified and counted as previously described (Regal et al., 2001).
Preparation of biotinylated target RNA for Affymetrix GeneChip analysis
Procedures described in the Affymetrix GeneChip Expression Analysis Manual were used. Total RNA was isolated from lung lobes of six to eight animals in each of the four treatment groups and hybridized at one animal per chip. Lung lobes for RNA were thawed, homogenized with a Kontes Pellet Pestle motor and a 1.5-ml pellet pestle in RLT buffer (RNeasy Qiagen), and then further disrupted by passing through 19-, 23- and finally 25-gauge needles. The Qiagen protocol was followed to prepare the total RNA from the homogenized lung tissue, and spectrophotometric measurements were used to quantify yield. First- and second-strand cDNAs were synthesized from 15 μg of total RNA using a HPLC-purified T7-(dT)24 primer as recommended by Affymetrix and the SuperScript Choice kit from Invitrogen Life Technologies. The double-stranded cDNA was further purified using phase-lock gel phenol/chloroform extraction. Size distribution and yield of the cDNA were determined on a 1% agarose gel prior to the synthesis of biotin-labeled cRNA. The synthesis of biotin-labeled cRNA was as described in the Affymetrix manual, using the Bioarray High Yield RNA Transcript labeling ENZO Kit. Products of the in vitro transcription (from both before and after purification with the Qiagen RNeasy columns) were analyzed by gel electrophoresis. The cRNA was fragmented at 94°C for 35 min in Fragmentation buffer (Affymetrix) and checked by gel electrophoresis prior to delivery to the Biomedical Image Processing Laboratory of the University of Minnesota for hybridization with the murine Affymetrix GeneChip U74Av2.
Quality control, normalization, and statistical analysis of gene expression data
Microarray Suite 5.0 was used for initial processing of the data. For each chip, we examined the chip image for gross flaws such as bubbles or streaks and for the alignment and intensity of both the corner signals and “landing lights” on the border of the chip. The Chip Report was inspected for each chip and the 5′/3′ ratios, percent present calls, background level, and presence of the spike controls were compared to Affymetrix standards. In addition, dChip (Li and Wong, 2001) was used to identify outlier chips. GeneTraffic (Iobion) was used to normalize the data using Robust Multichip Analysis (RMA; Irizarry et al., 2003). RMA-normalized, log-scale intensities from Gene Traffic were exported to SAS for identification of candidate genes by statistical analysis. Data were log transformed to insure a more normal distribution for statistical tests to be valid. Because of observed unequal variances in the gene expression data, significant treatment effects were tested using Satterthwaite's method for error degrees of freedom with PROC MIXED in SAS. In addition to a treatment effect, the model also included an effect to adjust for the date the chips were processed at the central array facility. The RMA method for normalization is designed to adjust for some differences in intensities, but there still remained a significant date effect after RMA normalization. Hence, we included the date effect in our ANOVA model (Iobion and Irizarry, personal communication).
Confirmation of selected microarray results using quantitative real-time PCR (qRT-PCR)
Total RNA was isolated according to the manufacturer's protocol (RNeasy, Qiagen) from lung tissue processed as described above. Lung samples were chosen from animals used for the gene expression studies, as well as from animals that had lung EPO measurements only. Two μg of total RNA were reverse transcribed using random hexamer primers (pf(N)6, Amersham Pharmacia Biotech Inc.) in a 20-μl reaction volume with 4 units of reverse transcriptase (Omniscript RT Kit, Qiagen), 0.4 units of RNase OUT Ribonuclease Inhibitor (Invitrogen Life Technologies), and a final concentration of 500 μM dNTPs in 1× reverse transcriptase buffer. The reverse transcription reaction was held at 37°C for 1 h. Subsequently, 0.5 μl of the 20-μl reverse transcription reaction for each animal was used in quantitative real-time PCR reactions (qRT-PCR) with each of seven primer sets (Integrated DNA Technologies, Inc., Table 1). A standard 15-μl qRT-PCR reaction mix contained 0.5 μl template (i.e., reverse transcription reaction product) and 7.5 μl SyberGreen Master Mix (Applied Biosystems). The qRT-PCR reactions were amplified in a 96-well plate format on a 7900HT Sequence Detection System (Applied Biosystems). A separate plate was used for each primer and contained experimental samples in duplicate for each animal. In addition to a no-template control, a control of reverse transcription reaction from standard lung RNA (Ambion) with G6pt1 (glucose-6-phosphate transporter protein 1) primers and a third control of reverse transcription reaction from standard lung RNA with test primers were included on each plate. The PCR profile was as follows: 50°C for 2 min, 10 min at 95°C, and then 40 cycles of 15 s at 95°C and 1 minute extensions at 60°C.
TABLE 1. Quantitative Real-Time PCR Primer Sets.
| Probe set(s) | Gene | Forward primer | Reverse primer |
|---|---|---|---|
| 97430_at | G6pt1 | ATGAACCTGCTGATGTTGGA | TGGACAGCACCCAGAGATAG |
| 93097_at | Arg1 | GGCAACCTGTGTCCTTTCTC | ACACGATGTCTTTGGCAGAT |
| 98473_at | Arg2 | AGGAACTGGCTGAAGTGGTT | GGCGTGACCGATAATGGTA |
| 92742_at | Ccl11 | AGCTCCACAGCGCTTCTATT | GTCATGATAAAGCAGCAGGAAGT |
| 92459_at | Ccl8 | GGCCAGATAAGGCTCCAGT | TAGCTTTTCAGCACCCGAAG |
| 104099_at & 162475_f_at | Pglyrp1 | CAATGTGCAGCATTACCACA | ACATGACCGTCCTCTCCAAT |
| 96336_at | Gatm | CTGTGTGCCACCATTCACA | CCTCAGCAACAGCCTTCTTC |
| 162287_r_at | Gob-5 | GGACCTTTGTCCATGAGTGG | CGGTAATGGCTGCTGAACA |
| 92694_at | Chi3l3 | CCTTTGCTGGAATGCAGAAT | CCAATGGCCAGGAGAGTTT |
| 160509_at | AMCase | TACCAGACAGGCTGGGTTCT | GCCACTCGTTGGCCTTATAG |
| 92559_at & 92560_g_at | Vcam1 | TCAAAGAAAGGGAGACTGTCAA | AAGCCTTGTGGAGGGATGTA |
The qRT-PCR results were analyzed using Applied Biosystems SDS 2.1 software. The number of cycles to reach threshold (Ct value) was determined from the ΔRn versus Cycle plot with the threshold set at 0.08 and the baseline configured automatically. The average of the Ct values for duplicate samples was calculated for each reverse transcription sample/primer pair. Ct values greater than 35 were omitted from averages and considered as lacking reverse transcription sample. The dissociation curve was monitored for each reaction, and reactions with bimodal dissociation curves were omitted from further analysis. The relative quantity of starting template was calculated according to the Applied Biosystems formula for Comparative Ct Method (User Bulletin No. 2, ABI Prism Sequence Detector 7700, P/N 4303859. Foster City, CA: The Perkin-Elmer Corporation, December 11, 1997). qRT-PCR products were also confirmed by sequence analysis (University of Minnesota Advanced Genetic Analysis Center).
Arginase assay
Arginase activity was measured by the method of Özer (1985). Briefly, lung samples were homogenized in 0.05 M Tris and 0.5% Triton-X 100 and then centrifuged 15 min at >900 × g (3000 rpm) in a refrigerated centrifuge (Jouan) to pellet debris. The supernatants were removed, and the MnCl2 concentration was adjusted to 1 mM. The samples were then heated to 55°C for 10 min to activate arginase. Assay conditions were as reported by Özer (1985) with the following changes: 200 μl reactions per well were run in microtiter plates, maintaining reagent concentrations as reported in Özer (1985). NADH was substituted for NADPH, and 0.89 mM ADP was present in each reaction well. Serial dilutions of each sample were assayed with and without L-arginine substrate to control for background NADH consumption in the tissue extracts. A standard curve was determined from serial dilutions of bovine liver arginase (Calzyme).
Statistical analysis of cellular infiltration data, qRT-PCR and arginase activity
All data were log transformed to insure a more normal distribution of the data and the valid application of statistical tests. Figures show the geometric mean +1 standard error, with significant comparisons indicated by an asterisk. Statistical significance was defined as p < 0.05. Throughout this study, four groups of animals were considered: (1) OVA sensitized, MSA challenged (OVAc), (2) OVA sensitized, OVA challenged (OVA), (3) TMA sensitized, MSA challenged (TMAc), (4) TMA sensitized, TMA-MSA challenged (TMA). Because of unequal variances, significances of treatment effects were tested using Satterthwaite's method for error degrees of freedom with PROC MIXED in SAS (SAS Institute Inc., Cary, NC). The following comparisons were made: OVAc versus OVA; TMAc versus TMA; difference in OVAc versus OVA to the difference in TMAc versus TMA; OVAc versus TMAc; and OVA versus TMA.
Results
Both OVA and TMA Sensitization and Challenge Cause Increased Lung Eosinophilia
Identical routes of administration were used for both OVA and TMA to insure that any difference in biological response was due to the allergen rather than the sensitization/challenge regimen. EPO of homogenized lung was used as an indicator of the number of eosinophils in the tissue plus the airspace. Preliminary studies demonstrated that a single challenge of 400 μg OVA or TMA-MSA resulted in similar changes in lung EPO. Furthermore, increasing the TMA-MSA dose to 4000 μg did not result in greater lung EPO than that seen after challenge with 400 μg TMA-MSA. As depicted by the lower set of brackets in Figure 1A, mice sensitized and challenged with either 400 μg OVA or TMA-MSA had significant increases in EPO in the lung over their corresponding controls. The focus of this study was on the change in eosinophilia (OVA-OVAc vs. TMA-TMAc), rather than the maximum eosinophilia attained (OVA vs. TMA), in order to relate the biological response to the change in gene expression from control. When the change in EPO between OVA and OVAc was compared to the change in EPO between TMA and TMAc animals, no statistical difference was detected, as indicated by the top bracket in Figure 1A. The maximum EPO for OVA was significantly greater than the maximum EPO for TMA (OVA vs. TMA), as was the OVAc compared to TMAc animals. That is, the lung EPO started at a higher point for the OVAc (lung EPO of 56 OD/min/g dry lung for OVAc vs. 35 for TMAc) and achieved a greater maximum for OVA (218 for OVA and 110 for TMA), but the changes in lung EPO for either allergen were not significantly different. Lungs from a subset of these animals were used for gene expression analysis, and the lung EPO values from these animals are shown in Figure 1B. Analysis of this subset showed identical statistically significant effects as for the data in Figure 1A. Sensitization and challenge with either OVA or TMA did not significantly increase lung myeloperoxidase activity as an indicator of the number of tissue neutrophils (data not shown).
FIG. 1.

Eosinophil peroxidase (EPO) activity in lung tissue 72 h after challenge. OVA-sensitized animals were challenged with either 400 μg MSA (OVAc) or OVA (OVA), and TMA-sensitized animals were challenged with either 400 μg MSA (TMAc) or TMA-MSA (TMA). (A) All sensitized and challenged animals. Values represent the geometric mean + SE for n = 18, 23, 28, and 21 animals for the four groups from left to right. (B) Subset of animals used for lung RNA isolation and microarray analysis. Values represent the geometric mean + SE for n = 8, 6, 6, and 7 animals for the 4 groups from left to right. NS = not significant. *p < 0.05. The lower set of brackets depicts the comparison of each allergen to its respective control. The upper bracket indicates the statistical comparison of the change in lung EPO (OVA-OVAc vs. TMA-TMAc). Geometric mean values are listed under respective treatment groups.
Examination of lung sections from additional animals in each of the four treatment groups (OVAc, OVA, TMAc, TMA) also demonstrated increases in eosinophils in the lungs of treated animals compared to their respective controls (data not shown). The eosinophil response was more robust in the OVA treatment group compared to the TMA treatment group, similar to the difference in maximal lung EPO noted in Figure 1. In addition, we noted significant increases in PAS staining for both allergens, indicating mucus production (data not shown).
A separate group of animals was anesthetized and lungs lavaged to assess cellular infiltration into the airspace with this sensitization and challenge regimen. Total white blood cells in the BAL were significantly increased over control in the TMA-sensitized and -challenged animals, but not in the OVA-sensitized and -challenged animals (data not shown). The total number of white blood cells in TMAc compared to OVAc did not differ. The change in white blood cells seen with TMA was primarily due to increases in eosinophils and macrophages. As seen in Figure 2, eosinophils were significantly increased in the bronchoalveolar lavage of the TMA group, but not in the OVA group, compared to their respective controls (lower brackets). The percent of eosinophils in the lavage increased from 10.5 to 23.5% for OVAc to OVA, and from 1.5% to 28.3% for TMAc to TMA. The eosinophil numbers in the OVAc group were significantly higher than the TMAc group, indicating a difference in the number of eosinophils between animals sensitized with OVA versus TMA at the time of challenge. Eosinophils in naïve animals were similar to the TMAc treatment group (data not shown), indicating that the elevated eosinophils in OVAc as compared to TMAc may have been due to residual effects of the intratracheal sensitization dose for OVAc. The number of macrophages in the BAL was also significantly increased in the TMA group compared to TMAc, but no significant increase was detected comparing OVA to OVAc. This represents a decrease in the percent of macrophages from 75% to 65% for OVAc to OVA and from 86% to 63% for TMAc to TMA. No treatment effect was seen for neutrophils (data not shown).
FIG. 2.

Total eosinophils and macrophages in BAL 72 h after challenge. OVA-sensitized animals were challenged with either 400 μg MSA (OVAc) or OVA (OVA), and TMA-sensitized animals were challenged with either 400 μg MSA (TMAc) or TMA-MSA (TMA). Values represent the geometric mean + SE for n = 7, 8, 9, and 10 animals for the four groups from left to right. NS = not significant. *p < 0.05. The lower set of brackets depicts the comparison of each allergen to its respective control. The upper bracket indicates the statistical comparison of the change (OVA-OVAc vs. TMA-TMAc). Geometric mean values are listed under respective treatment groups.
Identification of Candidate Genes by Statistical Analysis
Differential gene expression in the lung following sensitization and challenge with different allergens would suggest that different mechanistic pathways were instrumental in generating the asthma phenotype. Gene expression profiles in experimentally induced asthma were measured in the 27 animals shown in Figure 1B using the Affymetrix murine U74Av2 chip. Initial statistical analysis focused on the identification of probe sets with a difference in any of the four treatment groups (OVA, OVAc, TMA, TMAc). After RMA normalization, the values were exported to SAS, and ANOVA p values were computed for the F test. This statistical comparison detects genes differentially expressed between the four treatments. The q-values (false discovery rate) from the F test p values were computed in R (R Foundation) following Storey and Tibshirani (2003). Genes with a q-value of less than 0.10 were selected to ensure at least 9:1 odds that the expression of the genes was statistically different in the respective groups (Storey and Tibshirani, 2003). A q-value cutoff of 0.1 guarded against false positives, yet resulted in a candidate gene pool of 853 genes. For each of the 853 probe sets, the intensity of OVA/intensity of OVAc was plotted against the intensity of TMA/intensity of TMAc. A plot of the natural log of these ratios is shown in Figure 3. A ratio of greater than one indicates an increase in expression, and a ratio of less than one reflects a reduction in expression between the treated and control animals. Although these 853 probe sets demonstrate a statistically significant difference in expression among the four treatment groups, expression ratios between 1/1.2 and 1.2 (shown in gray in Fig. 3) would be difficult to reproduce by other measures in the lung tissue milieu due to a lack of sensitivity. For this reason we focus further discussion to those 392 probe sets which exhibit an experimental/control ratio of greater than 1.2 or less than 1/1.2 for at least one of the two allergens (OVA/OVAc or TMA/TMAc). The 392 probe sets (Supplementary Table 1) represent 376 genes, as 16 genes of the 392 were represented by duplicate probe sets.
FIG. 3.
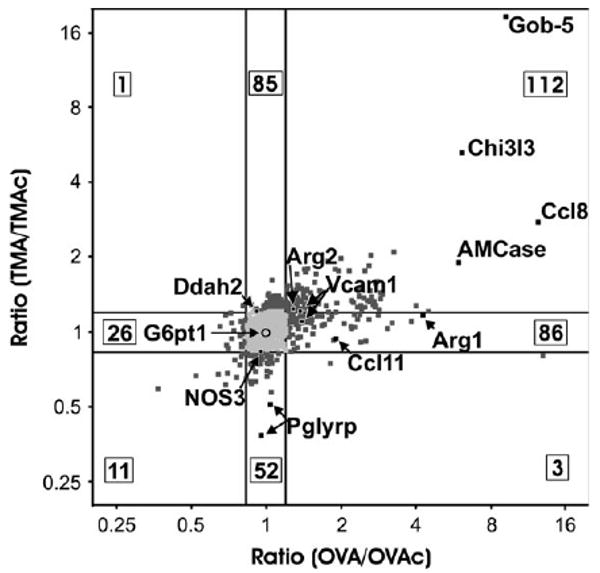
Relative changes in gene expression signal intensity for candidate genes. All 853 probe sets with a q-value less than 0.1 were plotted as the ratio of (OVA/OVAc) versus (TMA/TMAc) on a log2 scale. Values in the central grey box region represent 479 genes identified by the F test with a significant difference amongst the four treatment groups, but the experimental/control ratio was between 1/1.2 and 1.2 (lined in bold) for either OVA or TMA versus the respective control. The black points represent the 392 probe sets (376 genes) with a ratio greater than 1.2 (increased expression) or less than 1/1.2 (decreased expression). The boxed numbers designate the number of genes in the given region of the graph. The genes analyzed by qRT-PCR are indicated on the graph.
Two hundred thirty nine of the 376 genes depicted in Figure 3 had an expression ratio of greater than 1.2 (112 +86+ 3 = 201) or less than 1/1.2 (26 + 1 + 11) for the OVAc (sensitized only) versus OVA animals (sensitized and challenged). A subset of the genes up-regulated (ratio of ≥1.2) in response to OVA treatment is categorized in Table 2. Many of the 201 genes up-regulated in response to OVA sensitization and challenge have been described previously (Daheshia et al., 2002; Scheerens et al., 2002; Zhu et al., 2004; Zimmermann et al., 2003; Zuhdi Alimam et al., 2000). Genes up-regulated in response to OVA and novel to this study include the C3 complement regulator Daf2, T lymphocyte antigen CD2, and collagen alpha-1. Thus, our study confirms and extends previous studies demonstrating up-regulation of numerous genes in the OVA asthma model.
TABLE 2. Subset of Genes Up-Regulated in OVA-Induced Asthma.
| Functional classification | Gene name | Affymetrix probe set ID | OVA/OVAc ratio |
|---|---|---|---|
| Chemokines and cytokines | Ccl8 (MCP-2) | 92459_at | 12.40 |
| Ccr5 (CD195) | 161968_f_at | 2.62 | |
| Ccl9 (MIP-1 gamma) | 104388_at | 2.58 | |
| Ccl2 (MCP-1) | 102736_at | 2.21 | |
| Ccl11 (eotaxin) | 92742_at | 1.91 | |
| Cxcl13 | 102025_at | 1.78 | |
| Ccl7 (MCP-3) | 94761_at | 1.67 | |
| Ccl12 (MCP-5) | 93717_at | 1.47 | |
| Ccr1 (MIP-1 alphaR) | 99413_at | 1.43 | |
| Ccl6 (MRP-1) | 162198_f_at | 1.33 | |
| Cxcl9 (Mig) | 101436_at | 1.31 | |
| Ccl17 | 97783_at | 1.30 | |
| Il1r2 (interleukin 1 receptor, type II) | 161689_f_at | 1.21 | |
| Secretion and remodeling | Gob-5 (Clca3) | 162287_r_at | 9.16 |
| Gob-4 (Agr2) | 101075_f_at | 3.83 | |
| Tff2 (Trefoil factor 2) | 93302_at | 3.52 | |
| MMP12 (Matrix metalloproteinase 12) | 95339_r_at | 2.63 | |
| Muc5AC | 101803_at | 2.42 | |
| Timp1 | 101464_at | 1.52 | |
| Col3a1 (collagen alpha-1)a | 98331_at | 1.34 | |
| Cell adhesion and lectins | Chi3l3 (Chitinase 3-like 3) | 92694_at | 6.11 |
| AMCase | 160509_at | 5.91 | |
| CD53 | 94939_at | 1.56 | |
| CD83 | 103040_at | 1.53 | |
| Vcam1 | 92559_at | 1.39 | |
| Vcam1 | 92560_g_at | 1.37 | |
| CD2a | 95373_at | 1.26 | |
| Complement | C1qb | 96020_at | 2.78 |
| C1qc | 92223_at | 2.38 | |
| C1qa | 98562_at | 2.19 | |
| C3 | 93497_at | 1.74 | |
| Pcf (Properdin) | 101468_at | 1.52 | |
| Daf2a | 92198_s_at | 1.35 | |
| Arginine metabolism | Arg1 (Arginase1) | 93097_at | 4.27 |
| Gatm (glycine amidinotransferase) | 96336_at | 2.50 | |
| Arg2 | 98473_at | 1.28 |
Novel to this study.
Expression Differences between OVA- and TMA-Induced Asthma
As seen in Figure 3, 112 genes were up-regulated with both allergens (ratio OVA/OVAc and TMA/TMAc > 1.2), and 11 were down-regulated with both allergens (ratio OVA/OVAc and TMA/TMAc < 1/1.2). Our continued analysis focused on the 249 genes (85 + 86 + 52 + 26) that were uniquely up- or down-regulated for OVA or TMA (i.e., differentially expressed). A subset of the genes up-regulated in response to TMA treatment is presented in Supplementary Table 4. As seen in Figure 3, 86 genes were uniquely up-regulated for OVA and 85 uniquely up-regulated for TMA (i.e., a ratio of greater than 1.2 for one of the allergens and not the other). Also, 26 and 52 genes were uniquely down-regulated for OVA and TMA, respectively.
A subset of the uniquely up- or down-regulated genes is categorized in Table 3. The OVA animals display an up-regulation of numerous chemokines and receptors that show no change or decreased expression in TMA-treated animals. Ccl11 (eotaxin 1) is a mediator of eosinophil infiltration in OVA-induced asthma (Ganzalo et al., 1996). Eotaxin 2, a second eosinophil chemoattractant, is not represented on the U74Av2 chip. Two other chemoattractants up-regulated in both OVA- and TMA-induced asthma include the lectin YM1 (Chi3l3), which is identical to the lymphocyte-derived eosinophil chemotactic factor (ECF-L; Owhashi et al., 2000), and AMCase, which has 67% protein identity to mouse YM1/ECF-L/Chi3l3 (Owhashi et al., 2000). It should also be noted that many chemokines and receptors were coordinately up-regulated for OVA and TMA including Ccl8, Ccr5, Ccl9, Ccr1, and Ccl6 (Table 2 and Supplementary Table 4).
TABLE 3. Subset of Genes Differentially Regulated in OVA- versus TMA-Induced Asthmaa.
| Functional classification | Gene | Affymetrix probe set ID | Ratiob OVA/OVAc | Ratiob TMA/TMAc |
|---|---|---|---|---|
| Chemokines and cytokines | Ccl2 (MCP-1) | 102736_at | 2.21 | 1.02 |
| Ccl11 (eotaxin) | 92742_at | 1.91 | 0.93 | |
| Cxcl13 | 102025_at | 1.78 | 1.20 | |
| Ccl7 (MCP-3) | 94761_at | 1.67 | 1.05 | |
| Ccl12 (MCP-5) | 93717_at | 1.47 | 1.14 | |
| Socs1 (suppressor of cytokine signaling 1) | 92832_at | 1.37 | 0.95 | |
| Cxcl9 (Mig) | 101436_at | 1.31 | 1.12 | |
| Ccl17 | 97783_at | 1.30 | 1.09 | |
| Il1r2 (interleukin 1 receptor, type II) | 161689_f_at | 1.21 | 1.07 | |
| Pglyrp (peptidoglycan recognition protein) | 104099_at | 1.03 | 0.51 | |
| Pglyrp (peptidoglycan recognition protein) | 162475_f_at | 0.96 | 0.38 | |
| Cxcl12 (SDF-1) | 100112_at | 0.79 | 1.09 | |
| Secretion and remodeling | Sprr2a | 101024_i_at | 13.00 | 0.80 |
| Gob-4 (Agr2) | 101075_f_at | 3.83 | 1.09 | |
| RELM beta | 93755_at | 3.21 | 1.08 | |
| Sprr2b | 99701_f_at | 2.50 | 0.91 | |
| Sprr2a | 101025_f_at | 2.40 | 0.91 | |
| Scinderin | 103715_at | 2.27 | 1.04 | |
| pIgR | 99926_at | 1.81 | 0.74 | |
| Timp1 | 101464_at | 1.52 | 1.1 | |
| Cathepsin B | 94831_at | 1.36 | 1.13 | |
| Sfrp2; secreted frizzled-related sequence protein 2 | 93503_at | 1.35 | 1.12 | |
| Col3a1 (collagen) | 98331_at | 1.34 | 0.86 | |
| Cathepsin E | 104696_at | 1.33 | 1.13 | |
| Tfpi2 (tissue factor pathway inhibitor 2) | 94383_at | 1.26 | 1.08 | |
| Niemann Pick type C2 | 160344_at | 1.09 | 1.31 | |
| Stxbp2 (syntaxin binding protein 2) | 101398_at | 1.05 | 1.22 | |
| Vamp8 | 93305_f_at | 0.93 | 1.21 | |
| Krt2–7 (keratin) | 97920_at | 0.83 | 1.11 | |
| Cell adhesion and lectins | Intelectin | 102755_at | 3.07 | 1.02 |
| A430096B05Rik (Fc fragment of IgG binding protein) | 97826_at | 2.60 | 1.11 | |
| Vcam1 | 92559_at | 1.39 | 1.09 | |
| Vcam1 | 92560_g_at | 1.37 | 1.20 | |
| Col3a1 (collagen) | 98331_at | 1.34 | 0.86 | |
| Klra3 (killer cell lectin-like receptor) | 93894_f_at | 0.98 | 1.23 | |
| Sh3d4 (SH3 domain protein 4) | 98848_at | 0.96 | 0.77 | |
| Jup (junction plakoglobin) | 104121_at | 0.88 | 0.71 | |
| Complement | Properdin | 101468_at | 1.52 | 1.13 |
| Daf2 (decay accelerating factor 2) | 92198_s_at | 1.35 | 1.02 | |
| Arginine metabolism | Arg1 (arginase 1) | 93097_at | 4.27 | 1.15 |
| Ddah2 | 92887_at | 0.91 | 1.20 | |
| Nos3 (endothelial Nitric Oxide Synthase) | 94167_at | 0.95 | 0.83 |
Experimental/control ratio is >1.2 or <1/1.2 (0.83).
Ratio of relative change in chip intensity as depicted in Figure 3.
Genes involved in secretion and remodeling (Cathepsins B & E, Gob-4, RELM β, Scinderin, Sprr2a, Sprr2b, Tfpi2, and Timp1) were also generally up-regulated to a greater degree in OVA-treated animals than TMA-treated animals (Table 3). However, the exceptions were Niemann Pick type C2, syntaxin binding protein (Stxbp2), and Vamp8, which were up-regulated uniquely in TMA-induced asthma. Stxbp2/Munc 18–2 acts coordinately with target or t-SNAREs (soluble N-ethylmaleimide-sensitive fusion factor attachment protein receptor) and vesicular or v-SNAREs during mast cell degranulation (Martin-Verdeaux et al., 2002). Vamp8 is a v-SNARE and was up-regulated in TMA-induced asthma but not OVA-induced asthma, predicting a preferential degranulation of mast cells in TMA-induced asthma 72 h after challenge. Three genes previously described as asthma signature genes (Gob-4, RELM β, and Sprr2a; Zimmermann et al., 2003) were among the differentially expressed genes involved in secretion and remodeling. In contrast to previous studies with OVA (Stütz et al., 2003; Zimmermann et al., 2003), we found no change in expression of Gob-4 and RELM β in our TMA asthma model and a down-regulation of Sprr2a (Table 3).
The greatest difference in expression of cell adhesion molecules was seen for the gene intelectin (three-fold difference between OVA and TMA). This gene is expressed in intestinal Paneth cells and is up-regulated in response to Trichinella infection (Pemberton et al., 2004). Its up-regulation in OVA-induced asthma has been previously described (Zimmermann et al., 2003), but its role in asthma is unknown.
The complement component C5 has been described as a susceptibility locus for the development of airway hyperresponsiveness in OVA models of asthma (Hawlisch et al., 2004). In addition, C3a, a product of complement system activation, has been implicated as a critical mediator of airway hyperresponsiveness in OVA-induced asthma (Hawlisch et al., 2004). Thus, we looked for differential expression of components of the complement system. As seen in Table 3, the complement genes Properdin and Daf2 were up-regulated in OVA-induced asthma but showed no change in TMA-treated animals. However, other complement genes C1qa, C1qb, C1qc, and C3 were coordinately up-regulated for both OVA- and TMA-induced asthma (Table 2 and Supplementary Table 4). Thus, expression of complement system genes does not fit a consistent pattern.
A very limited number of genes (3 + 1) demonstrated oppositional regulation in OVA- versus TMA-induced asthma (Fig. 3). Three genes (Sprr2a, pIgR and Fv-4) were up-regulated in OVA and down-regulated in TMA, whereas cytosolic acyl-CoA thioesterase 1 (Cte1) was significantly up-regulated in TMA and down-regulated in OVA. Sprr2a is a member of a family of small proline-rich proteins which form the precursor to the cornified envelope (CE) in stratified squamous epithelial cells and may be important in airway remodeling (Zimmermann et al., 2005). pIgR is the polymeric immunoglobulin receptor whose function includes the transport of IgA and IgM class polymeric immunoglobulins across the glandular and mucosal epithelia (reviewed in Mostov, 1994). Sprr2a and pIgR are both STAT6-regulated proteins (Schjerven et al., 2000; Zimmermann et al., 2005). The differential regulation of Sprr2a and pIgR with OVA and TMA suggests coregulators interacting with STAT6 vary with allergen.
Confirmation of Microarray Analysis by qRT-PCR
To confirm the microarray results, a subset of genes was analyzed by qRT-PCR using the primers shown in Table 1. Lungs from six to eight animals were used for qRT-PCR determinations and included at least three animals in each treatment group that were also analyzed by microarrays. Genes with a variety of levels of expression were chosen, including genes involved in arginine metabolism. The gene G6pt1 (glucose-6-phosphate transporter protein 1) was selected as the housekeeping control by selecting the probe set from the microarray data with the smallest treatment effect (data not shown). qRT-PCR confirmed that G6pt1 expression does not significantly change with treatment (data not shown). Data from all primer sets were normalized to G6pt1 expression. The correlation (r) between microarray and qRT-PCR data for animals used in both assays ranged from 0.68 to 0.97 and was statistically significant (p < 0.05) for Gob-5, Ccl8, Chi3l3, AMCase, Arg1, Gatm, and Ccl11. In the case of Vcam1, Arg2, and Pglyrp1, the correlation was not significant. This lack of significance may be explained by the low expression levels observed for these genes in the microarray analysis, as Etienne et al. (2004) noted a reduced correlation between microarray and RT-PCR data with reduced expression levels, increasing number of absent calls, and increased distance between probes.
The relative RNA quantity for each animal determined by qRT-PCR was plotted by treatment for the selected genes (Fig. 4). In Panel A, statistical analysis indicated a significant and similar increase in Chi3l3, Ccl8, and Gob-5 for both allergens, consistent with the microarray data. In Panel B, statistical analysis of the qRT-PCR data indicated a significant increase in AMCase, Arg1, and Ccl11 for each allergen, with the increase for OVA/OVAc being significantly greater than that seen for TMA/TMAc. Gatm expression levels, shown in Panel C, significantly increased with OVA/OVAc, but not with TMA/TMAc. For each of the genes in Panels A, B, and C, the experimental/control ratio from microarray analysis showed the same relative changes as seen with qRT-PCR. In general, our data are consistent with Yuen et al. (2002) demonstrating qRT-PCR estimates of mRNA levels are consistently larger than those measured by microarray. No significant differences were detected by qRT-PCR analysis with Vcam1, Arg2, and Pglyrp1 (data not shown).
FIG. 4.

The relative quantity of RNA as determined by quantitative real-time PCR in lungs 72 h after challenge. OVA-sensitized animals were challenged with either 400 μg MSA (OVAc) or OVA (OVA), and TMA-sensitized animals were challenged with either 400 μg MSA (TMAc) or TMA-MSA (TMA). Genes with a similar increase in expression for both allergens are shown in A. Genes of interest with an increase in expression for both allergens, and a significantly greater increase in expression with OVA treatment are shown in B. Category C includes Gatm, a gene of interest with a significant increase in expression for OVA treatment only. Values represent the geometric mean + SE for n = 6 to 8 animals for each of the four treatment groups. NS = not significant. *p < 0.05 for the comparisons indicated. The lower set of brackets depicts the comparison of each allergen to its respective control. The upper bracket indicates the statistical comparison of the change (OVA-OVAc vs. TMA-TMAc).
Differences in Arginine Metabolism in OVA- and TMA-Induced Asthma
The treatment-specific expression differences observed for Arg1 were further addressed by assessment of enzymatic activity. Previous work (Zimmermann et al., 2003) indicated that arginase gene expression and enzyme activity are up-regulated as part of the asthmatic response induced by OVA in the mouse. Since the microarray results had indicated that arginase 1 expression was not up-regulated (≤1.2) in TMA-induced asthma, and the qRT-PCR data indicated a significant difference in up-regulation between OVA- and TMA-induced asthma, we hypothesized that pathways of arginine metabolism may differ in OVA- and TMA-induced asthma. As shown in Figure 5, arginase enzyme activity in the lung was significantly increased in both OVA- and TMA-induced asthma. However, the increase in arginase enzyme activity was significantly greater in OVA-induced asthma compared to TMA-induced asthma, consistent with the gene expression data.
FIG. 5.

Arginase activity in lungs 72 h after challenge. OVA-sensitized animals were challenged with either 400 μg MSA (OVAc) or OVA (OVA), and TMA-sensitized animals were challenged with either 400 μg MSA (TMAc) or TMA-MSA (TMA). Values represent the geometric mean + SE for n = 6 animals for each of the four treatment groups. *p < 0.05. The lower set of brackets depicts the comparison of each allergen to its respective control. The upper bracket indicates the statistical comparison of the change (OVA-OVAc vs. TMA-TMAc). Geometric mean values are listed under respective treatment groups.
Given the observed differences between OVA- and TMA-induced arginase gene expression and enzyme activity, we examined the microarray data for other probe sets involved in arginine metabolism and/or NO production. For these data, statistical analysis involved ANOVA analysis with specific contrasts, without correction for multiple comparisons. A significance level of 0.05 in combination with a gene expression fold change on the microarrays of greater than 1.2 or less than 1/1.2 were the criteria used. Those genes involved in arginine metabolism with a significant change in expression for either allergen are labeled on Figure 3. By these criteria, Arg1, Arg2, and Gatm (glycine amidinotransferase) gene expression were increased in OVA-induced asthma, but not TMA-induced asthma (see experimental/control ratios on Fig. 3). Two additional genes involved in arginine metabolism, Ddah2 (dimethylarginine dimethylaminohydrolase 2) and NOS3 (eNOS), were also differentially regulated in TMA- versus OVA-challenged animals. Ddah2 was significantly increased in TMA-induced asthma but not in OVA-induced asthma. Conversely, NOS3 was down-regulated in TMA-induced asthma but not in OVA-induced asthma. No significant changes were detected for NOS2 (iNOS), NOS1 (nNOS), the L-arginine transporter CAT2, ornithine decarboxylase, ornithine aminotransferase, argininosuccinate lyase, and argininosuccinate synthase. In addition, four probe sets on the chip related to arginine methyltransferase (Carm1-pending, Hrmt 1l3, Hrmt1l2, and Skb1/JBP1) showed no significant increase in expression with either allergen.
Discussion
Asthma is a multifaceted lung disorder at both the physiologic and genetic levels. Using microarray analysis, we tested the hypothesis that different allergens evoke unique patterns of gene expression suggestive of unique effector mechanisms leading to the asthma phenotype, hence presenting distinctive “transcriptional signatures.” Although the same change in tissue eosinophilia was seen using identical sensitization and challenge regimens with two different allergens, we observed distinct transcriptional changes for mice challenged with OVA versus mice challenged with TMA. One hundred twenty three genes were coordinately expressed in OVA and TMA (i.e., up-regulated or down-regulated in both OVA and TMA), albeit to varying degrees of up- or down-regulation. Four genes showed opposing expression for OVA and TMA, and the remaining 249 genes (2% of the genes on the array) had differential regulation for either OVA or TMA but not both treatments. Our conclusions are based on microarray analysis, qRT-PCR of candidate genes, and assessment of the enzymatic activity of a gene product, i.e., arginase. These data support the hypothesis that different allergens cause the same asthmatic phenotype via alternate mechanisms in the organism.
The lung contains numerous cell types: endothelial, epithelial, dendritic, white blood cells, nerve cells, etc. Thus, gene expression changes noted in this study could be the result of changes in any one cell type, or changes in the percent of a single cell type, or a combination thereof. All of the cell types have been implicated as being important in the asthmatic response. Studying each individual cell type in vitro yields useful information, as does studying isolated airways and their reactivity. However, the interaction of cell types and mediators ultimately leads to the response in the whole lung with eosinophilia and airway obstruction. For example, gene expression changes in a purified macrophage population may be quite different from gene expression changes in a macrophage residing in the lung due to interactions with adjacent cell populations within the lung tissue. Thus, it is important to consider the whole lung and the complex interactions occurring in vivo, rather than just a single cell type.
In compiling a list of candidate genes from the array data, we utilized both a test of statistical significance and an expression ratio cutoff. Fold-change cutoffs used alone tend to result in a larger fraction of false positives due to outlier responses. After using a q-value cutoff of 0.1, we further narrowed our candidate gene list by declaring that an experimental-to-control ratio of 1.2 or greater (or 1/1.2 and lower) was necessary for us to consider the gene as potentially biologically significant. This further reduced our candidate gene list to 249 genes. A twofold change was initially an arbitrary change reported as significant in many early microarray studies. However, this standard has changed considerably with advances in (a) the normalization of data (e.g., RMA) and (b) improved statistical analysis of array data. Our choice of 1.2 as an expression ratio cutoff with biological significance was based on a review of the literature and the fact that we were using a whole tissue where important changes in gene expression in a single cell type may be diluted. Because we were using a mixed tissue, we wanted to insure that any gene expression changes that might have been diluted due to the variety of cell types present were also still considered. Therefore, we used an experimental/control ratio of 1.2 that was both statistically significant and broad. Studies of Jin et al. (2001) and Pierce et al. (2003) demonstrated that gene expression changes as low as 1.2-fold were biologically significant in a Drosophilia model and a colorectal cancer cell line, respectively. The actual biological significance of any change in gene expression can only be determined by critically testing focused hypotheses using more classical methods, including inhibitors and measurements of protein.
Our study demonstrated that many genes are up-regulated to a greater extent with TMA than OVA (e.g., Gob-5), just as many genes are up-regulated more with OVA than TMA (e.g., arginase). This suggests that differences in gene expression noted for OVA versus TMA are not likely due to differences in the dose of allergen or the extent of the allergic reaction. Additional unpublished studies using a different OVA sensitization and challenge regimen resulted in maximum lung EPO values comparable to the TMA treatment group in this study. In these unpublished studies with OVA as the allergen and a lower maximum lung EPO response, arginase up-regulation was also evident (experimental/control ratio of 10.3), suggesting that the extent of the eosinophilia does not determine the up-regulation of arginase. One would expect genes important in cellular infiltration to correlate in some manner with that biological endpoint. Clearly the differentially expressed genes are not all critical mediators of the eosinophil infiltration component of the asthmatic response, and the extent of up- or down-regulation for any given gene does not necessarily correlate with the magnitude of the allergic endpoint being measured. Our long-term goal is to link allergen-specific gene expression patterns with allergen-specific effector pathways or profiles of asthma symptoms. The differences in gene expression could reflect either differences in effector pathways leading to the same profile of asthma symptoms or differences in effector pathways leading to a different profile of asthma symptoms, i.e., asthma heterogeneity.
A single time point (72 h after challenge) was chosen to compare gene expression changes in the effector phase of asthma induced by the two different antigens, OVA and TMA. This 72 h time point was chosen because, in preliminary studies examining lung eosinophilia at 48 and 72 h after a single 400 μg TMA-MSA or OVA challenge, the change in lung EPO was as great or greater at 72 h than 48 h for both allergens. Similar time courses in cell infiltration are expected with these identical sensitization and challenge regimens, and the differences in gene expression are not attributable to different routes of administration of the antigens, the presence of different adjuvants, or different mouse strains. The phenotypes are similar in that the change in eosinophilia in the lung tissue is no different for the two allergens (Fig. 1). Unlike some more vigorous sensitization and challenge regimens, our method of sensitization and challenge with OVA and TMA did not result in any increase in neutrophils in the lung or BAL. However, we saw a significant increase in BAL macrophages and eosinophils with TMA sensitization and challenge, but not OVA sensitization and challenge (Fig. 2). We have not investigated whether gene expression profiles in TMA-induced asthma suggest greater macrophage chemotaxis and/or extravasation or instead result from greater expression of local myeloid progenitor cells. In a study by Tomkinson et al. (2001) using a single intranasal OVA challenge in sensitized animals, tissue eosinophilia peaked 48 h after OVA challenge, and BAL eosinophils peaked 48–96 h after challenge and were sustained for at least 8 days. Thus, there is clear precedent for differences in tissue eosinophilia and BAL eosinophilia at any particular time point. In addition, studies of Pope et al. (2005) have suggested that eotaxin 1 and 2 are mediators important in determining eosinophil accumulation in the airspace versus the tissue. Thus, the difference in BAL eosinophilia noted with the two allergens may reflect different effector pathways contributing to the heterogeneity of the allergic response with the two allergens. Clearly, further studies are indicated to rigorously examine the time course of changes in gene expression with the time course of each asthma symptom, e.g., airway hyperresponsiveness, mucus production, BAL and tissue eosinophilia, etc., for each allergen.
Of the 249 genes differentially regulated for OVA and TMA, we chose to focus on genes that were uniquely up- or down-regulated in arginine metabolism. Previous studies of others had implicated arginase as an important step in asthma pathogenesis (Zimmermann et al., 2003). A simplified diagram of our hypothetical working model of arginase action in the cell and potential fates of L-arginine in OVA- versus TMA-induced asthma is shown in Figure 6. Our gene expression results predict that OVA-induced asthma would increase levels of ornithine and perhaps the downstream products of spermine and polyamines due to increased expression of arginase 1, arginase 2, and Gatm and decreased production of NO. L-ornithine is a precursor element for production of collagen and polyamines, both of which are important for airway remodeling. This is consistent with the up-regulation of other remodeling genes noted in OVA-induced asthma but not TMA-induced asthma (Table 3). The decreased NO production predicted in the OVA model could be explained by a reduction in L-arginine availability due to increased activities and/or expression of arginase 1, arginase 2, and Gatm. Lower concentrations of L-arginine would ultimately favor the competitive inhibition of NOS by endogenously formed asymmetric dimethylarginine (ADMA) and N-monomethylarginine (NMMA). In contrast, the animals with TMA-induced asthma present a gene expression pattern favoring increased production of NO through the stable expression of arginase 1 and arginase 2 and the up-regulation of Ddah2, which will decrease the levels of competitive inhibitors of NOS (ADMA and NMMA). These data are consistent with results in a guinea pig model of TMA-induced asthma demonstrating an up-regulation of Ca2+-independent iNOS activity in the lung tissue and increased nitrite levels in BAL (Yan et al., 1995). Whether the hypothetical NO production results in bronchodilation in the airway or increased reactive nitrogen species with protein nitration, inflammation, and cytotoxicity is unknown. The literature on NO in the lung is extensive and conflicting, and the end result of NO production depends on the concentration of NOS substrates and the site of NO generation (Ricciardolo et al., 2004). Certainly, continued studies beyond gene expression are required to adequately test the hypotheses presented in this working model.
FIG. 6.

Hypothetical working model of differences in arginine metabolism in the mouse models of OVA- and TMA-induced asthma. This hypothetical model incorporates the differences in gene expression observed in this study and will serve as a testable model for future studies. Arg 1, arginase 1; Arg 2, arginase 2; ADMA, asymmetric dimethylarginine; CAT2, cationic amino acid transporter; Ddah2, dimethylarginine dimethylaminohydrolase; Gatm, glycine amidinotransferase; NOS, nitric oxide synthase.
Besides differential expression of genes involved in arginine metabolism, we also categorized a subset of the genes that were uniquely up- or down-regulated in OVA- versus TMA-induced asthma as chemokines or cytokines and their receptors, genes involved in secretion and remodeling or adhesion, and genes involved in the complement system (Table 3). Results for OVA confirmed previously reported studies (Fulkerson et al., 2004; Scheerens et al., 2002; Zimmermann et al., 2003). Considering chemokines and cytokines, many were coordinately up-regulated in both OVA- and TMA-treated animals. However, a subset of chemokines was differentially regulated. Noteworthy among the differentially regulated chemokines was Ccl11 (eotaxin 1). Although no significant increase in eotaxin 1 (Ccl11) expression was seen in TMA-induced asthma on the microarray, a three-fold increase in expression was detected by qRT-PCR (Fig. 4). Since OVA and TMA treatments elicited similar changes in eosinophilia (Fig. 1), additional chemoattractants must help mediate eosinophilia with TMA. Of the known eosinophil chemoattractants present on the microarray chip (Ccl3, Ccl5, Ccl7, Ccl11, Ccl22), none were up-regulated in response to TMA challenge, while two (Ccl7 and Ccl11) showed an increase in expression with OVA-induced asthma. Recent studies have indicated that AMCase is a critical mediator of eosinophilia in a mouse model of OVA-induced asthma (Zhu et al., 2004), with an indication that AMCase acts through eotaxin to induce eosinophilia (Donnelly and Barnes, 2004). AMCase was significantly up-regulated with both allergens in the present study and may contribute to the eosinophilia seen in the TMA model of asthma. Finally, the unique chemokine expression profiles seen with the two different allergens may reflect the different cell profiles seen in the BAL.
Remodeling and secretion may comprise a larger component of the response in OVA-induced asthma than in TMA-induced asthma, as suggested by the difference in gene expression profiles for Sprr2a, Sprr2b, RELM β, scinderin, and Gob-4 between OVA and TMA and as noted in the working model in Figure 6. Sprr2a and RELM β are produced in goblet cells (He et al., 2003; Komiya et al., 1999) and contain STAT binding sites, consistent with many other genes involved in airway inflammation. Several other genes expressed in goblet cells (Gob-5, Muc5AC, and Tff2) have been previously described in OVA-induced asthma (Nakanishi et al., 2001; Nikolaidis et al., 2003; Zuhdi Alimam et al., 2000) and were up-regulated in response to both OVA and TMA in our study.
The largest difference in expression detected between OVA and TMA was noted with Sprr2a, a member of a family of small proline-rich proteins which form the precursor to the cornified envelope (CE) in stratified squamous epithelial cells. This protein has been reported as STAT6-dependent for both OVA- and Aspergillus-induced asthma models (Zimmermann et al., 2004) and may be important in airway remodeling (Hohl et al., 1995; Zimmermann et al., 2005). However, in our TMA-induced model of asthma, we do not observe an increase in Sprr2a expression. This difference indicates that the regulation or timing of regulation for Sprr2a in TMA-treated animals differs from OVA-treated animals. If the regulation of Sprr2a is STAT6-dependent, then there may be coregulators of Sprr2a that differ between these asthma models. Further analysis is necessary to determine what if any coregulators exist for Sprr2a.
A group of 291 genes, proposed as “asthma signature genes” by Zimmermann et al. (2003), increased in expression to OVA- and Aspergillus-induced airway inflammation and included the genes Sprr2a, RELM β, arginase 1, Scinderin, cathepsin B, and Ccl11. In the present model of OVA- and TMA-induced asthma, this particular subset of “asthma signature genes” were either increased in OVA- but not TMA-induced asthma (Sprr2a, RELM beta, Scinderin, cathepsin B) or elevated in both but to significantly different levels (Arg1, Ccl11). The disparities in gene expression between our study and that of Zimmermann et al. (2003) may be due in part to different treatment regimens, samples sizes, or statistical analysis. Zimmermann et al. (2003) used an acute and chronic treatment regimen for OVA and Aspergillus, respectively, whereas we used the same acute treatment regimen for both OVA and TMA. In addition, the sample sizes and statistical analysis differed in the two studies, with Zimmermann et al. (2003) using sample sizes of two to four for treatment groups and selection criteria of p < 0.05 and fold change >2. In contrast, our study used an n of 6 or more for each treatment group as recommended by Cui and Churchill (2003) and statistical analysis included computation of q-values to guard against false positives.
A cross-comparison of the results of our study and Zimmermann et al. (2003) results in a set of 62 genes that were up-regulated in response to all 3 allergens; OVA, TMA, and Aspergillus (Supplementary Table 2). Five of these genes, Ccl8, Chi3l3, Gob-5, Mmp12, and Tff2, have a 2-fold or greater increase in expression with all three allergens (this study and Zimmermann et al., 2003). Together these data suggest that the effector mechanism of TMA-induced asthma may converge with the OVA and Aspergillus effector pathways at a point downstream of the asthma signature genes described by Zimmermann et al. (2003). Only continued studies with additional allergens will determine whether up-regulation of this set of 62 genes is indicative of asthma and suggestive of a common final effector pathway for all allergens. Nonetheless, TMA-induced allergic airway inflammation does not completely fit the model of asthma signature genes presented by Zimmermann et al. (2003).
Asthma is a complex lung disorder involving multiple cell types and signaling from within and beyond the lung. Although the phenotype of different allergens may appear similar, the mechanisms by which the asthmatic phenotype is presented appear to be quite different. This may have significant impact on potential therapeutic interventions in asthma. We have used microarray analysis and subsequent confirmation by qRT-PCR and an enzymatic assay to show that two occupational allergens, OVA and TMA, present different gene expression profiles, although their respective biological phenotypes are similar. Specifically, L-arginine utilization may be quite different in the two models due to contrasting levels of arginase activity. Such differences may result in alternate signaling pathways and cellular involvement producing the same constellation of asthma symptoms.
Supplementary Material
Acknowledgments
The excellent statistical assistance of Mingqian Duan is acknowledged. This work was supported in part by a grant from the U.S. Army Medical Research Acquisition Activity DAMD 17–02–1–0191. The U.S. Army Medical Research Acquisition Activity, 820 Chandler Street, Fort Detrick MD 21702– 5014 is the awarding and administering acquisition office for this award. In addition, the support of the Supercomputing Institute of the University of Minnesota was essential to completion of this project.
Footnotes
Supplementary Data: Supplementary data are available online at www.toxsci.oupjournals.org. Files in this data supplement:
- Supplementary Data—Summary
- Supplementary Table 1—Differentially expressed genes
- Supplementary Table 2—62 asthma genes
- Supplementary Table 3—Normalized microarray data
- Supplementary Table 4—Subset of genes up-regulated in TMA-induced asthma
Conflict of interest: none declared.
References
- Arakawa H, Kawikova I, Skoogh B, Hayes J, Morikawa A, Lofdahl C, Lotvall J. Role of arachidonic acid metabolites in airway responses induced by trimellitic anhydride in actively sensitized guinea pigs. Am Rev Respir Dis. 1993;147:1116–1121. doi: 10.1164/ajrccm/147.5.1116. [DOI] [PubMed] [Google Scholar]
- Bel EH. Clinical phenotypes of asthma. Curr Opin Pulm Med. 2004;10:44–50. doi: 10.1097/00063198-200401000-00008. [DOI] [PubMed] [Google Scholar]
- Boeniger MF, Lummus ZL, Biagini RE, Bernstein DI, Swanson MC, Reed C, Massoudi M. Exposure to protein aeroallergens in egg processing facilities. Appl Occup Environ Health. 2001;16:660–670. doi: 10.1080/10473220118319. [DOI] [PubMed] [Google Scholar]
- Cui X, Churchill GA. Statistical tests for differential expression in cDNA microarray experiments. Genome Biol. 2003;4:210–219. doi: 10.1186/gb-2003-4-4-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daheshia M, Tian N, Connolly T, Drawid A, Wu Q, Bienvenu JG, Cavallo J, Jupp R, De Sanctis GT, Minnich A. Molecular characterization of antigen-induced lung inflammation in a murine model of asthma. Ann NY Acad Sci. 2002;975:148–159. doi: 10.1111/j.1749-6632.2002.tb05948.x. [DOI] [PubMed] [Google Scholar]
- Donnelly LE, Barnes PJ. Acidic mammalian chitinase— a potential target for asthma therapy. Trends Pharmacol Sci. 2004;25:509–511. doi: 10.1016/j.tips.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Etienne W, Meyer MH, Peppers J, Meyer RA., Jr Comparison of mRNA gene expression by RT-PCR and DNA microarray. Biotechniques. 2004;36:618–626. doi: 10.2144/04364ST02. [DOI] [PubMed] [Google Scholar]
- Fraser DG, Regal JF, Arndt ML. Trimellitic anhydrideinduced allergic response in the lung: Role of the complement system in cellular changes. J Pharmacol Exp Ther. 1995;273:793–801. [PMC free article] [PubMed] [Google Scholar]
- Fulkerson PC, Zimmermann N, Brandt EB, Muntel EE, Doepker MP, Kavanaugh JL, Mishra A, Witte DP, Zhang H, Farber JM, et al. Negative regulation of eosinophil recruitment to the lung by the chemokine monokine induced by IFN-γ (Mig, CXCL9) Proc Natl Acad Sci USA. 2004;101:1987–1992. doi: 10.1073/pnas.0308544100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganzalo JA, Jia GQ, Aguirre V, Friend D, Coyle AJ, Jenkins NA, Lin GS, Katz H, Lichtman A, Copeland N, et al. Mouse eotaxin expression parallels eosinophil accumulation during lung allergic inflammation but it is not restricted to a Th2-type response. Immunity. 1996;4:1–14. doi: 10.1016/s1074-7613(00)80293-9. [DOI] [PubMed] [Google Scholar]
- Hawlisch H, Wills-Karp M, Karp CL, Köhl J. The anaphylatoxins bridge innate and adaptive immune responses in allergic asthma. Mol Immunol. 2004;41:123–131. doi: 10.1016/j.molimm.2004.03.019. [DOI] [PubMed] [Google Scholar]
- He W, Wang ML, Jiang HQ, Steppan CM, Shin ME, Thurnheer MC, Cebra JJ, Lazar MA, Wu GD. Bacterial colonization leads to the colonic secretion of RELMbeta/FIZZ2, a novel goblet cell-specific protein. Gastroenterology. 2003;125:1388–1397. doi: 10.1016/j.gastro.2003.07.009. [DOI] [PubMed] [Google Scholar]
- Hohl D, de Viragh PA, Amiguet-Barras F, Gibbs S, Backendorf C, Huber M. The small proline-rich proteins constitute a multigene family of differentially regulated cornified cell envelope precursor proteins. Dermatology. 1995;104:902–909. doi: 10.1111/1523-1747.ep12606176. [DOI] [PubMed] [Google Scholar]
- Humbles AA, Lu B, Friend DS, Okinaga S, Lora J, Al-Garawi A, Martin TR, Gerard NP, Gerard C. The murine CCR3 receptor regulates both the role of eosinophils and mast cells in allergen-induced airway inflammation and hyperresponsiveness. Proc Natl Acad Sci USA. 2002;99:1479–1484. doi: 10.1073/pnas.261462598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of affymetrix genechip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W, Riley RM, Wolfinger RD, White KP, Passador-Gurgel G, Gibson G. The contributions of sex, genotype and age to transcriptional variance in Drosophila melanogaster. Nat Genet. 2001;29:389–395. doi: 10.1038/ng766. [DOI] [PubMed] [Google Scholar]
- Komiya T, Tanigawa Y, Hirohashi S. Cloning of the gene gob-4, which is expressed in intestinal goblet cells in mice. Biochim Biophys Acta. 1999;1444:434–438. doi: 10.1016/s0167-4781(99)00010-x. [DOI] [PubMed] [Google Scholar]
- Li C, Wong WH. Model-based analysis of oligonucleotide arrays: Expression index computation and outlier detection. Proc Natl Acad Sci USA. 2001;98:31–36. doi: 10.1073/pnas.011404098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Bryce PJ, Humbles AA, Laouini D, Yalcindag A, Alenius H, Friend SS, Oettgen HC, Gerard C, Geha RS. CCR3 is essential for skin eosinophilia and airway hyperresponsiveness in a murine model of allergic skin inflammation. J Clin Invest. 2002;109:621–628. doi: 10.1172/JCI14097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Verdeaux S, Pombo I, Iannascoli B, Roa M, Varin-Blank N, Rivera J, Blank U. Evidence of a role for Munc18–2 and microtubules in mast cell granule exocytosis. J Cell Sci. 2002;116:325–334. doi: 10.1242/jcs.00216. [DOI] [PubMed] [Google Scholar]
- Mostov KE. Transepithelial transport of immunoglobulins. Ann Rev Immunol. 1994;12:63–84. doi: 10.1146/annurev.iy.12.040194.000431. [DOI] [PubMed] [Google Scholar]
- Nakanishi A, Morita S, Iwashita H, Sagiya Y, Ashida Y, Shirafuji H, Fujisawa Y, Nishimura O, Fujino M. Role of gob-5 in mucus overproduction and airway hyperresponsiveness in asthma. Proc Natl Acad Sci USA. 2001;98:5175–5180. doi: 10.1073/pnas.081510898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaidis NM, Zimmermann N, King NE, Mishra A, Pope SM, Finkelman FD, Rothenberg ME. Trefoil factor-2 is an allergen-induced gene regulated by Th2 cytokines and STAT6 in the lung. Am J Respir Cell Mol Biol. 2003;29:458–464. doi: 10.1165/rcmb.2002-0309OC. [DOI] [PubMed] [Google Scholar]
- Owhashi M, Arita H, Hayai N. Identification of a novel eosinophil chemotactic cytokine (ECF-L) as a chitinase family protein. J Biol Chem. 2000;275:1279–1286. doi: 10.1074/jbc.275.2.1279. [DOI] [PubMed] [Google Scholar]
- Özer N. A new enzyme-coupled spectrophotometric method for the determination of arginase activity. Biochem Med. 1985;33:367–371. doi: 10.1016/0006-2944(85)90012-2. [DOI] [PubMed] [Google Scholar]
- Pemberton AD, Knight PA, Gamble J, Colledge WH, Lee JK, Pierce M, Miller HR. Innate BALB/c enteric epithelial responses to Trichinella spiralis: Inducible expression of a novel goblet cell lectin, intelectin-2, and its natural deletion in C57BL/10 mice. J Immunol. 2004;173:1894–1901. doi: 10.4049/jimmunol.173.3.1894. [DOI] [PubMed] [Google Scholar]
- Pierce M, Wang C, Stump M, Kamb A. Overexpression of the β-catenin binding domain of cadherin selectively kills colorectal cancer cells. Int J Cancer. 2003;107:229–237. doi: 10.1002/ijc.11372. [DOI] [PubMed] [Google Scholar]
- Pope SM, Fulkerson PC, Blanchard C, Akei HS, Nikolaidis NM, Zimmermann N, Molkentin JD, Rothenberg ME. Identification of a cooperative mechanism involving interleukin-13 and eotaxin-2 in experimental allergic lung inflammation. J Biol Chem. 2005;280:13952–13961. doi: 10.1074/jbc.M406037200. [DOI] [PubMed] [Google Scholar]
- Regal JF. Immunologic effector mechanisms in animal models of occupational asthma. J Immunotoxicol. 2004;1:25–38. doi: 10.1080/15476910490438351. [DOI] [PubMed] [Google Scholar]
- Regal JF, Fraser DG. Systemic complement system depletion does not inhibit cellular accumulation in antihistamine pretreated allergic guinea pig lung. Int Arch Allergy Appl Immunol. 1996;109:150–160. doi: 10.1159/000237214. [DOI] [PubMed] [Google Scholar]
- Regal JF, Mohrman ME, Sailstad DM. Trimellitic anhydrideinduced eosinophilia in a mouse model of occupational asthma. Toxicol Appl Pharmacol. 2001;175:234–242. doi: 10.1006/taap.2001.9251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricciardolo FLM, Sterk PJ, Gaston B, Folkerts G. Nitric oxide in health and disease of the respiratory system. Physiol Rev. 2004;84:731–765. doi: 10.1152/physrev.00034.2003. [DOI] [PubMed] [Google Scholar]
- Scheerens J, van Gessel SB, Nijkamp FP, Folkerts G. Eotaxin protein levels and airway pathology in a mouse model for allergic asthma. Eur J Pharmacol. 2002;453:111–117. doi: 10.1016/s0014-2999(02)02364-6. [DOI] [PubMed] [Google Scholar]
- Schjerven H, Brandtzaeg P, Johansen FE. Mechanism of IL-4-mediated up-regulation of the polymeric Ig receptor: Role of STAT6 in cell type-specific delayed transcriptional response. J Immunol. 2000;165:3898–3906. doi: 10.4049/jimmunol.165.7.3898. [DOI] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stütz AM, Pickart LA, Trifilieff A, Baumruker T, Prieschl-Strassmayr E, Woisetschläger M. The Th2 cell cytokines IL-4 and IL-13 regulate found in inflammatory zone 1/resistin-like molecule α gene expression by a STAT6 and CCAAT/enhancer-binding protein-dependent mechanism. J Immunol. 2003;170:1789–1796. doi: 10.4049/jimmunol.170.4.1789. [DOI] [PubMed] [Google Scholar]
- Tomkinson A, Ciellewicz G, Duez C, Larson KA, Lee JJ, Gelfand EW. Temporal association between airway hyperresponsiveness and airway eosinophilia in ovalbumin-sensitized mice. Am J Respir Crit Care Med. 2001;163:721–730. doi: 10.1164/ajrccm.163.3.2005010. [DOI] [PubMed] [Google Scholar]
- Van Kampen V, Merget R, Baur X. Occupational airway sensitizers: An overview on the respective literature. Am J Ind Med. 2000;38:164–218. doi: 10.1002/1097-0274(200008)38:2<164::aid-ajim7>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Yan ZQ, Hannsson GK, Skoogh BE, Lötvall JO. Induction of nitric oxide synthase in a model of allergic occupational asthma. Allergy. 1995;50:760–764. doi: 10.1111/j.1398-9995.1995.tb01221.x. [DOI] [PubMed] [Google Scholar]
- Yuen T, Wurmbach E, Pfeffer RL, Ebersole BJ, Sealfon SC. Accuracy and calibration of commercial oligonucleotide and custom cDNA microarrays. Nucleic Acids Res. 2002;30:e48. doi: 10.1093/nar/30.10.e48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeiss CR, Mitchell JH, Peenen V, Harris J, Levitz D. A twelve year clinical and immunologic evaluation of workers involved in the manufacture of trimellitic anhydride (TMA) Allergy Proc. 1990;11:71–77. doi: 10.2500/108854190778993245. [DOI] [PubMed] [Google Scholar]
- Zeiss CR, Patterson R, Pruzansky JJ, Miller MM, Rosenberg M, Levitz D. Trimellitic anhydride-induced airway syndromes: Clinical and immunologic studies. J Allergy Clin Immunol. 1977;60:96–103. doi: 10.1016/0091-6749(77)90033-1. [DOI] [PubMed] [Google Scholar]
- Zhu Z, Zheng T, Homer RJ, Kim YK, Chen NY, Cohn L, Hamid Q, Elias JA. Acidic mammalian chitinase in asthmatic Th2 inflammation and IL-13 pathway activation. Science. 2004;304:1678–1682. doi: 10.1126/science.1095336. [DOI] [PubMed] [Google Scholar]
- Zimmermann N, Doepker MP, Witte DP, Stringer KF, Fulkerson PC, Pope SM, Brandt EB, Mishra A, King NE, Nikolaidis NM, et al. Expression and regulation of small proline-rich protein 2 in allergic inflammation. Am J Respir Cell Mol Biol. 2005;32:428–435. doi: 10.1165/rcmb.2004-0269OC. [DOI] [PubMed] [Google Scholar]
- Zimmermann N, King NE, Laporte J, Yang M, Mishra A, Pope SM, Muntel EE, Witte DP, Pegg AA, Foster PS, et al. Dissection of experimental asthma with DNA microarray analysis identifies arginase in asthma pathogenesis. J Clin Invest. 2003;111:1863–1874. doi: 10.1172/JCI17912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann N, Mishara A, King NE, Fulkerson PC, Doepker MP, Nikolaidis NM, Kindinger LE, Moulton EA, Aronow BJ, Rothenberg ME. Transcript signatures in experimental asthma: Identification of STAT6-dependent and -independent pathways. J Immunol. 2004;172:1815–1824. doi: 10.4049/jimmunol.172.3.1815. [DOI] [PubMed] [Google Scholar]
- Zuhdi Alimam M, Piazza FM, Selby DM, Letwin N, Huang L, Rose MC. Muc-5/5AC mucin messenger RNA and protein expression is a marker of goblet cell metaplasia in murine airways. Am J Respir Cell Mol Biol. 2000;22:253–260. doi: 10.1165/ajrcmb.22.3.3768. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.