

Abstract
Next-generation sequencing is limited to short read lengths and by high error rates. We systematically analyzed sources of noise in the Illumina Genome Analyzer that contribute to these high error rates and developed a base caller, Alta-Cyclic, that uses machine learning to compensate for noise factors. Alta-Cyclic substantially improved the number of accurate reads for sequencing runs up to 78 bases and reduced systematic biases, facilitating confident identification of sequence variants.
Next-generation sequencers are revolutionizing biological research1,2. They impact many aspects of genomics, including studies of sequence and structural variation in genomes3,4, and studies of the epigenome5. Nevertheless, these platforms are error-prone and suffer from short read lengths as compared to conventional sequencers4. We sought to improve the base-calling procedure for Illumina Genome Analyzers to obtain more accurate and longer sequence reads. Such an improvement would boost overall output per run, increasing genomic coverage and the ability to detect sequence variants. Longer reads also increase mapping precision6 and may even enable de novo genome assembly7.
Inspired by ideas from communication theory, we analyzed the sequencing platform’s nonstationary noise factors, as these accumulate throughout the run and reduce accuracy in later sequencing cycles (Supplementary Data, Supplementary Figs. 1–5 and Supplementary Table 1 online). We found that three noise factors were dominant (Fig. 1 and Supplementary Figs. 1–5). The first one was phasing, which is a well-known source of noise in sequencers such as the Illumina Genome Analyzer that use cyclic reversible termination (CRT)8,9. Briefly, CRT involves repetitive cycles of three steps: (i) extension of a nascent strand with addition of a single extension-blocked, fluorophore-labeled nucleotide, (ii) imaging and (iii) removal of the block and fluorophore in preparation for the next synthesis cycle. Ideally, after any number of cycles, the lengths of all nascent strands within the DNA cluster would be the same and would generate a strong, coherent signal (Fig. 1a). Imperfections in the chemistry of CRT, however, cause stochastic failures in nucleotide incorporation or block removal, or incorporation of more than one nucleotide in a particular cycle. This results in heterogeneity in nascent strand lengths, introducing lagging (too short) and leading (too long) nascent strands within the cluster and reduces the purity of signal output from the interrogated position by contamination with signals from adjacent nucleotides. This is referred to as phasing noise (Fig. 1b). The variation in nascent-strand length increases with every cycle, and consequently the precision of base calling drops, which limits maximal useful read lengths. The second dominant noise factor is fading, an exponential decay in fluorescent signal intensity as a function of cycle number (Fig. 1c). This is likely attributable to material loss during sequencing. The third nonstationary noise factor is a cycle-dependent change in fluorophore cross-talk, which induces a substantial bias toward certain base calls in later cycles (Fig. 1d). The physical basis of this observation remains to be understood, but it might be attributable to accumulation of a specific fluorophore as background on the flow cell or to changes in laser intensities over time. Taking into account these three noise factors, we constructed and tested a parametric model that describes signal distortion as a function of cycle (Supplementary Data). Our model suggests that by accounting for both phasing and changes in the cross-talk matrix, we can enhance the signal-to-noise ratio and improve the quality of sequence reads.
Figure 1.

Schematic representation of main Illumina noise factors. (a–d)A DNA cluster comprises identical DNA templates (colored boxes) that are attached to the flow cell. Nascent strands (black boxes) and DNA polymerase (black ovals) are depicted. In the ideal situation, after several cycles the signal (green arrows) is strong, coherent and corresponds to the interrogated position (a). Phasing noise introduces lagging (blue arrows) and leading (red arrow) nascent strands, which transmit a mixture of signals (b). Fading is attributed to loss of material that reduces the signal intensity (c). Changes in the fluorophore cross-talk cause misinterpretation of the received signal (teal arrows; d). For simplicity, the noise factors are presented separately from each other.
Based on our analyses, we built a base caller, named Alta-Cyclic, which we designed to specifically address these noise factors (Fig. 2). Alta-Cyclic works in two stages: the training stage and the base-calling stage. During the training stage, Alta-Cyclic learns run-specific noise patterns according to our model and finds an optimized solution that reduces the effect of these noise sources. The optimization is mainly achieved by supervised learning using a rich DNA library with a known reference genome. Alta-Cyclic then enters the base-calling stage and reports all the sequences from the run with the optimized parameters. Alta-Cyclic uses the fluorescence intensity values that are generated by Firecrest, the Illumina image-processing software. The training process is transparent, although computationally intensive. The main requirement is that one sample on each flow cell have an available reference genome. We defined this sample as the training library. Although in most cases, we used the bacteriophage phi X genome, which is a common Illumina control, theoretically, any sample can be used provided that there is a corresponding reference genome. Initially, only the lane containing training library is base-called using the Illumina base-calling software. Both the original intensity data and the reference sequences mapped to the genome after standard base calling are supplied to train Alta-Cyclic. Once trained, Alta-Cyclic base-calls all flow-cell lanes, ultimately providing sequence files and quality scores in the same format as the standard base caller.
Figure 2.
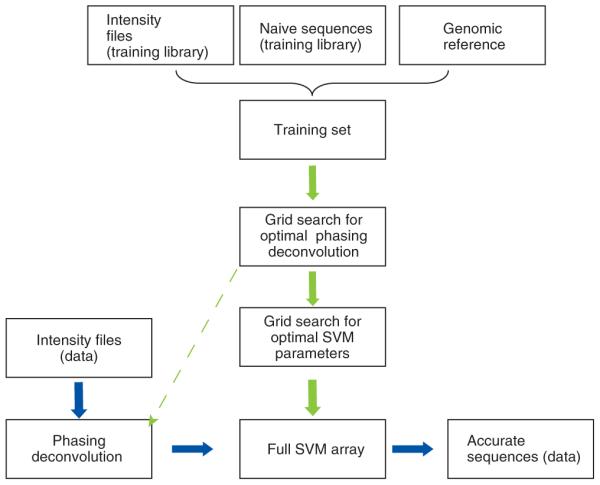
Alta-Cyclic base caller data flow. The training process (green arrows) starts with creation of the training set, beginning with sequences generated by the standard Illumina pipeline, by linking intensity reads and a corresponding genome sequence (the ‘correct’ sequence). Then, two grid searches are used to optimize the parameters to call the bases. After optimization, a final SVM array is created, each of which corresponds to a cycle. In the base-calling stage (blue arrows), the intensity files of the desired library undergo deconvolution to correct for phasing noise using the optimized values and are sent for classification with the SVM array. The output is processed, and sequences and quality scores are reported.
Alta-Cyclic treats DNA sequencing as a classification problem, in which the analog intensity signals that are generated by detection of the four fluorophores must be designated as adenine, cytosine, guanine or thymine (A, C, G or T). The training process starts by random sampling of reads from the training library, base called by the Illumina pipeline. The ‘correct answer’ for each of the reads is determined by alignment to the reference genome, allowing several mismatches. This process generates the training set: fluorescence intensities and their corresponding correct base calls.
Alta-Cyclic then uses an iterative two-dimensional grid search to find the best values that describe phasing noise (see Fig. 2, Supplementary Data and Supplementary Fig. 6 online). The first grid search finds near-optimal phasing-deconvolution parameters. In particular, Alta-Cyclic focuses on optimizing the solution for phasing in later cycles, as these data give better resolution and reflect the overall accumulated noise. Unfortunately, this is somewhat confounded by extensive changes in fluorophore cross-talk. Therefore, at each grid point, Alta-Cyclic first deconvolutes the phasing effect from the intensity values of the training set according to the grid coordinate. Then, it picks the intensity values of the last few cycles and their corresponding correct base calls, and uses a support vector machine (SVM) for each cycle to find the optimal margins that separate the fluorophores. Only half of the training set is used for SVM training and the other half is used for cross-validation. The average success rate in the cross-validation of the SVMs is used as a feedback to the grid search. Hence, in each grid-search iteration, Alta-Cyclic converges to more accurate phasing parameters that increase correct classification rates. After phasing optimization, Alta-Cyclic uses additional grid searches to optimize the SVM learning process by scanning a two-dimensional grid for the SVM cost parameter and gamma kernel parameter. Alta-Cyclic is then ready for final training of the full array of SVMs (one per sequencing cycle), which will be used for base calling of the full sequencing run.
During base calling, intensity files are deconvoluted using the optimized phasing parameters, and resulting intensity values for each sequencing cycle are sent for prediction to the SVM corresponding to that cycle. Alta-Cyclic converts the results to sequences and quality scores that are based on the reported probability of classification from the SVM (see Supplementary Methods online for software architecture).
Alta-Cyclic has several notable differences from the standard Illumina base caller that should improve read accuracy. First, all the calling parameters are optimized empirically and tested to enhance the accuracy of the base calling for each run, whereas in the Illumina base caller parameters are statically derived, though in a sophisticated manner, and are not evaluated. Second, Alta-Cyclic calculates phasing parameters based on a parametric model using data from the latest cycles, whereas the Illumina base caller relies on a numerical method and data from early cycles. Finally, Alta-Cyclic dynamically tracks changes in fluorophore cross-talk, which severely disrupts signals in later cycles, whereas the Illumina base caller statically determines cross-talk based on values from early cycles.
We tested Alta-Cyclic using both Genome Analyzer I (the ‘classic’) and II (GAI and GAII) output for long runs. On the GAII, we ran test samples for 78 cycles. The training set contained 100,000 randomly chosen reads from the phi X control, which we trimmed to 25 nucleotides and aligned back to the phi X genome, allowing 5 mismatches. Using the trained program, we base-called two human libraries representing small RNAs of <200 bases (Supplementary Methods). As these libraries were enriched for microRNAs, we clipped known adaptors and removed sequences smaller than 19 nt. Alta-Cyclic reported about 1,000,000 more reads that could be perfectly matched to the consensus human genome than did the Illumina base caller (Fig. 3a). As expected, the accuracy of longer reads improved most dramatically. The use of Alta-Cyclic increased the number of fully correct >30-base reads by ~3.5-fold and the number of fully correct reads of >60 bases by 18-fold. There was also an improvement in adaptor clipping with Alta-Cyclic. With a library of Tetrahymena thermophila micronuclear DNA, the average improvement for fully correct reads was only about twofold for 60-bp reads (Fig. 3b). This is likely due to the lack of a reference micronuclear genome. There is substantial rearrangement that occurs between the micronuclear DNA and the macronuclear genome10 used in its place.
Figure 3.

Comparison between Alta-Cyclic and Illumina base caller on the GAII platform. (a) Analysis of the HepG2 RNA library using Alta-Cyclic. The absolute number of additional fully correct reads (in addition to those generated by the Illumina base caller) is indicated by the red line; the fold change of the improvement is indicated by the blue bars. (b) A comparison of fully correct reads for the Tetrahymena micronuclear library by the Illumina base caller and Alta-Cyclic. (c) The average error rate in calls of the artificial SNP locations in the phi X library as a function of the cycle in which they were called. The dashed line represents 1% error rate (Q20). The plot on the right shows the last 18 cycles in a different scale. (d) A comparison of fully correct reads for the phi X library with 1% artificial SNPs. (e) Phi X sequences generated by Alta-Cyclic or Illumina were exhaustively aligned to the reference genome (allowing up to 53 mismatches out of 78). The distribution of alignment scores is shown beginning with an identical number of raw reads for input into each base caller.
A common hope for next-generation sequencers is that they will be able to identify sequence variants. For variant calls, sequence data would normally have a reference genome that could be used for training. One caution is that the presence of variants could cause over-fitting of the data or could degrade the accuracy of a learning-based method. To simulate a scenario in which there might be substantial variation between a training set and the actual sequencing target (such as in cross-species comparisons), we retrained Alta-Cyclic using a phi X library into which we had introduced 1% artificial single-base changes. After training, we base-called the phi X sample and matched the results back to the artificially mutated phi X genome. We examined the error rate for artificial single-nucleotide polymorphism (SNP) sites as a function of cycle number after filtering high-quality reads (see Supplementary Methods). Alta-Cyclic called SNPs with <1% error rate in the first 62 cycles, whereas for the Illumina base caller, this low error rate was restricted to the first 48 cycles. Furthermore, the mean error rate of SNP calling in the last 10 cycles by Alta-Cyclic was ~5%, but it was 40% for the Illumina base caller (Fig. 3c). When we compared the reads to the intact phi X genome, we found that Alta-Cyclic reported ~2,600,000 correct 78-nt reads, which represents 22% of all reads. In contrast, the Illumina base caller reported only ~600,000 correct 78-nt reads, which represents 5% of all reads (Fig. 3d).
In addition, we used this model to check whether Alta-Cyclic improves the data globally, even in very noisy reads. We aligned the total Alta-Cyclic and Illumina base-caller output to the phi X genome under highly permissive conditions, allowing up to 53 mismatches in a 78-base run, just slightly above random matching. The average number of mismatches in Alta-Cyclic–called sequences was 9, whereas it was 16 for the Illumina base caller. This corresponds to more than 100 million additional correct individual base calls with Alta-Cyclic as compared to that with the Illumina base caller. The distribution of the number of mismatches in the library emphasizes that Alta-Cyclic improves even the very noisy reads that would normally be discarded during quality filtering (Fig. 3e). Considering these results together, we conclude that Alta-Cyclic does not over-fit the data in the training process and that the approach enhances overall base calling. We observed similar improvements for base calls of runs on GAI machines (Supplementary Data and Supplementary Fig. 7 online).
The strategies that we developed both improve the accuracy of base calling and allow the production of longer reads. By extending the accurate read lengths to as many as 78 bases on the GAII, we substantially increased potential sequence output. Moreover, longer reads allow more accurate mapping and may allow de novo assembly of complex genomes. Undoubtedly, the hardware underlying next-generation platforms will continue to improve, but it is equally likely that the general strategies described here will continue to allow the limits of these platforms to be maximized. Alta-Cyclic is available for nonprofit use at http://hannonlab.cshl.edu/Alta-Cyclic/main.html.
Supplementary Material
ACKNOWLEDGMENTS
We thank M. Rooks, E. Hodges, K. Fejes-Toth and C. Malone for help in preparing libraries. We thank M. Regulski, D. Rebolini and L. Cardone for Illumina sequencing, and T. Heywood for assistance with cluster computing. F. Chen, D. Hillman and J. Eisen (Lawrence Berkeley National Lab) provided the Tetrahymena micronuclear library. Y.E. is a Goldberg-Lindsay Fellow of the Watson School of Biological Sciences. P.P.M. is a Crick-Clay Professor. G.J.H. is an investigator of the Howard Hughes Medical Institute. This work was supported by grants from the US National Institute of Health, the National Science Foundation and the Stanley Foundation.
Footnotes
Note: Supplementary information is available on the Nature Methods website.
References
- 1.Pennisi E. Science. 2007;318:1842–1843. doi: 10.1126/science.318.5858.1842. [DOI] [PubMed] [Google Scholar]
- 2.Chi KR. Nat. Methods. 2008;5:11–14. doi: 10.1038/nmeth1154. [DOI] [PubMed] [Google Scholar]
- 3.Korbel JO, et al. Science. 2007;318:420–426. doi: 10.1126/science.1149504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hillier LW, et al. Nat. Methods. 2008;5:183–188. doi: 10.1038/nmeth.1179. [DOI] [PubMed] [Google Scholar]
- 5.Cokus SJ, et al. Nature. 2008;452:215–219. doi: 10.1038/nature06745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whiteford N, et al. Nucleic Acids Res. 2005;33:e171. doi: 10.1093/nar/gni170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chaisson M, Pevzner P. Genome Res. 2008;18:324–330. doi: 10.1101/gr.7088808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Metzker M. Genome Res. 2005;15:1767–1776. doi: 10.1101/gr.3770505. [DOI] [PubMed] [Google Scholar]
- 9.Metzker M, Raghavachari R, Burgess K, Gibbs R. Biotechniques. 1998;25:814–817. doi: 10.2144/98255st01. [DOI] [PubMed] [Google Scholar]
- 10.Eisen JA, et al. PLoS Biol. 2006;4:e286. doi: 10.1371/journal.pbio.0040286. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.