

Abstract
Orthotopic liver transplantation was performed 15 months to 20 years ago in 126 recipients, all of whom were under 18 years of age. Eighty-six of these pediatric recipients were treated before 1980 with azathioprine (or cyclophosphamide) and prednisone, to which antilymphocyte globulin (ALG) usually was added. One-year patient survival was 40%. In the last 40 cases, the new drug cyclosporine has been given with low doses of steroids. The one-year patient survival increased to 65%. Both in the pre-cyclosporine era and more recently, the survival of patients with biliary atresia has been lower than in the next largest category of patients, namely, those with liver-based inborn metabolic errors. The difficulty of operation in patients with biliary atresia has been greater than in recipients with other diagnoses, partly because of previous operations such as portoenterostomy (Kasai procedure). Hepatic portoenterostomy, worthwhile as it is, has posed technical difficulties for eventual liver transplantation, particularly when complicated Roux limb techniques or venting procedures have been applied. In our total experience the longest survival after liver replacement in a child whose original diagnosis was biliary atresia is 13⅔ years.
Although hepatic portoenterostomy is the first operation to be performed on children with biliary atresia [1], the majority of such Kasai procedures fail, leaving hepatic transplantation as the only option. The use of liver transplantation in infants and children has been questioned because of the high morbidity and mortality rates. Even after “successful” transplantation, the quality of life has been too often degraded by the side effects of high-dose steroid therapy.
We report here the recent progress in liver transplantation in infants and children that has been made possible with the new immunosuppressive drug regimen of cyclosporine and low-dose steroids. The results have been compared to those obtained in the past with conventional immunosuppression. The special focus in this communication will be on the subgroup of pediatric recipients who had biliary atresia.
Materials and Methods
The first human liver transplantations were performed in 1963 [2]. Since then we have attempted more than 300 liver replacements, first at the University of Colorado and since 1981 at the University of Pittsburgh. The first 170 patients were treated before March, 1980, with the “conventional” immunosuppressive regimen of azathioprine (or cyclophosphamide), prednisone, and usually antilymphocyte globulin (ALG) [3]. Since March, 1980, liver allograft recipients were treated with a new immunosuppressive regimen of cyclosporine and prednisone [3, 4]. Steroids are a necessary addition to cyclosporine to achieve adequate immunosuppression, but the doses used are much less than those required when given with azathioprine.
Among the 170 patients given conventional immunosuppressive therapy, 86 were pediatric recipients who were age 18 or under. Of 100 consecutive liver recipients under cyclosporine-steroid therapy between March, 1980, and December, 1982, forty were pediatric recipients.
Indications for Liver Transplantation
The reasons for liver transplantation in pediatric patients are listed in Table 1. Patients with biliary atresia or hypoplasia accounted for 71 of the 126 total pediatric liver recipients. Almost all had failed hepatic portoenterostomy. The second largest recipient group was made up of several inborn errors of metabolism, including alpha-1-antitrypsin deficiency disease, Wilson's disease, tyrosinemia, glycogen storage disease, and the sea blue histiocyte syndrome.
Table 1.
Indications for transplantation in pediatric patients (⋜18 years).
| Precyclosporine era (before 1980) Number |
Cyclosporine era (after 1980) Number |
Total number | |
|---|---|---|---|
| Biliary atresia and hypoplasia | 51 | 20 | 71 |
| Inborn metabolic errors | 13a | 10d | 23 |
| Chronic aggressive hepatitis | 13 | 3 | 16 |
| Hepatoma | 3b | 0e | 3 |
| Neonatal hepatitis | 2 | 1 | 3 |
| Congenital hepatic fibrosis | 2 | 0 | 2 |
| Secondary biliary cirrhosis | 2c | 1f | 3 |
| Byler's disease | 0 | 3g | 3 |
| Budd-Chiari syndrome | 0 | 1 | 1 |
| Cellular inflammatory pseudotumor | 0 | 1 | 1 |
| Total | 86 | 40 | 126 |
Inborn errors: Alpha-1-antitrypsin deficiency (9); Wilson's disease (2); Tyrosinemia (1); Type IV glycogen storage disease (1).
Five other patients had incidental malignancies (4 hepatomas and 1 hepatoblastoma) in their excised liver. The principal diagnoses in these 5 cases were biliary atresia (3 examples), alpha-1-antitrypsin deficiency (1 example), and congenital tyrosinemia (1 example). The diagnosis of the neoplastic change was known in advance only in 2 of the 5 cases.
Secondary to trauma or choledochal cyst (one each).
Inborn errors: Alpha-l-antitrypsin deficiency (6); Wilson's disease (1); Tyrosinemia (1); Type I glycogen storage disease (1); Sea-blue histiocyte syndrome (1).
Two patients had incidental hepatomas: one with tyrosinemia, another with sea-blue histiocyte syndrome.
Secondary to choledochal cyst.
Diagnosis equivocal in one case.
Donor Operation
The liver, heart, and both kidneys can be procured from a single donor without compromising the anatomy and effective preservation of any of the organs [5]. In an infant or child whose liver is being removed, it is often advisable to keep the celiac axis in continuity with the upper abdominal and thoracic aorta. The donor thoracic aorta can be turned down 180° for anastomosis to the recipient's lower abdominal aorta. The use of the aortic anastomotic technique is particularly valuable when the liver graft receives part of its blood supply from the superior mesenteric artery; then both the celiac axis and the superior mesenteric artery can be vascularized via the single aortic anastomosis [6, 7].
Recipient Operation
The techniques of orthothopic liver transplantation have become well standardized [2, 3, 6, 7].
In infants and young children, a bilateral subcostal incision or upper abdominal transverse incision using the previous incision (when present) gives adequate exposure. Recipient hepatectomy in children is generally easier than in adults. On the other hand, the vascular anastomoses (particularly of the artery and portal vein) and biliary reconstruction are more difficult, or at least more sophisticated, in pediatric recipients than in adults.
We employ a continuous suture using monofilament polypropylene (Prolene®) for all vascular anastomoses. To avoid the purse-stringing that can easily occur with this technique, care is taken to prevent undue traction on the sutures. When the sutures are completed, the knots are placed away from the vessel wall in order to leave extra Prolene® which slowly works its way back into the suture line. This “growth factor” technique [8] is an important means of eliminating strictures in smallcalibre anastomoses.
The ideal form of biliary duct reconstruction is a choledochocholedochostomy with a T-tube stent, but in patients with biliary atresia, that option does not exist. Cholecystoenterostomy, which depends on bile drainage through the cystic duct, is not used because of the high incidence of subsequent obstruction of the cystic duct [3, 9]. End-to-side anastomosis of graft common duct to a Roux-en-Y loop of jejunum with an internal stent (choledochojejunostomy) has been the reconstruction of choice in cases of biliary atresia and for other disorders in which the recipient common duct is inadequate; it has proven to be one of the most satisfactory of all the methods of biliary reconstruction [9].
In patients with biliary atresia, the Roux limb construction for hepatic portoenterostomy can be reused at the time of liver transplantation providing the limb is not irreparably damaged during the hilar dissection of recipient hepatectomy. The risk of Roux limb injury has been great when skin jejunostomy has been used with the Kasai procedure in hopes of decreasing cholangitis. Even if the ostomy had been closed by the time of transplantation, this portion of the Roux limb frequently has to be resected, and often with great difficulty.
Results
Causes of Death
Fifty-three of the 86 pediatric liver recipients between 1963 and early 1980 and 14 of the 40 between March, 1980, and December, 1982, died within a year after transplantation. Although the causes of death in these patients were usually multiple, a single most important factor was selected in each case (Table 2).
Table 2.
Main causes of death within a year after liver transplantation among 86 children in precyclosporine era and 40 children in cyclosporine era.
| Precyclosporine era (86 children) |
Cyclosporine era (40 children) |
|
|---|---|---|
| Intra- and Perioperative death | 3 | 1 |
| Surgical technical complication | 15 | 7 |
| Unsatisfactory liver graft | 3 | 1 |
| Graft rejection | 9 | 4 |
| Infection | 20 | 0 |
| Recurrent malignancy | 1 | 0 |
| Other | 2a | 1b |
| Total | 53 | 14 |
One died from pulmonary emboli at 52 days and another from respiratory insufficiency due to an oversized graft.
One died from graft hypoxia due to marked pulmonary arteriovenous shunt.
During the precyclosporine era, bacterial, fungal, and viral infections were the main cause of death within a year after transplantation (20 of 53 deaths). There were 9 deaths from abdominal sepsis, 5 from viral infection (adenovirus, chickenpox, and herpes), 4 from systemic bacterial and fungal sepsis, and 2 from pulmonary infection. Although infectious complications continue to be a major threat to life after transplantation, none of the 14 deaths under cyclosporine therapy was thought to have been primarily from infection.
Significant numbers of deaths were caused by surgical technical complications of arterial, venous, or biliary anastomoses. In the precyclosporine era, there were 15 deaths in this category. Eight of the 15 were failures in biliary duct reconstruction, 4 in hepatic artery anastomosis, 3 in portal vein anastomosis, and 1 in the suprahepatic inferior vena caval anastomosis. In the cyclosporine era, surgical technical complications were the most frequent direct cause of death (7 of 14). There were 3 failures in hepatic artery anastomosis, and one each of the hepatic artery anastomosis, the portal venous anastomosis, the suprahepatic vena caval anastomosis, and biliary duct reconstruction.
Acute or chronic rejection was the third main cause of death in the precyclosporine era, and the second most common cause in the cyclosporine era (Table 2). Despite better immunosuppression and improved survival with cyclosporine, approximately 10% of liver graft recipients died from rejection in both the precyclosporine era and the cyclosporine era.
Survival
The 2-year actual survival of 86 pediatric recipients in the precyclosporine era and actuarial survival of 40 pediatric recipients in the cyclosporine era are shown in Fig. 1. The shortest follow-up period in the cyclosporine-treated group is 12 months, and the longest is more than 3 years. After the introduction of cyclosporine, the 1-year survival after liver transplantation improved from 40% to 65%.
Fig. 1.
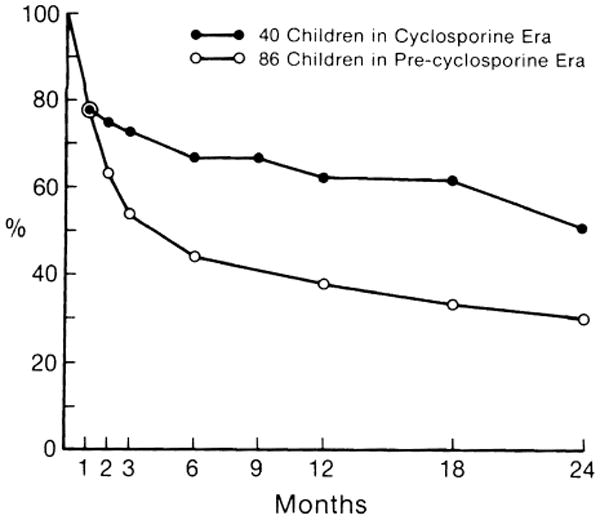
A comparison of survival after liver transplantation between 86 pediatric liver recipients under conventional immunosuppressive therapy (see text for description) and 40 pediatric recipients under cyclosporineprednisone therapy.
The influence of original liver disease on survival was analyzed for the 2 most common indications for transplantation (biliary atresia and inborn metabolic errors) in the precyclosporine and cyclosporine eras (Figs. 2, 3). The survival of children with biliary atresia was substantially lower than overall survival in the precyclosporine era (Fig. 2), but it was only slightly worse than the overall survival in the cyclosporine era (Fig. 3). The survival of children with biliary atresia was inferior to that of children with inborn metabolic errors during both the early and later phases of our experience.
Fig. 2.
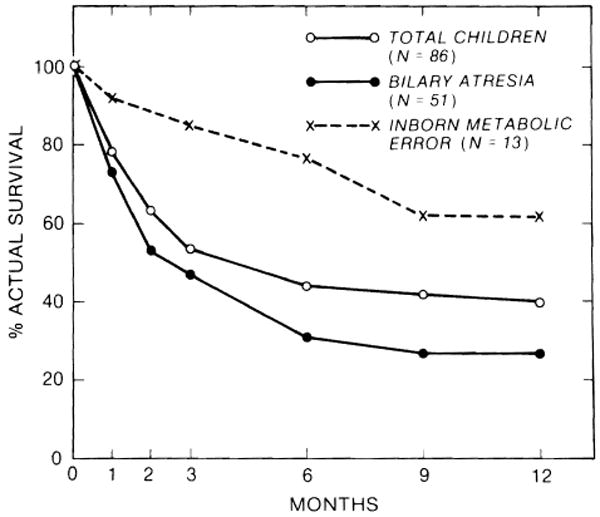
Influence of original liver disease on 1-year survival under conventional immunosuppression.
Fig. 3.
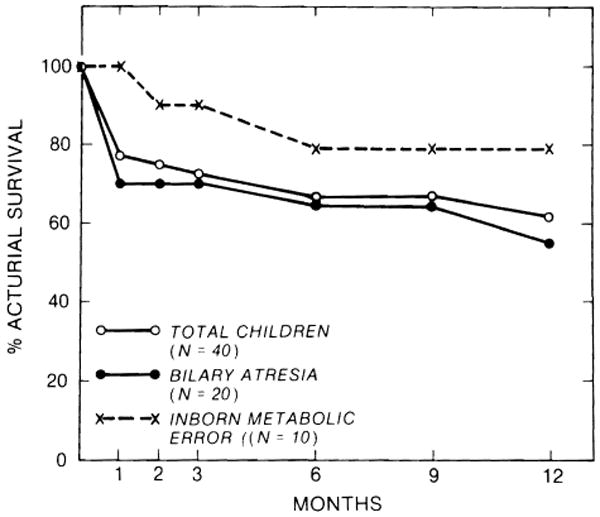
Influence of original liver disease on 1-year survival under cyclosporine-steroid therapy.
The influence of age at the time of liver transplantation on survival was studied. In the precyclosporine era, 28 (53%) of 53 infants and preschool children (less than 6 years), 8 (50%) of 16 school age children (between 6 and 12 years), and 13 (76%) of 17 adolescents lived more than 3 months (Fig. 4). In the cyclosporine era, 16 (76%) of 21 infants and preschool children, 8 (67%) of 12 school age children, and 6 (86%) of 7 adolescents lived more than 3 months (Fig. 4). Thus, the 3-month survival of infants and preschool children was similar to that of school age children both in the precyclosporine era and in the cyclosporine era. It is noteworthy that 5 (71%) of 7 infants less than a year old lived more than 3 months. The technical difficulties of liver transplantation in infants have been fairly well overcome. The age of children should not be a consideration in accepting them as candidates for liver transplantation, although liver donors for infants are scarce.
Fig. 4.
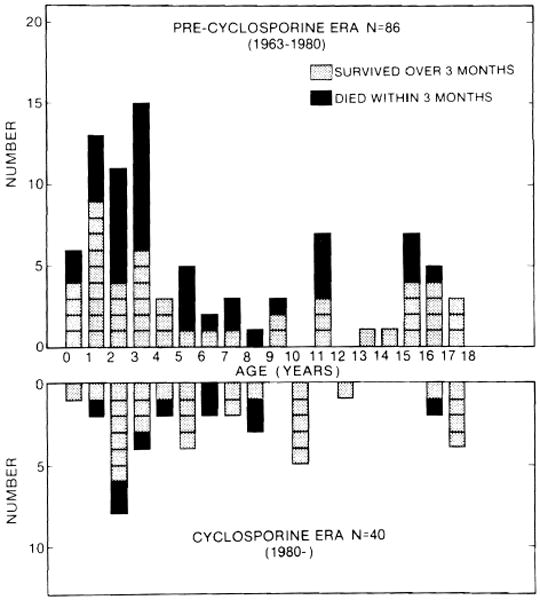
Age distribution of 126 pediatric liver recipients. A shaded square represents a child who survived more than 3 months, and a black square represents a child who died within 3 months. All of the surviving patients treated with cyclosporine have follow-ups of at least 15 months.
Discussion
Despite the continued efforts over the last 2 decades to improve the survival and the quality of life after liver transplantation, real progress had to await the discovery [10] and clinical use [4, 11, 12] of cyclosporine. In combination with low-dose steroids, cyclosporine provides more effective and safer immunosuppression than conventional double or triple drug therapy [4, 12]. With this new immunosuppressive agent, the results after transplantation of kidneys, liver, and heart have improved.
In pediatric liver transplantation, the 1-year survival improved from 40% to 65% with cyclosporine and low-dose steroid therapy. Infectious complications, which had been the most common direct cause of death after liver transplantation, became less frequent and more treatable. The absence of significant morbidity has been remarkable [12]. The complications of high-dose steroids, such as growth retardation, Cushingoid features, and bone disease that plagued efforts at pediatric transplantation, have been virtually eliminated.
Survival after liver transplantation of children with biliary atresia has been inferior to that of children with liver-based inborn metabolic errors in both the precyclosporine era and the cyclosporine era. This is partly due to the fact that previous major procedures such as hepatic portoenterostomy make the transplant operation more difficult. In addition, anatomical anomalies, such as hypoplastic, sclerotic, and sometimes thrombosed portal veins are more common in children with advanced biliary atresia [3, 6]. However, in the cyclosporine era the survival rate of children who undergo liver transplantation for biliary atresia has increased almost to the level of overall survival in the pediatric cases as a whole.
The contention that infants and small children are not good candidates for liver transplantation because of age and small size has been proven incorrect. The opinion that the quality of life will be substandard has been held only by those who have not seen these infants and children at play.
Acknowledgments
Supported by research grants from the Veterans Administration; and the National Institutes of Health Bethesda, grant AM-29961.
References
- 1.Kasai M, Suzuki H, Ohashi E, Ohi R, Chiba T, Okamoto A. Technique and results of operative management of biliary atresia. World J Surg. 1978;2:571. doi: 10.1007/BF01556048. [DOI] [PubMed] [Google Scholar]
- 2.Starzl TE, Marchioro TL, von Kaulla KN, Hermann G, Brittain RS, Waddell WR. Homotransplantation of the liver in humans. Surg Gynecol Obstet. 1963;117:659. [PMC free article] [PubMed] [Google Scholar]
- 3.Starzl TE, Iwatsuki S, Van Thiel DH, Gartner JC, Zitelli BJ, Malatack JJ, Schade RR, Shaw BW, Jr, Hakala TR, Rosenthal JT, Porter KA. Evolution of liver transplantation. Hepatology. 1982;2:614. doi: 10.1002/hep.1840020516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Starzl TE, Klintmalm GBG, Porter KA, Iwatsuki S, Schroter GP. Liver transplantation with use of cyclosporin A and prednisone. N Engl J Med. 1981;305:266. doi: 10.1056/NEJM198107303050507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shaw BW, Jr, Hakala T, Rosenthal JT, Iwatsuki S, Broznick B, Starzl TE. Combination donor hepatectomy and nephrectomy and early functional results of allografts. Surg Gynecol Obstet. 1982;155:321. [PMC free article] [PubMed] [Google Scholar]
- 6.Starzl TE, Putnam CW. Experience in Hepatic Transplantation. Philadelphia: W.B. Saunders; 1969. [Google Scholar]
- 7.Shaw BW, Jr, Iwatsuki S, Starzl TE. Alternative methods of hepatic graft arterialization. Surg Gynecol Obstet. in press. [PMC free article] [PubMed] [Google Scholar]
- 8.Starzl TE, Iwatsuki S, Shaw BW., Jr A “growth factor” in fine vascular anastomoses. Surg Gynecol Obstet. in press. [PMC free article] [PubMed] [Google Scholar]
- 9.Iwatsuki S, Shaw BW, Jr, Starzl TE. Biliary tract complications in liver transplantation under cyclosporine-steroid therapy. Transplant Proc. 1983;15:1288. [PMC free article] [PubMed] [Google Scholar]
- 10.Borel JF, Feurer C, Gubler HU, Stahelin H. Biological effects of cyclosporin A: A new antilymphocytic agent. Agents Actions. 1976;6:468. doi: 10.1007/BF01973261. [DOI] [PubMed] [Google Scholar]
- 11.Calne RY, Rolles K, White DJG, Thiru S, Evans DB, McMaster P, Dunn DC, Craddock GN, Henderson RG, Aziz S, Lewis P. Cyclosporin A initially as the only immunosuppressant in 34 recipients of cadaveric organs: 32 kidneys, 2 pancreases, and 2 livers. Lancet. 1979;2:1033. doi: 10.1016/s0140-6736(79)92440-1. [DOI] [PubMed] [Google Scholar]
- 12.Starzl TE, Iwatsuki S, Malatack JJ, Zitelli BJ, Gartner JC, Hakala TR, Rosenthal JT, Shaw BW., Jr Liver and kidney transplantation in children under cyclosporine A and steroids. J Pediatr. 1982;100:681. doi: 10.1016/s0022-3476(82)80564-7. [DOI] [PMC free article] [PubMed] [Google Scholar]