

Abstract
Basement membranes are sheet-like cell-adherent extracellular matrices that serve as cell substrata and solid-phase agonists, contributing to tissue organization, stability and differentiation. These matrices are assembled as polymers of laminins and type IV collagens that are tethered to nidogens and proteoglycans. They bind to cell surface molecules that include signal-transducing receptors such as the integrins and dystroglycan and form attachments to adjacent connective tissues. The cell receptors, in turn, provide links between the matrix and underlying cytoskeleton. Genetic diseases of basement membrane and associated components, collectively the basement membrane zone, disrupt the extracellular matrix and/or its linkages to affect nerve, muscle, skin, kidney and other tissues. These diseases can arise due to a loss of matrix integrity, adhesion strength and/or receptor-mediated signaling. An understanding of the mechanisms of basement membrane zone assembly and resulting structure can provide insights into the development of normal tissues and the pathogenic mechanisms that underlie diverse disorders.
Keywords: Extracellular matrix, laminin, collagen, nidogen, perlecan, integrin, dystroglycan, agrin, muscular dystrophy, peripheral neuropathy, Alport’s syndrome, Goodpasture syndrome, epidermolysis bullosa, Pierson syndrome
INTRODUCTION
Basement membranes (basal laminae) are sheet-like cell-adherent extracellular matrices (ECMs) that contribute to tissue organization and differentiation in vertebrate and invertebrate organisms. In mature tissues, they underlie epithelia and vascular endothelia; form the limiting boundaries of many organs, including the nervous system; surround individual cells or functional units in tissues such as muscle, kidney, pancreas, adipose tissue, and peripheral nerve; and mediate specialized attachments between tissues, including neuromuscular synapses and myotendinous junctions. In most if not all cases, basement membranes serve as both supportive cell substrata and solid-phase agonists.
The process of basement membrane formation is largely one of self-assembly on cell surfaces. The resulting matrix architecture is one of enmeshed polymers of laminins and type IV collagens that are bound to nidogens -1 and -2 and to the heparan sulfate proteoglycans agrin and perlecan. Basement membranes appear to be attached primarily through the laminins to cell surface sulfated glycolipids and transmembrane receptors. Signal transduction is mediated by matrix-ligated integrins, dystroglycan, and receptor kinases modulated by the mechanical properties of the matrix. The matrix components, receptors, and cytoskeletal binding interactions are illustrated in Figs. (1–3) and their binding interactions summarized in Tables 1–2.
Fig. (1). Basement Membrane Components and Interactions.

Examples are shown for major laminins. Each possesses three subunits and possesses globular, rod-like and coiled-coil domains (these are indicated for laminin-111). Various proteolytic cleavages (jagged lines) generate additional diversity. Each type IV collagen is a heterotrimer with three different chain compositions (most common is α12α2[IV]). In addition are found two nidogens (Nd1, Nd2), and the heparan sulfate proteoglycans agrin (Ag) and perlecan (perl). Major binding and minor (heavy and thin dashed arrows) binding interactions are indicated among the structural components (black lines) and with cell surface components (green lines to integrins, α-dystroglycan (αDG), hemidesmosomal (HD) BP180, sulfated glycolipids (SGL) and the Lutheran (Lu) antigen. In squamous epithelia, laminin 3A32 is processed by several proteases. It binds to the α6β4 integrin and BP180 and binds to type VII collagen.
Fig. (3). Cytoskeletal Linkages.
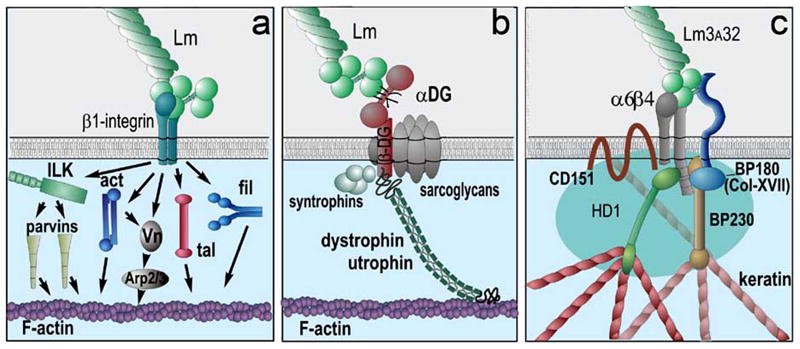
Basement membrane ligands can establish links to the actin and keratin cytoskeletons by binding to integrins and dystroglycan. (a) β1-integrins: Nearly all basement membrane components bind to β1-integrins. These integrins bind to cytoskeletal intermediates that bind to F-actin. The intermediates shown are integrin linked kinase (ILK) and α- and β-parvin, α-actinin (act), talin (tal) vinculin (Vn) and Arp2/3, and filamin (fil) (drawing after [45]). (b) Dystroglycan: The LG domains of laminins, agrin and perlecan bind to α-dystroglycan (αDG). αDG binds to β-dystroglycan, a transmembrane protein that binds to F-actin through dystrophin and utrophin. In muscle, dystroglycan is part of a complex that includes the sarcoglycans and other proteins. Homologues of dystrophin and utrophin exist in other tissues. (c) α6β4-integrin: Laminin 332 binds to this unique integrin that forms part of hemidesmosome complexes and that forms a linkage with keratin filaments [14].
Table 1.
Basement Membrane Anchoring and Receptor Interactions
| Receptor/Anchor | Ligands | Nature of Interaction | References |
|---|---|---|---|
| Integrin α1β1 | Col-I, Col-IV, α1-laminins (α1LN) and α2-laminins (α2LN). | [173, 174] | |
| Integrin α2β1 | Col-I, Col-IV, α2-laminin LN domains, Perlecan LG domains (endorepellin) | [156, 173, 175] | |
| Integrin α3β1 | α3- and α5-laminins, nidogens. | KD: α5-laminin (3.4 nM*) and α3-Lm (14 nM*); Nidogen-2 > Nidogen-1. | [28, 138] |
| Integrin α6β1 | α1-, α3-, α5-laminins. The integrin interacts with LG domains 1–3 & the distal coiled-coil. | KD: α5-Lms (0.73 nM*) > α3-Lms (7.5 nM*) ≈ α1-Lms (9.5 nM*) ≫ α4-Lms; α2- and α4-laminin interaction detected by cell adhesion only. | [28, 176] |
| Integrin α6β4 | α3- and α5-laminins. Mediates attachment to hemidesmosomes. | α3-Lms (12 nM*) > α5-Lms (25 nM*) (LG1–3 domains). | [28] |
| Integrin α7X1β1 | α2-laminins (0.6 nM) > α5-Lms (1.2 nM*) (LG1–3/distal coiled-coil) | Integrin expressed in muscle, peripheral nerve and other tissues. | [28] |
| Integrin α7X2β1 | α1- and α2-laminins. | KD: α1-Lms ( 1 nM*) > α2-Lms (2.6 nM*) | [28] |
| Integrin αvβ3/β1 | α5-Lms (domain L4b) | Possible role in vasculature | [177] |
| α-dystroglycan (αDG) | α1-, α2-, α5-laminins, agrin, perlecan. αDG binding is dependent upon O-linked carbohydrate. Reduced/absent DG binding adversely affects only some (e.g. muscle and brain) tissues. | KD (muscle agrin) = 2 nM; perlecan-LG= 16 nM; α1-Lm ≈ 20 nM; α2-Lm = 43 nM; α5-Lm = 150 nM; α4-Lm4 ≥ 1 μM (activity resides in LG domains). | [152, 178–184] |
| heparin/heparan sulfates of various cellular proteoglycans | Laminins (LG and αLN domains), collagen-IV, agrin (LG), perlecan (LG) | Heparan sulfate chains tether growth factors (e.g. FGF-2, TGFβ family members). | [30, 31, 141, 185, 186] |
| galactosyl sulfatide (and related sulfated glycolipids) | Laminin-, agrin-, perlecan-LG domains; laminin-LN domains. These glycolipids, expressed in Schwann cell, kidney and other tissues, are thought to mediate and/or enhance anchorage of laminins. The LN interactions may enable the short arms to bind the cell surface in addition to the LG domains. |
|
[17, 27, 30, 31, 181, 182, 184, 187–192]; Capizzi & Yurchenco, unpublished data (agrin and charge-density effects) |
| galectins | Laminin-111 and other ECM molecules | May contribute to cell adhesion and signaling. | [193] |
| receptor tyrosine kinase discoidin-domain receptor-1 (DDR1) | Collagens (including collagen-IV) | Absence of receptor associated with GBM thickening and hearing loss. | [194, 195, 196] |
| CD44 (chondroitin sulfate PG) | Collagen-IV | May contribute to cell adhesion. | [197] |
| Extracellular β1,4-gal-transferase | Laminin-111 and other ligands | May contribute to cell adhesion and signaling. | [198] |
detected in solid-phase binding assay with soluble clasped integrins in 1 mM Mn++.
Table 2.
Basement Membrane Scaffold Inter-Component Binding Interactions
| Bond | Ligand Domain(s) | Nature of Interaction(s) | References |
|---|---|---|---|
| laminin - laminin | LN domains of the α, β and γ short arms interact with corresponding LN domains. | Polymerization (crit. conc. ≈ 0.1 μM) requiring three short arm LN domains. Individual LN+LEa | [10, 16–19] |
| nidogen - laminin | Nidogen-G3 to Lmγ1(LEb3), γ2, γ3 |
|
[134, 138] |
| agrin - laminin | Agrin NtA to Lm-γ1 coiled-coil | Lm-111: KD ≈ 2 nM | [79, 151] |
| laminins - heparin/heparan sulfate |
|
|
[17, 179, 199, 200] |
| nidogen - collagen IV | Nd (G3>G2 domains) to collagenous chain |
|
[134, 138] |
| nidogen - perlecan | Nd1-G2 and Nd2 to perlecan-domain IV | KD ≈ 10 nM | [142, 191] |
| perlecan - fibronectin | Perlecan domain IV | KD ≈ 10 nM | [142] |
| perlecan - laminin | Perlecan domain-I (likely heparan sulfate chains) to laminin LG and LN domains | Likely relatively low affinity and specificity. | [142] |
| perlecan - collagen IV | Perlecan domain-I (likely heparan sulfate chains) to collagen-IV | Likely relatively low affinity and specificity. | [142] |
| perlecan - heparin | Perlecan domain IV | see above | [142] |
| nidogen - fibulin 2 | KD ≤ 0.5 nM | [138] | |
| collagen IV - collagen IV |
|
|
[6, 107, 109, 112, 120, 201] |
A wide array of human disorders result from, or are associated with, defects in basement membrane assembly or composition. Some are acquired through autoimmune and metabolic means, including Goodpasture syndrome of kidney and lung, and primary complications of chronic diabetes mellitus, which affects the microvasculature of kidney, nerve and retina. Others arise from mutations of the genes coding for laminins, collagens or proteoglycans. These include MDC1A congenital muscular dystrophy, a disorder of muscle, nerve and brain; junctional epidermolysis bullosa; a severe blistering disease of skin and mucous membranes; Pierson syndrome, a disease of kidney and eye; Alport’s syndrome, a defect of renal function and hearing; and Schwartz-Jampel syndrome, a myotonia and chondrodysplasia. The pathogenesis of these disorders depends upon loss of function and structure through such mechanisms as destabilization of matrix integrity, loss of cell anchorage, loss of critical ligands, and/or loss of receptor interactions.
BASEMENT MEMBRANE ARCHITECTURE
Basement membranes are found between cell surfaces and interstitium or between cells. By light microscopy they are seen in sections as periodic acid Schiff- and silver-staining linear entities that immunostain with antibodies specific for the four primary basement membrane components (laminin, collagen IV, nidogen, and perlecan). These elements topographically coincide with α- and β-dystroglycan, various integrins, and their receptor-associated partners within and immediately beneath the plasma membranes. Type XV and type XVIII collagens are concentrated at or near the interstitial face of the basement membrane. Fibronectin, a widely-distributed ECM glycoprotein, while detected in some (e.g. early embryonic) basement membranes, is usually not considered a basement membrane component per se.
At the ultrastructural level, single basement membranes seen in metal-impregnated thin sections typically range from ~50 to ~100 nm in thickness. They usually possess a distinctive electron-lucent layer (lamina lucida) sandwiched between the plasma membrane and an outer electron-dense layer (lamina densa). However, basement membranes appear homogenous when tissues are fixed by freeze-substitution, suggesting that the lamina lucida represents a retraction of the ECM from the cell surface during chemical fixation [1–3].
Transmission electron microscopy of thin (≤ 1 nm) high angle (≥ 45°) platinum/carbon metal replicas has allowed investigators to visualize the supramolecular architecture of basement membranes. Using this approach, distinct type IV collagen and laminin polymeric networks were identified in the basement membrane of the placental amnion and the EHS tumor whose organization was similar to that of the corresponding polymers assembled in vitro [4–8]. The laminin polymer had the appearance of network of short (~35 nm) interconnecting struts whereas the collagen polymer had the appearance of branching thin fibrils, many running in parallel for short distances as coils containing two or more intertwined collagenous filaments.
THE LAMININS
This family of glycoproteins exist as α-β-γ heterotrimers. Isoforms in vertebrates are assembled from a palette of five α-, three β-, and three γ-subunits, whose expression varies between cell types and with development. New nomenclature designates laminins according to their chain composition (e.g. laminin-111 corresponds to α1β1γ1) [9]. Laminins have a branched structure. The N-terminal portions of the subunits are separate, forming short arms. Most short arms begin with an N-terminal globular LN domain, followed by a rod-like region consisting of multiple laminin-type EGF repeats (LE domains) in combination with two additional globular domains (L4, LF). However, the α3A, α4, β3 and γ2 subunits have truncated short arms, and (with the exception of β3) lack an LN domain. The more distal sequences of the three subunits participate in an extended coiled-coil domain that joins the subunits and forms a long arm. The α-subunit projects beyond the end of the coiled-coil in a series of five β-sandwich LG domains with a hinge-like region between LG3 and LG4. While there is little evidence of alternative splicing of laminin subunits, proteolytic processing mediated by furin-like and other proteases is seen in α2, α3, α4, α5, and γ2 subunits, with dissociation of the cleaved segments occurring in msot cases.
Distinct Role in Basement Membrane Formation
Studies in culture and in vivo suggest laminins are principally responsible for organizing basement membrane assembly on cell surfaces. Alone among basement membrane components, purified laminins are uniquely able to assemble sheet-like ECMs on cell surfaces in the absence of other components [10]. All basement membranes contain laminin as a principal component, and genetic mutations taht eliminate laminin expression in specific tissues prevent basement membranes from forming in those tissues specifically. As one example, early embryos assemble two prominent basement membranes, the sub-visceral endodermal basement membrane at the embryonic plate, and Reichert’s membrane, each of which contains two laminin isoforms (laminins-111 and -511). Knockout of either the γ1 or β1 subunits, which precludes expression of both laminins, resulted in peri-implantation embryos that lacked any basement membrane, leading to developmental arrest and involution [11, 12]. In contrast, knockout of type IV collagen subunits [13], nidogens, perlecan, and agrin, or of compensated laminin subunits was not found to substantially affect the appearance of basement membranes until much later in development (reviewed in [14]). Thus, it appears laminins are critical to the general scaffolding of basement membranes, while type IV collagen, the combined nidogens, and perlecan enhance basement membrane stability, at least during development [13, 15].
Polymerization and Scaffold Construction
Most laminins self-assemble to form a polymer, a two-step interaction consisting of a calcium-independent nucleation step followed by a calcium-dependent propagation step first identified with laminin-111 [16]. Laminin polymerization is reversible, entropy-driven, and cooperative with a critical conc. of ~0.14 and ~0.28 μM for laminin-111 and laminin-211, respectively [8, 16–20]. The laminin polymer that forms is a gel whose stiffness depends upon the laminin concentration or the presence of other modifying components [21]. The laminin molecules bind to each other through the LN domain located at the end of the three short arms such that one finds at each interacting locus one α, one β and one γ LN domain [10, 18]. Combined with the cooperative activation and polygonal structure of the resulting network [8], the observations indicate that polymerization is driven by the formation of a particularly stable tripartite complex of interlocked LN domains in which the α-, β-, and γ- short arms from neighboring laminin molecules are obligate partners. Laminin and laminins containing the α3B or α4 subunits that lack the α-subunit short arm (and hence the αLN domain), or that contain the truncated γ2 subunit, are thought not to polymerize [8, 10, 18, 19]. The rod-like LE domains that constitute much of the mass of the short arms likely provide spacing between LN-domain complexes, facilitating the formation of sheet-like polygonal networks on cell surfaces. However, the α-LN-LEa short arm segments have also been shown to bind to each other [22]. Reinterpreted in the face of evidence that each laminin must possess three different LN domains to polymerize, this additional short-arm interaction might represent part of a mechanism to explain how polymer layers may in some cases attach to each other to become multilayered.
Netrins comprise a family of extracellular proteins that include axonal guidance factors. They possess N-terminal globular domains that are homologous to the laminin LN domains (reviewed in [23]). While it has been suggested that netrins may interact with basement membranes through their LN domains, this has been demonstrated only for netrin-4 [24].
Anchorage, Cytoskeletal Linkage, and Signaling
Whereas laminins and other basement membrane components are secreted as soluble forms, a cardinal characteristic of basement membranes that they form in association with cell surfaces. A prediction arising from cell surface laminin adhesion (that increases the local laminin concentration) and cooperative polymerization (that prohibits assembly in dilute environments) is that the cell surface provides a nucleation surface for laminin, favoring assembly on that surface. At concentrations below the (solution) critical concentration, laminin would form a molecular monolayer.
Basement membranes are often found on only one of several adjacent cell surfaces. Thus, basement membranes form along the basal side of epithelial cells, but not adjacent fibroblasts; basement membranes selectively form on the abaxonal surfaces of Schwann cells, without incorporating components from the adjacent basement membranes forming in the perineurial sheath; each skeletal muscle fiber forms a single basement membrane over its entire surface that seamlessly incorporates unique isoforms of the principal components at junctional sites with nerve and tendon cells. The specificity with which inherently soluble components form basement membranes on a subset of the adjacent cell surfaces suggests that certain integral cell surface molecules serve as critical receptors in determining which surfaces are competent to form basement membranes.
The identity of the cell surface anchoring molecules that serve to initiate laminin-binding and basement membrane formation has been controversial. Plasma membranes contain multiple binding partners for laminins and other basement membrane components. Moreover, the surface expression and distribution of these receptors is often heavily influenced by basement membrane contact, and the avidity of receptors for their extracellular ligands is regulated by intracellular agents and interactions with the cytoskeleton. Thus, an interplay between receptor expression and activation, accumulation of initial basement membrane components, and co-organization of the subjacent cytoskeleton may cooperatively control sites of basement membrane assembly.
Primary candidates for receptors contributing to basement membrane assembly are those that support the initial accumulation of laminin, the essential component. More specifically, the initial process of laminin deposition appears to substantially depend upon interactions with the LG domains located at the α chain c-terminus. For example, disruption of LG domain interactions through deletion or competition results in a failure of basement membrane assembly in several culture models [10, 25–27]. The LG domains contain binding sites for integrins, α-dystroglycan, sulfated glycolipids, and heparan sulfate chains. Integrins including α3β1, α6β1, α6β4 and α7β1 bind principally to epitopes located along the LG1–3 domains of the different laminins; binding also requires participation of the adjacent coiled-coil domains, with a critical contribution from a distal glutamic acid in the γ1 subunit [28, 29]. Binding to the glycoconjugate-type receptors, including heparan sulfates, galactosylsulfatide (a glycolipid), and α-dystroglycan (for which a dense, mucin-like glycosylation is required to bind laminin), is focused at sites on the LG4 and LG5 domains that bear a high density of surface-exposed lysines and arginines, as revealed by crystallography of the laminin α1 LG4 and laminin α2 LG4–5 domains [30, 31]. Further dissection of unique binding site requirements may lead to mutational studies that define whether specific receptors determine basement membrane assembly, or if several overlap by simply providing anchorage sites. For example, a promising approach to dissecting primary and secondary laminin/receptor interactions in specific tissues involves the transgenic expression of truncated and chimeric laminin α-chain variants in mutant strains of mice lacking the normally expressed laminin α-chain [32].
To date, genetic perturbation studies have failed to support a specific receptor as the sole anchor for basement membrane assembly in animal tissues. Basement membranes form independent of β1-, β4- and other of the main basement membrane integrins, α-dystroglycan in most tissues (Reichert’s membrane may be an exception), galactosyl-3-sulfate ceramide (the most common sulfatide), and heparin/heparan sulfates. In contrast, studies of cultured cells have implicated the importance of individual anchor candidates. In one study, cultured epithelial cells lost their ability to accumulate surface-bound laminin when rendered null for dystroglycan, and regained this ability when dystroglycan expression was restored by transfection [33]. In another study, β1 integrin-null and dystroglycan-null embryoid bodies were found to be capable of assembling basement membranes, provided a laminin was present [26]. In a third study, selective removal of sulfatides, but not removal of dystroglycan, β1-integrins or heparan sulfates, prevented basement membrane assembly in Schwann cells, with the complementary observation that addition of galactosyl sulfatide to the plasma membranes of fibroblasts enabled basement membrane assembly [27]. It may be that redundancy of, and/or compensation by, anchorage molecules obscures their separate contributions in the genetic studies and that different cells utilize different anchors or combinations of anchors. If so, then anchorage may depend on integrated contributions arising from sulfated glycolipids, dystroglycan and integrins with one serving as a principal (but replaceable) anchor for a particular cell by virtue of its preponderance and affinity. Such a hypothesis also suggests a basis for the cell surface binding specificity by different laminins, based upon their different affinities for dystroglycan, sulfatides, integrins, and (in the case of α5 laminins) the Lutheran (Lu) antigen (see Table 1). This hypothesis may also partially account for differential compensation by alternative isoforms of laminin. For example, α2-laminins are the principal laminins in muscle and nerve. Mutations that decrease expression of α2-laminins cause severe muscular dystrophy and defects in nerve myelination, despite a substantial reactive increase in the expression and accumulation of laminins containing the alternative α4 chain. Clearly α4-laminins do not functionally compensate for α2-laminins in muscle or nerve. Interestingly, while α2-laminins show the highest affinities among laminins for sulfatide and dystroglycan, the laminin α4 subunit is particularly weak in its ability to bind to α-dystroglycan or almost any integrin. Further, while laminin α4 exhibits moderate sulfatide-binding, it lacks an aminoterminal LN domain to sustain laminin-laminin interactions or provide additional receptor anchorage. An ability to link the upregulated α4-laminins to dystroglycan has been the presumptive explanation for the ability of a shortened “mini” form of agrin to compensate loss of α2-laminins in the muscles of dystrophic (dy-strain) mice [34].
Integrins and dystroglycan couple the basement membrane to intracellular events through two mechanisms. They serve as signaling receptors by activating receptor-associated components, which regulates such cell functions as proliferation, migration, protection from apoptosis, and differentiated state (reviewed in [35, 36]). They also directly link the basement membrane to the underlying cytoskeleton, an attribute that may be particularly important where strong mechanical stresses exist (e.g., skeletal muscle), but which also supports the activity and stability of motile processes. Integrins and dystroglycan form such transmembrane linkages by indirectly binding to the actin cytoskeleton. Integrin-associated adaptor proteins that mediate cytoskeletal linkage include integrin linked kinase (ILK), α- and β-parvins, talin, α-actinin and filamin, whereas dystroglycan-associated proteins that participate in this linkage include dystrophin and utrophin [37–45]. The laminin polymer is thought to directly contribute to basement membrane signaling, in part through the organization of the cytoskeleton, and in part by mechanosensory transduction mechanisms that are influenced by the viscoelastic properties of an ECM [46–48].
Laminin 3A32 and Junctional Epidermolysis Bullosa
This laminin (α3Aβ3γ2) is found in epithelial basement membranes such as that present at the epidermal-dermal junction. The laminin binds to nidogen (which can attach it to the type IV collagen network) but is a non-polymerizing laminin due to its short arm truncations [19]. It binds to the α6β4 and α3β1 integrins and to type VII collagen, enabling it to form strong adhesions between cell hemidesmosomes and anchoring fibrils [49]. Mutations affecting expression of any of its three subunits are causes of junctional epidermolysis bullosa (JEB) in which a split develops within the epidermal/dermal basement membrane leading to blister formation [50, 51]. Deletions of the α6 integrin subunit, β4 integrin subunit, and α3 laminin subunit all result in related epithelial detachment phenotypes while mutations of type VII collagen cause blistering defects within the stroma adjacent to basement membrane [52–55].
Pierson Syndrome
This disease characteristically affects the renal glomerulus, eye, and neuromuscular junction. It results from mutations in the LAMB2 gene that codes for the laminin β2 subunit, present in laminins -221, -421, and -521 [56, 57]. In the mouse models of the disease, in which the pathologies of the neuromuscular junction and kidney have been well described, there is a compensatory incorporation of orthologous laminin isoforms containing the β1 subunit; however, these forms do not prevent pathogenesis [58–60]. Studies to date have focused on unique activities associated with the laminin β2 chain, although a possibility that insufficient expression of the β1 chain contributes to pathogenesis in Pierson syndrome has not been excluded.
Laminin α2-Deficient Congenital Muscular Dystrophy
α2-laminins are major components of the basement membranes of Schwann cell (endoneurium), skeletal muscle fiber (sarcolemma), neuromuscular junction, and myotendinous junction. Nonsense and missense mutations of the LAMA2 gene coding for the laminin α2 subunit cause about 1/3 of cases of congenital muscular dystrophy (clinically classified as type MDC1A). The syndrome is characterized by generalized muscle necrosis with variable levels of atrophy and regeneration, in combination with reduced peripheral nerve conduction velocity (mostly motor neuron), and thinning of white matter of the brain as detected by nuclear magnetic resonance imaging [61]. LAMA2 mutations that eliminate expression of α2-laminins uniformly result in a severe muscular dystrophy; patients are floppy at birth, fail to ambulate, and typically die of extreme muscle wasting and respiratory failure in the second decade. Dogs, cats, and mice lacking laminin α2 are similarly affected. The series of dystrophic (dy) mouse LAMA2 mutations provide a strong experimental model of the disease that accurately reflects the variable extent of muscle and peripheral nerve pathology in the affected human patient population. Specific strains include dy2J (in-frame deletion of the α2 LN domain), dy3K (α2 knockout), dyW (severe hypomorph of the α2 subunit), and nmf417 (dy7J; point mutation in α2 LN domain) [62–65].
Mutations affecting the laminin α2 subunit, dystroglycan (the core protein itself as well as its O-linked mannosyl carbohydrate required for binding to LG domains in laminin, agrin, and perlecan), the dystrophin-associated sarcoglycans, dystrophin, α7-integrin, and integrin-linked kinase (ILK) all cause muscular dystrophy [44, 61, 66–70]. In skeletal muscle, these components and other submembrane-cytoskeletal components vinculin and β-spectrin are co-localized within a transmembrane adhesion complex called the costamere. Co-stameres are organized as an orthogonal surface array across the sarcolemma, and it has been thought that they might mediate the transmission of lateral forces generated by contraction across the sarcolemma membrane and between neighboring myofibers [71]. Two series of molecular links have been traced between basement membrane and actin cytoskeleton, with a complex of dystroglycan, dystrophin/utrophin, and dystrophin-associated glycoproteins in one case, and integrins, ILK, PINCH, parvins and additional adaptor proteins in the other [45, 72]. Thus, deficiency in the key components of the sarcolemmal basement membrane, their sarcolemmal receptor complexes, and their cytoskeletal-anchoring partners within the costamere are thought to represent a common pathogenic pathway to muscular dystrophy by destabilizing the overall linkage between basement membrane and myofiber cytoskeleton. In mouse models, the pathology is typically not appreciated until the affected animal begins to exercise, suggesting that the peri-sarcolemmal structural organization is too unstable to withstand the mechanical stress of muscle contraction against load [73, 74].
The sarcolemma basement membrane is a fundamental feature of this model of sarcolemmal pathogenesis in dystrophy. The sarcolemmal lipid bilayer presumably lacks the inherent stability to withstand high levels of tension generated at sarcolemma/cytoskeleton attachment sites when the myofiber contracts. Instead, stresses are transferred across the sarcolemma via transmembrane receptor complexes that directly bind to both cytoskeleton and basement membrane. The laminin network within the basement membrane appears ideally suited to withstand the cytoskeletal stresses by virtue of its polygon polymer structure, which serves to evenly redistribute local tension into the surrounding matrix across the fiber surface, as well as into the interstitial matrix between fibers. Consistent with a central role for basement membrane stability, the dystrophy associated with mutations that eliminate expression of α2-laminins is severe. For comparison, laminin α2-null mice die from catastrophic myodegeneration between 5 and 8 weeks, while mice lacking dystrophin survive well into adulthood.
Accordingly, therapeutic approaches to ameliorate muscular dystrophy have been directed at repairing the transmembrane bridge between cytoskeleton and basement membrane. Several experimental approaches to inhibiting the dystrophic muscle phenotype and pathology have been evaluated in recent years, using the LAMA2 mutant dy-strains of mice. A direct approach of restoring laminin expression by germline transgenesis is effective [75, 76], but faces difficulties in translation to human patients. Thus, there is considerable interest in an alternative strategy which attempts to compensate deficiency for laminin α2 by providing supplementary means to sustain high-affinity of binding of basement membrane to the sarcolemmal surface. One approach has been to upregulate receptor activity through over-expression of the glycosyl-transferase LARGE and α7-integrins. A second approach has been to supplement the basement membrane with smaller components such as mini-agrin and NtA-perlecan/LG, which are designed to not just bind to the laminin α2 receptors, but restore linkage to the residual components of the basement membrane [41, 77, 78].
Miniagrin is a shortened version of non-neural agrin, consisting of a fusion of agrin’s N-terminal NtA domain and C-terminal cell LG domains [34]. The NtA domain binds to a site in the coiled-coil domain of laminins (primarily through the γ1 subunit), and the LG domains bind to α-dystroglycan, β1 integrins, and galactosyl sulfatide ([79] and Yurchenco & Capizzi, unpublished observations). Shortening the DNA construct to eliminate unneeded intervening sequences facilitates widespread delivery in postnatal muscles in affected individuals. Transgenic expression of miniagrin, and of a related NtA-perlecan LG fusion protein, substantially ameliorated the muscle pathophysiology of mouse models of laminin α2 deficient muscular dystrophy [34, 77, 80–82]. Similar benefits occurred when the gene for miniagrin was delivered to the dystrophic mouse tissues using an adeno-associated virus delivery system [83]. The basis for this improvement in muscle structure and function has been attributed to the ability of agrin to simultaneously bind with high affinity to α-dystroglycan and the α4-laminins that are upregulated in α2-deficient muscles. This interpretation emphasizes the weak binding of the laminin α4 LG domains to dystroglycan. Accordingly, miniagrin restores this link to dystroglycan, while the α4-laminins maintain links to other basement membrane components, including the type IV collagen network.
Nevertheless, there remains some uncertainty regarding the mechanism by which miniagrin stabilizes muscle fibers in dystrophic mice. First, miniagrin dramatically rescued the structural integrity of the basement membrane. This result is consistent with the notion that basement membrane stability is critical to muscle fiber survival. However, it is not consistent with the notion that mini-agrin acts primarily through interactions with α4-laminins, which entirely lack the ability to polymerize, organize, and maintain a structurally integrated sheet of basement membrane. In principle, this result could be taken as evidence that there are alternative mechanisms to form and maintain basement membrane architectures, apart from the tripartite LN-domain interactions between laminins. However, miniagrin expression also resulted in high level incorporation of laminin-511 into the sarcolemma basement membrane [34]. Laminin-511 (laminin-10) contains the α5 subunit, which contains an LN polymerization domain. The α5-laminins are fully competent to form and maintain basement membranes; a synapse-specific isoform (laminin-521) maintains the synaptic cleft in dy-strain mouse muscles [84, 85]. Thus, an alternative possibility is that miniagrin restores the integrity of the sarcolemma basement membrane by increasing the incorporation of α5-laminins. The basis for this increase is not certain, but could include effects on laminin assembly, secretion, transcription, or surface “capturing” of a laminin that otherwise would dissociate from the cell surface.
While the principal contributors to muscle stabilization studied to date have focused on receptor-associated linkages, the state of the matrix structure is important as well. The dy2J mice, which lack most of the α2-LN domain due to a splice donor defect, are characterized by a moderate skeletal muscle dystrophy that includes a disrupted organization of the sarcolemmal costameric array [86]. An analysis of laminin extracted from the skeletal muscle of these mice and of a recombinant laminin fragment containing the same deletion revealed that the dy2J α2-laminins have increased proteolytic instability of the α2 LN domain and defective polymerization [62, 87]. The dy2J phenotype therefore estimates the contribution of the LN-domain-mediated laminin network to muscle fiber stability, and most directly implicates a loss of basement membrane strength in the increased fragility of muscle fibers in dy2J mice. However, even a modest (~three-fold) decrease of otherwise normal laminins cause dystrophic muscle changes (Yang, McKee, Patton, Chen, Strickland and Yurchenco, unpublished observations), suggesting that a certain density of laminin interactions are necessary to stabilize basement membrane structure. In contrast, a point-mutation disruption of the α2-LN domain was recently discovered, which causes dystrophy with subtle defects in basement membranes and no decrease in laminin levels [65]. The muscle basement membranes were not interrupted, as with other α2 mutations, but were significantly thicker in some regions, possibly destabilizing myofibers through a lack of flexibility under contraction. Viewed in broad strokes, mutations that fully or nearly eliminate α2-laminins from the sarcolemma greatly destabilize myofibers, while mutations that preserve moderate levels of α2-laminins but disrupt the organization of the basement membrane cause moderate or mild forms of dystrophy.
Laminins and Peripheral Myelination
Laminins play critical roles in the developmental myelination of peripheral nerves. Embryonic nerves consist of naked axons tightly fasciculated into several distinct bundles; each bundle is covered by the flat, wide processes of immature Schwann cells. During myelination, each axon becomes isolated by a dedicated cohort of Schwann cells. Large axons are myelinated (wrapped dozens of times by their Schwann cells), but many small sensory axons are ensheathed without becoming myelinated. The developmental process by which the population and type of Schwann cells is matched to the population and type of axon is called axonal sorting, or “radial sorting”, for the processive nature by which the embryonic axon fascicles are reduced to separate nerve fibers. The ability of Schwann cells to participate in axonal sorting is dependent on the laminin concentrated in the endoneurial basement membrane that Schwann cells assemble along their outer (abaxonal) surface. This matrix is composed principally of laminins-211 and -411, collagen IV (α1α2), nidogen-1, and perlecan. In mutant mice in which Schwann cells lack both isoforms of laminin, through targeted disruption of the α2+α4 genes or cell-specific down-regulation of the γ1 gene, the basement membrane fails to properly assemble, and axonal sorting fails completely [88, 89]. While the mutant mice survive postnatal development, their nerves remain essentially embryonic: nearly all axons remain tightly bundled in their original fascicles, and each bundle remains surrounded by the processes of immature Schwann cells (e.g., lacking nuclear localization of Krox-20). Thus, complete loss of Schwann cell laminins arrests axonal sorting and results in complete “amyelination”.
These findings established a baseline for assessing the individual contributions of the two laminin isoforms, 211 and 411, to myelination. In brief, the discovery that deficiency for each individual laminin isoform causes a partial amyelination implicates distinct, non-redundant roles for each. Moreover, combined with patent evidence that only one of the laminins (211) supports basement membrane stability, the comparative analysis of phenotypes unexpectedly challenges long-held views that axonal sorting depends on the ability of promyelinating Schwann cells to assemble basement membranes, and that the critical role of laminin during axonal sorting is mediated by its special role in organizing and/or stabilizing basement membranes.
The original idea that Schwann cell basement membrane structure is essential to promyelinating differentiation of immature Schwann cells began with observations of the nerves in laminin α2-mutant dy mice [90–93]. Of the two laminins expressed by developing Schwann cells, only laminin-211 possesses three intact LN domains (in α, β, and γ) and is therefore competent to scaffold basement membrane assembly. Thus, loss or mutation of the α2 subunit in the dy-strains of mice prevent Schwann cells from forming or maintaining stable basement membranes [93] despite high levels of laminin-411, which lack the α-subunit LN domain [94, 95]. Each mutation of laminin α2 causes a characteristic “partial” amyelination: spinal roots are severely amyelinated, and half the axons in the sciatic nerve are amyelinated. Of particular interest, mutations targeted to the LN domain of the α2 (dy2J and nmf417) almost completely inactivate laminin-211 for axonal sorting. For example, the LN-domain point mutation in nmf417 causes the same severe amyelination in mouse nerve spinal roots as the α2-null allele (dy3K). Further, a combination of the dy2J α2-mutation with the laminin α4-null mutation results in severe amyelination, revealing that when the dy2J-variant of laminin-211 is isolated on Schwann cells it has almost no inherent activity. In short, the ability of laminin-211 to promote axonal sorting is fully dependent on the LN-domain interactions that underlie the laminin network and basement membrane assembly. Complementing these findings, cell culture studies found that Schwann cells require media conditions which stimulate basement membrane assembly in order to begin myelinating axons [96–99]. Thus, the integrity of the Schwann cell basement membrane appears linked to the onset of myelination both in vivo and in vitro. The combination led to a proposal extrapolated from epithelial differentiation, that the endoneurial basement membrane polarizes the immature Schwann cells in preparation for differentiation into mature myelinating Schwann cells [96].
However, there is considerable evidence which is not neatly accommodated by a simple basement membrane model of axonal sorting. First, while loss of laminin α2 disrupts endoneurial basement membranes throughout the mouse PNS, many nerves are normally myelinated, including the large brachial nerves supplying the arms. Second, non-mammals complete myelination before endoneurial basement membranes form. Thus, a more comprehensive assessment suggests that basement membrane formation (as assessed by conventional fixed EM preparations) is not tightly correlated with the ability of Schwann cells to undergo promyelinating differentiation and complete axonal sorting.
Third, perhaps the strongest contradictions to the initial basement membrane hypothesis are provided by the discovery that laminin-411, containing the α4 subunit, has an independent and indispensible role in axonal sorting. A null mutation in the mouse laminin -α4 gene causes partial amyelination of peripheral nerves very similar to that caused by loss of α2-laminins, including nearly identical deficits in myelinated axons and distributions of naked axon bundles [89, 100]. Loss of non-polymerizing α4-laminins and polymerizing α2-laminins are equally disruptive. Importantly, loss of α4 laminin had no effect on levels of α2-laminins, and no apparent effect on the ultrastructural appearance of the Schwann cell basement membrane. That is, α4-laminins appear to be required for Schwann cells to complete axonal sorting and myelination in sciatic nerves, even though they provide no added value to the appearance of the basement membranes. Conversely, by reference to the complete lack of sorting in α2/α4-double null mutant mice, it is apparent that laminin-411 is responsible for all of the sorting and myelination that occurs in α2-mutant dy-strains of mice, in the absence of laminin-211 and without the support of well-formed basement membranes. Finally, observations in the spinal roots of dy2J/α4-null double mutant mice reveal that α2-laminins also activate axonal sorting and myelination without ultrastructurally-detectable basement membrane [89]. Together, the results reveal little correlation exists between the appearance of the endoneurial basement membrane and the ability of Schwann cells to proceed with axonal sorting.
The discovery that both laminins-211 and -411 are required to complete sorting in mouse sciatic nerves implies they have non-redundant activities, either mediated by distinct receptors or mediated by different structures that result in different types of information transmitted to the Schwann cell. Because α4-laminins lack the ability to support basement membrane assembly, they most likely serve to activate signaling receptors on the surface of immature Schwann cells. β1 integrins would logically represent a candidate receptor class for α4-laminins in axonal sorting given the strong overlap in the characteristic distribution of amyelination that results from Schwann cell specific knockdown of the integrin β1 subunit [89, 100–102]. Since the loss of β1-integrins caused a more extensive amyelination than that seen in the absence of the laminin α4 subunit, this suggests a non-exclusive interaction between α4-laminin and β1-integrins. Receptors which selectively mediate laminin-211 in axonal sorting have not been strongly implicated. Schwann cell specific knockdown of dystroglycan caused little or no defect in axonal sorting or basement membrane structure [103]. Laminin-211 activity relies heavily on LN-domain function, but appears less dependent on the broader superstructure of the basement membrane that results from laminin-polymerization. The LN domain might have direct signaling activities. Alternatively, laminin-211 might stabilize multi-receptor/cytoskeletal signaling complexes through short-range LN-domain polymer interactions.
A question that arises is whether laminins represent the principal integrin ligand involved. Given laminin-integrin binding specificities, this may be unlikely. First, the principal integrin receptor for α2-laminins is the α7X1β1 integrin, and not α6β1 (enriched in the endoneurium), α6β4 or α3β1 as once thought, whereas α4-laminins can support Schwann cell attachment through the α6β1 integrin even though they at best bind only weakly to integrins in general [28, 89, 104]. The α7β1 integrin is expressed quite late [28] in peripheral nerve development and knockout of the α7 subunit is not associated with a developmental nerve phenotype [105]. One can question whether the laminin-specific α6β1 integrin is the key receptor (analysis of the null phenotype in nerve will require a conditional knockout approach) given that knockout of its cognate laminin α4 ligand produces a more restricted peripheral nerve myelination defect compared to that seen with knockout of the laminin α2 subunit [89]. It should be emphasized, however, that loss of laminin in a basement membrane is generally associated with a loss of the other BM components as well [11, 102]. A simple explanation of the apparent conundrum may be that it is the composite binding between β1-integrin and most or all basement membrane components (i.e. α2- and α4-laminins through α7β1 and α6β1 integrins respectively, collagen-IV through α1β1 and α2β1, nidogens and agrin through α3β1, and perlecan through α2β1) that provide the relevant set of interactions rather than selective interactions with laminins. A related issue concerns the role of dystroglycan in basement membrane – Schwann cell interactions. Dystroglycan, similar to integrin α7β1, is expressed late in peripheral nerve development just prior to myelination [106] and knockout of DG produced milder defects of slowed nerve conduction with abnormal Schwann sheath folding and nodal architecture [103].
In summary, our review of the current data provides an alternative model with the view that laminins promote axonal sorting and myelination as autocrine signaling factors that act independent of the basement membrane structure. However, no study in other tissues provides a similar example of laminin function divorced from the context of basement membrane integrity. It therefore seems wise to consider whether a proxied view of basement membrane structure limits our ability to discern more essential matrix characteristics on which laminin function and axonal sorting does depend. We offer a primary caveat to the above consideration of the relationship of basement membrane assembly and receptor interactions to radial sorting, i.e. the absence or presence of a basement membrane has been generally referred primarily to its ultrastructural appearance. It should be noted that basement membrane-like extracellular matrices can be detected by immunofluorescence in the absence of a linear lamina densa (Yurchenco, unpublished observations) while modifications of staining techniques (e.g. tannic acid or permanganate treatment) can affect the sensitivity of detection of a lamina densa by electron microscopy. Furthermore, it can be argued that the presence of a continuous lamina densa does not guarantee the stability or functionality of the basement membrane. Proper functionality may depend upon the integration of molecular architecture with sufficient cellular anchorage and correct coding of receptor-ligand interactions in which defects of one can affect the other. An alternative hypothesis to be considered, then, is as follows: The structural contributions of α2-, α4- and α5-laminins to the endoneurial basement membrane differ such as α2 assembly leads to a prominent lamina densa, α4 assembly leads to an alternate architecture in which type IV collagen and nidogen are tethered to non-polymerized laminin, and α5 assembly, which can provide partial functional compensation, leads to formation of an attenuated laminin/collagen network structure (the nature of which remains to be determined). Each laminin contributes different signaling to the Schwann cell by virtue of its receptor specificities and different architectural characteristics. A combination of the separate α2 and α4 contributions are required to enable Schwann cells to undergo proper axonal sorting. Resolving these different models is an important goal, necessary to developing therapeutic modalities as much as understanding mechanism, and requiring new insights gained from the integration of genetic, cellular and molecular approaches.
TYPE IV COLLAGENS
Type IV collagen self-assembles into the only known covalently-stabilized network polymer. The first evidence for the molecular nature of this network was provided by an elegant biochemical and electron microscopic analysis of type IV collagen (α12α2[IV] composition) extracted from the EHS tumor matrix. Both N-terminal (7S domain) and C-terminal (NC1) domains that contributed to the network were characterized [107]. The 7S domains interact to form a multimeric complex, named for the sedimentation constant of the collagenase resistant fragment. The 7S complex forms spontaneously in solution through dimeric and trimeric intermediates, combining the N-terminal segments from four separate collagen chains to create a ~30 nm region of overlap that becomes further stabilized through disulfide and lysyl-oxidase derived covalent crosslinks [108–114]. At the C-terminal end of the collagen chain, the NC1 globular domain undergoes self-interactions that extend the collagen IV network indefinitely [115]. Since each NC1 domain consists of three C-terminal segments, the dimeric complex has also been described as a hexamer. The NC1 domain crystal structure revealed a flat interacting face for dimerization [116, 117]. Subsequent analysis of the domain structure revealed an unusual covalent crosslink between α1 Met93 and Hyl211 that stabilizes the domain complex [118, 119]. The NC1 domains of the less common α3α4α5[IV] undergo a similar self-assembly, and it has been shown that the NC1 subunits can define the specificity of intramolecular collagenous chain association [120–122].
A third contributing interaction to the formation of the collagenous network is that of lateral (side-by-side) associations. These were first identified morphologically, as segmental associations of parallel collagenous filaments with frequent branchings in low angle rotary shadowed Pt/C replicas of extracted collagen, and supported biochemically as part of the heat of gelation [109]. These networks were later visualized in high angle Pt/C replicas of freeze-dried amniotic, EHS, and corneal basement membranes [4, 6, 7]. The lateral associations were not detected in the absence of the N- and C-terminal interactions, and it is likely they represent low affinity associations that drive network complexity.
During development of the renal glomerular basement membrane (GBM), the type IV collagen α1 and α2 chains become replaced by type IV collagen α3, α4 and α5 chains. Analysis of collagen containing these alternative chains revealed a collagen network with disulfide crosslinks not found with the α1 and α2 chain type IV collagen, which renders portions of the network resistant to dissociation [123]. These covalent links likely stabilize the GBM network, and may help explain the GBM defect seen in Alport’s disease and in the Alport’s-like phenotype of the α3-collagen-IV null mouse [124].
Type IV collagen does not organize basement membrane assembly. The time-line of laminin and type IV collagen expression during development of the genetic model C. elegans, a nematode, provided early evidence for layered functions of the two types of basement membrane components [125–127]. Nematodes possess two laminins (differing in their α-subunit) that appear in basement membranes at the time of gastrulation, while type IV collagens does not appear until embryonic elongation. Inhibiting the expression of both laminin α subunits was found to result in loss of basement membranes and early embryonic lethality, providing evidence for an early requirement for laminins [127]. In contrast, mutations preventing type IV collagen expression did not prevent the initial assembly of basement membranes, but resulted in late embryonic lethality as muscle contractions commenced [125, 126]. The correlation of degeneration with the onset of activity suggests that type IV collagen is needed to enable the basement membrane to withstand mechanical stress. Primary evidence for a similar stabilizing role in vertebrates for type IV collagen comes from generation of a mouse null allele of the Col4α1/α2 locus (where the genes are paired) and analysis of the phenotype resulting from the loss of the two principal collagen chains [13]. Homozygous mutant embryos developed up to E9.5 with basement membrane proteins deposited at the expected sites. However, similar to events in C. elegans, lethality occurred between E10.5–E11.5 with structural deficiencies clearly evident in the basement membranes, including failure of integrity of Reichert’s membrane. Further evidence that basement membranes in early embryogenesis do not rely on collagens comes from analysis of knockout of the Hsp47 chaperone, which is required for procollagen (including type IV collagen) triple-helical folding [128, 129]. Embryos progress until ~E11.5, when they terminate with basement membrane disruptions accompanied by abnormal epithelial orientation and ruptured blood vessels.
Alport’s Syndrome
Mutations of the X-linked COL4A5 gene cause Alport’s syndrome in 85% of cases [130]. Less commonly mutations of the COL4A3, COL4A4 genes that encode the other chains of the α3α4α5[IV] collagen produce the disease. Classically, the disease presents in males as progressive renal failure with hematuria and proteinuria accompanied by hearing loss. The GBM contains the embryonic collagen isoform (α12α2[IV]) rather than the mature α3α4α5[IV] isoforms, and it is thought that this collagen degrades more readily due to the absence of stabilizing disulfide bonds. Histologically, the Alport’s GBM is thin and contains splits or laminations. Patients are often treated with dialysis, sometimes followed by transplantation. A small fraction of the transplanted patients develop anti-GBM autoantibodies.
Hereditary Porencephaly and Hemorrhagic Stroke
While null mutations of the COl4A1 and CoL4A2 genes cause embryonic lethality, mutations in the COL4A1 gene that lead to only a reduction of the widely expressed α1[IV] subunit through faulty chain assembly are a cause of hereditary porencephaly and have been linked to hemorrhagic stroke in humans [131, 132A]. Thus while null mutations of type IV collagen and laminin γ1, β1, and α5 may cause embryonic lethality, more subtle mutations in these components may be a cause of more common human diseases.
NIDOGENS
Nidogen-1 and nidogen-2, also known as entactins, are homologous glycoproteins (150 kDa) found widely distributed among basement membranes. Each consists of three globular domains (G1, G2, G3) with intervening rod-like regions [133–138]. G1 and G2 are located in the N-terminal moiety, separated by a small linkage region. EGF-like repeats dominate the central region. Domain G3 is located at the carboxyl terminal region. The nidogen-1 G3 domain binds strongly to laminins, through a specific sequence element residing in domain LEb3 of the laminin γ1 short arm, confirmed through solution of the crystal structure of a nidogen-laminin complex [139]. The nidogen G2 domain binds to type IV collagen (as well as G3), as well as perlecan and fibulins [140–142]. Nidogen thereby directly tethers the laminin and collagen IV networks. A recent analysis of basement membrane assembly on Schwann cells using various recombinant laminins confirmed this bridging role [10]. The laminin-nidogen-collagen linkage is thought to provide stability to basement membranes, particularly important in those tissues exposed to mechanical stress, such as skeletal muscle [13, 15]. While tissue/organ culture studies have supported a preeminent role for nidogen as the key bridge between laminins and type IV collagen, genetic studies in both mice and nematodes demonstrate that the absence of nidogen(s) or the nidogen-binding site in laminin γ1 does not adversely affect the accumulation of either laminin or type IV collagen into developing basement membranes [15, 143–147]. The findings argue there must be additional interactions that incorporate type IV collagen into assembling basement membranes. Possibilities include direct collagen-laminin interactions and binding through perlecan. However, although laminin isoforms may vary, collagen IV binds weakly to laminin-111, and perlecan interactions are dominated by heparan sulfates, which typically lack specificity and high affinity. Thus, an alternative possibility is that integrin-mediated binding suffices to concentrate the assembly of the collagen IV network to the cell surface.
AGRIN
Agrin is a large (>400 kDa) multi-domain heparan sulfate proteoglycan of basement membranes that exists in several variant forms generated through alternative splicing and transcription initiation sites [148]. The predominant form of agrin in basement membranes contains an N-terminal NtA domain, which binds to the laminin γ1 subunit in the coiled-coil of the laminin heterotrimer. In tissues lacking basement membranes, such as the central nervous system, the NtA domain of agrin is often replaced by an alternative aminoterminal sequence that provides a direct GPI-mediated linkage to the cell surface. All agrins contain C-terminal LG domains, which bind to α-dystroglycan and integrins [79, 149–152]. Agrin LG binding to dystroglycan has the highest affinity of all of the LG domains tested. The affinity of NtA-agrin for both laminin and dystroglycan may serve to increase laminin anchorage to cell surfaces in some tissues. As noted above, this role is generally consistent with the improvement in sarcolemmal basement membrane structure and function seen by over-expression of a shortened version of agrin in laminin α2-deficient mice [34]. Motor neurons secrete a unique set of agrin splice variants that regulate the organization and maintenance of the postsynaptic apparatus at the neuromuscular junction. The neuronal splice variants of agrin contain a short loop of residues inserted at the fringe of the beta-sheet sandwich of the LG domain. Interestingly, a conserved pattern of splice-generated sequence variations occurs at similar fringe sites in the LG domains of neurexins, which participate in synaptic maturation at interneuronal synapses.
PERLECAN
Perlecan is a heparan sulfate proteoglycan in basement membranes and a chondroitin sulfate proteoglycan in cartilaginous tissues [153–155]. Its core protein is large (~400 kDa) and consists of a tandem arrangement of five “domains” (each really a distinct region composed of one or more structural domains). The fourth and fifth “domains” (immunoglobulin repeats; and, LG domains separated by LE repeats, respectively) bind to a host of basement membrane components and BMZ receptors. These associations include nidogen-1 (domain IV), integrin α2β1, and α-dystroglycan (domain V). Perlecan heparan sulfate chains, located primarily in the N-terminal domain I, have the potential to bind to laminins and to type IV collagen, and to tether FGF2 and other growth factors. Iozzo and co-workers have found that domain V, renamed endorepellin, is a potent inhibitor of angiogenesis through an activity dependent upon its binding to the α2β1 integrin [156, 157].
Perlecan may have a critical role in basement membrane maintenance in some tissues, directly or through support to the underlying cells, but does not appear to have a principal role in basement membrane assembly. Basement membranes were found to develop normally in mutant mouse embryos homozygous for a null-allele for perlecan gene, which survived to between ~E10.5 and E19 (i.e., birth) [158, 159]. Embryos that died early a cardiac defect in which basement membranes developed disruptions through which blood exited to the developing pericardial space. Specific mutations of perlecan in humans have been found to cause Schwartz-Jampel syndrome, a disorder combining chondrodysplasia and myotonia. The latter is thought to occur because perlecan is required for binding to acetylcholinesterase at the neuromuscular junction [160–162]. The former may result because perlecan carries an oversulfated form of chondrotin sulfate (chondoitin 2,6 disulfate) in cartilage that accelerates the rate of assembly and increases the diameter of type II collagen fibers [163].
TYPE VI, VII, XV, AND XVIII COLLAGENS AND THE STROMAL INTERFACE
Basement membranes commonly lie adjacent to an extracellular matrix stroma that contains banded interstitial collagens. Extracellular matrix components, in particular several collagens, are thought to stabilize the interface by binding to both basement membrane and stromal components. However, no single collagen has been found to mediate this function.
In skin, anchoring fibrils reinforce attachment of the epidermal basement membrane to the dermal stroma. Type VII collagen, a major component of the fibrils that assembles into anti-parallel dimers, has been reported to bind to laminin-332 through its β3 subunit short arm, and to bind to both type IV collagen and to interstitial banded collagen fibrils [164–166]. Dominant and recessive mutations of this collagen are a cause of dystrophic epidermolysis bullosa, a blistering skin disease in which the split occurs in the dermis at the level of anchoring fibrils (reviewed in [167]).
In skeletal muscle, type VI collagen is thought to anchor basement membrane to adjacent connective tissue (reviewed in [168]). The collagen triple helical monomer has been shown to self-assemble into secreted disulfide-stabilized anti-parallel tetramers and to form beaded microfibrils and filamentous networks in tissues. The collagen has been found to interact with type IV collagen, type I collagen, type II collagen, type XIV collagen, perlecan, and the microfibril-associated glycoprotein MAGP1. Mutations of the collagen are a cause of both Betham myopathy and Ullrich congenital muscular dystrophy.
Type XV and XVIII collagens are also candidates for providing basement membrane-stromal linkage. Type XV collagen contains many interruptions of its gly-x-y sequence and is modified by chondroitin sulfate and heparan sulfate chains. It is present in most basement membrane zones where it is associated with large collagen fibers [169]. The function of type XV collagen is not well understood, in part because inactivation of the gene for this collagen was not found to be associated with basement membrane defects or a relevant phenotype [170]. Type XVIII collagen is also a heparan sulfate proteoglycan [171]. Proteolytic cleavage of its NC1 domain releases a fragment called endostatin that has been reported to inhibit angiogenesis and that has been studied for its ability to inhibit tumor growth. However, a general physiological vascular role for the collagen was not established from investigation of the phenotype generated by inactivation of the type XVIII collagen gene [172]. On the other hand, that study supported the concept that the collagen is involved in connecting the basement membrane to stromal collagen in a subset of tissues, especially vitreous vessels. Mutations of the gene for this collagen are associated with Knobloch syndrome that is characterized by age-dependent vitreoretinal degeneration and occipital encephalocele.
CONCLUSIONS
One can consider the basement membrane as a cross- linked heteropolymer that on the one hand provides lateral stability to the cell surface receptors and adjacent cytoskeleton, and that on the other that provides a series of stabilizing chain links that extend from the basement membrane to the cell cytoskeleton. Disruptions of these interconnecting components along either axis are destabilizing to tissues, affecting structure and signaling. While the diseases that result from such defects can theoretically be repaired by replacing the missing component, more workable solutions might be to provide simpler and smaller linking components derived from existing basement membrane components that are easier to express in tissues or to affect the expression of redundant/compensatory pathways. Whatever therapies ultimately emerge, their development will likely require detailed knowledge of the mechanisms of assembly, structure and function.
Fig. (2). Basement Membrane Assembly.
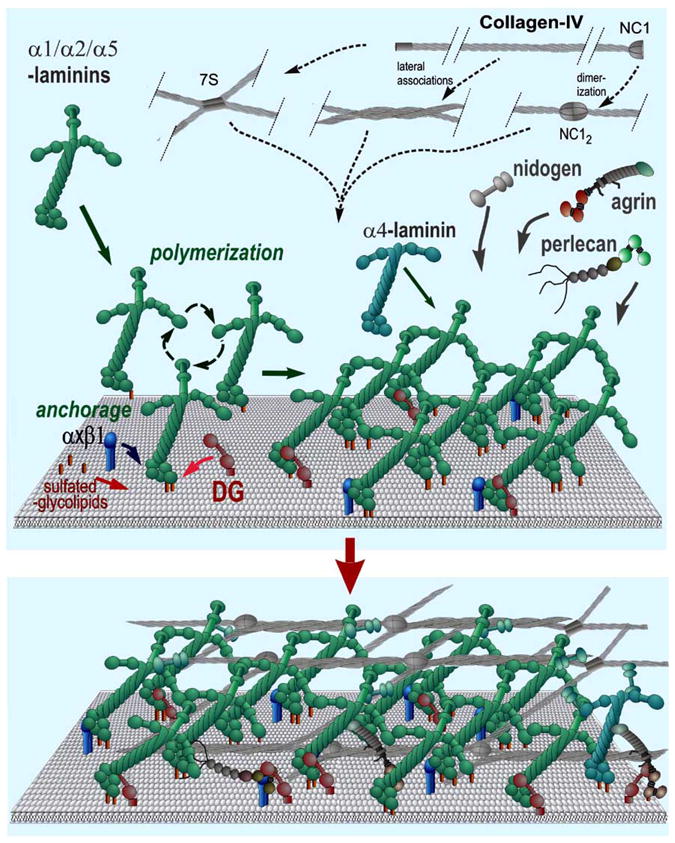
In this simplified model, laminins become anchored to the cell surface through their LG domains. Anchorage is further enhanced through binding of the α-LN domain to sulfated glycolipids. If the laminin has three LN domains it polymerizes, creating a “nascent” scaffolding. Nidogens, type IV collagens, perlecan, and agrin, are incorporated into this initial matrix by binding to laminin (or by binding through a nidogen bridge). The type IV collagen self-assembles into a covalently-crosslinked network. The non-laminin components provide crucial stability and increase ligand complexity. The basement membrane ligands interact with integrins and dystroglycan and the heparan sulfates of agrin and perlecan enable the tethering of tissue-specific growth factors.
Acknowledgments
This review was supported by grants R37-DK36425 (PDY), R01-NS38469 (PDY), R01-NS040759 (BLP) and R01-NS064397 (BLP) from the National Institutes of Health.
References
- 1.Chan FL, Inoue S. Lamina lucida of basement membrane: An artifact. Microsc Res Tech. 1994;28:48–59. doi: 10.1002/jemt.1070280106. [DOI] [PubMed] [Google Scholar]
- 2.Chan FL, Inoue S, Leblond CP. The basement membranes of cryo-fixed or aldehyde-fixed, freeze-substituted tissues are composed of a lamina densa and do not contain a lamina lucida. Cell Tissue Res. 1993;273:41–52. doi: 10.1007/BF00304610. [DOI] [PubMed] [Google Scholar]
- 3.Miosge N. The ultrastructural composition of basement membranes in vivo. Histol Histopathol. 2001;16:1239–48. doi: 10.14670/HH-16.1239. [DOI] [PubMed] [Google Scholar]
- 4.Ruben GC, Yurchenco PD. High resolution platinum-carbon replication of freeze-dried basement membrane. Microsc Res Tech. 1994;28:13–28. doi: 10.1002/jemt.1070280104. [DOI] [PubMed] [Google Scholar]
- 5.Yurchenco PD, Cheng YS, Ruben GC. Self-assembly of a high molecular weight basement membrane heparan sulfate proteoglycan into dimers and oligomers. J Biol Chem. 1987;262:17668–76. [PubMed] [Google Scholar]
- 6.Yurchenco PD, Ruben GC. Type IV collagen lateral associations in the ehs tumor matrix. Comparison with amniotic and in vitro networks. Am J Pathol. 1988;132:278–91. [PMC free article] [PubMed] [Google Scholar]
- 7.Yurchenco PD, Ruben GC. Basement membrane structure in situ: Evidence for lateral associations in the type IV collagen network. J Cell Biol. 1987;105:2559–68. doi: 10.1083/jcb.105.6.2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yurchenco PD, Cheng YS, Colognato H. Laminin forms an independent network in basement membranes. J Cell Biol. 1992;117:1119–33. doi: 10.1083/jcb.117.5.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aumailley M, Bruckner-Tuderman L, Carter WG, Deutzmann R, Edgar D, Ekblom P, et al. A simplified laminin nomenclature. Matrix Biol. 2005;24:326–32. doi: 10.1016/j.matbio.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 10.McKee KK, Harrison D, Capizzi S, Yurchenco PD. Role of laminin terminal globular domains in basement membrane assembly. J Biol Chem. 2007;282:21437–47. doi: 10.1074/jbc.M702963200. [DOI] [PubMed] [Google Scholar]
- 11.Smyth N, Vatansever HS, Murray P, Meyer M, Frie C, Paulsson M, et al. Absence of basement membranes after targeting the lamc1 gene results in embryonic lethality due to failure of endoderm differentiation. J Cell Biol. 1999;144:151–60. doi: 10.1083/jcb.144.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miner JH, Li C, Mudd JL, Go G, Sutherland AE. Compositional and structural requirements for laminin and basement membranes during mouse embryo implantation and gastrulation. Development. 2004;131:2247–56. doi: 10.1242/dev.01112. [DOI] [PubMed] [Google Scholar]
- 13.Poschl E, Schlotzer-Schrehardt U, Brachvogel B, Saito K, Ninomiya Y, Mayer U. Collagen iv is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development. 2004;131:1619–28. doi: 10.1242/dev.01037. [DOI] [PubMed] [Google Scholar]
- 14.Yurchenco PD, Amenta PS, Patton BL. Basement membrane assembly, stability and activities observed through a developmental lens. Matrix Biol. 2004;22:521–38. doi: 10.1016/j.matbio.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 15.Willem M, Miosge N, Halfter W, Smyth N, Jannetti I, Burghart E, et al. Specific ablation of the nidogen-binding site in the laminin gamma1 chain interferes with kidney and lung development. Development. 2002;129:2711–22. doi: 10.1242/dev.129.11.2711. [DOI] [PubMed] [Google Scholar]
- 16.Yurchenco PD, Tsilibary EC, Charonis AS, Furthmayr H. Laminin polymerization in vitro. Evidence for a two-step assembly with domain specificity. J Biol Chem. 1985;260:7636–44. [PubMed] [Google Scholar]
- 17.Garbe JH, Gohring W, Mann K, Timpl R, Sasaki T. Complete sequence, recombinant analysis and binding to laminins and sulphated ligands of the N-terminal domains of laminin [alpha]3b and [alpha]5 chains. Biochem J. 2002;362:213–21. doi: 10.1042/0264-6021:3620213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yurchenco PD, Cheng YS. Self-assembly and calcium-binding sites in laminin. A three-arm interaction model. J Biol Chem. 1993;268:17286–99. [PubMed] [Google Scholar]
- 19.Cheng YS, Champliaud MF, Burgeson RE, Marinkovich MP, Yurchenco PD. Self-assembly of laminin isoforms. J Biol Chem. 1997;272:31525–32. doi: 10.1074/jbc.272.50.31525. [DOI] [PubMed] [Google Scholar]
- 20.Paulsson M. The role of Ca2+ binding in the self-aggregation of laminin-nidogen complexes. J Biol Chem. 1988;263:5425–30. [PubMed] [Google Scholar]
- 21.Yurchenco PD, Cheng YS, Schittny JC. Heparin modulation of laminin polymerization. J Biol Chem. 1990;265:3981–91. [PubMed] [Google Scholar]
- 22.Odenthal U, Haehn S, Tunggal P, Merkl B, Schomburg D, Frie C, et al. Molecular analysis of laminin n-terminal domains mediating self-interactions. J Biol Chem. 2004;279:44505–12. doi: 10.1074/jbc.M402455200. [DOI] [PubMed] [Google Scholar]
- 23.Yurchenco PD, Wadsworth WG. Assembly and tissue functions of early embryonic laminins and netrins. Curr Opin Cell Biol. 2004;16:572–9. doi: 10.1016/j.ceb.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 24.Schneiders FI, Maertens B, Bose K, Li Y, Brunken WJ, Paulsson M, et al. Binding of netrin-4 to laminin short arms regulates basement membrane assembly. J Biol Chem. 2007;282:23750–8. doi: 10.1074/jbc.M703137200. [DOI] [PubMed] [Google Scholar]
- 25.Tsiper MV, Yurchenco PD. Laminin assembles into separate basement membrane and fibrillar matrices in Schwann cells. J Cell Sci. 2002;115:1005–15. doi: 10.1242/jcs.115.5.1005. [DOI] [PubMed] [Google Scholar]
- 26.Li S, Harrison D, Carbonetto S, Fässler R, Smyth N, Edgar D, et al. Matrix assembly, regulation, and survival functions of laminin and its receptors in embryonic stem cell differentiation. J Cell Biol. 2002;157:1279–90. doi: 10.1083/jcb.200203073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li S, Liquari P, McKee KK, Harrison D, Patel R, Lee S, et al. Laminin-sulfatide binding initiates basement membrane assembly and enables receptor signaling in Schwann cells and fibroblasts. J Cell Biol. 2005;169:179–89. doi: 10.1083/jcb.200501098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nishiuchi R, Takagi J, Hayashi M, Ido H, Yagi Y, Sanzen N, et al. Ligand-binding specificities of laminin-binding integrins: A comprehensive survey of laminin-integrin interactions using recombinant alpha3beta1, alpha6beta1, alpha7beta1 and alpha6beta4 integrins. Matrix Biol. 2006 doi: 10.1016/j.matbio.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 29.Ido H, Nakamura A, Kobayashi R, Ito S, Li S, Futaki S, et al. The requirement of the glutamic acid residue at the third position from the carboxyl termini of the laminin gamma chains in integrin binding by laminins. J Biol Chem. 2007;282:11144–54. doi: 10.1074/jbc.M609402200. [DOI] [PubMed] [Google Scholar]
- 30.Wizemann H, Garbe JH, Friedrich MV, Timpl R, Sasaki T, Hohenester E. Distinct requirements for heparin and alpha-dystroglycan binding revealed by structure-based mutagenesis of the laminin alpha2 LG4–5 domain pair. J Mol Biol. 2003;332:635–42. doi: 10.1016/s0022-2836(03)00848-9. [DOI] [PubMed] [Google Scholar]
- 31.Harrison D, Hussain SA, Combs AC, Ervasti JM, Yurchenco PD, Hohenester E. Crystal structure and cell surface anchorage sites of laminin alpha 1 LG4–5. J Biol Chem. 2007;282:11573–81. doi: 10.1074/jbc.M610657200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kikkawa Y, Virtanen I, Miner JH. Mesangial cells organize the glomerular capillaries by adhering to the g domain of laminin {alpha}5 in the glomerular basement membrane. J Cell Biol. 2003;161:187–96. doi: 10.1083/jcb.200211121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weir ML, Oppizzi ML, Henry MD, Onishi A, Campbell KP, Bissell MJ, et al. Dystroglycan loss disrupts polarity and {beta}-casein induction in mammary epithelial cells by perturbing laminin anchoring. J Cell Sci. 2006;119:4047–58. doi: 10.1242/jcs.03103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moll J, Barzaghi P, Lin S, Bezakova G, Lochmuller H, Engvall E, et al. An agrin minigene rescues dystrophic symptoms in a mouse model for congenital muscular dystrophy. Nature. 2001;413:302–7. doi: 10.1038/35095054. [DOI] [PubMed] [Google Scholar]
- 35.Higginson JR, Winder SJ. Dystroglycan: A multifunctional adaptor protein. Biochem Soc Trans. 2005;33:1254–5. doi: 10.1042/BST0331254. [DOI] [PubMed] [Google Scholar]
- 36.Roskelley CD, Bissell MJ. Dynamic reciprocity revisited: A continuous, bidirectional flow of information between cells and the extracellular matrix regulates mammary epithelial cell function. Biochem Cell Biol. 1995;73:391–7. doi: 10.1139/o95-046. [DOI] [PubMed] [Google Scholar]
- 37.Rybakova IN, Ervasti JM. Dystrophin-glycoprotein complex is monomeric and stabilizes actin filaments in vitro through a lateral association. J Biol Chem. 1997;272:28771–8. doi: 10.1074/jbc.272.45.28771. [DOI] [PubMed] [Google Scholar]
- 38.Rybakova IN, Patel JR, Davies KE, Yurchenco PD, Ervasti JM. Utrophin binds laterally along actin filaments and can couple co-stameric actin with sarcolemma when overexpressed in dystrophin-deficient muscle. Mol Biol Cell. 2002;13:1512–21. doi: 10.1091/mbc.01-09-0446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rybakova IN, Patel JR, Ervasti JM. The dystrophin complex forms a mechanically strong link between the sarcolemma and costameric actin. J Cell Biol. 2000;150:1209–14. doi: 10.1083/jcb.150.5.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Warner LE, DelloRusso C, Crawford RW, Rybakova IN, Patel JR, Ervasti JM, et al. Expression of dp260 in muscle tethers the actin cytoskeleton to the dystrophin-glycoprotein complex and partially prevents dystrophy. Hum Mol Genet. 2002;11:1095–105. doi: 10.1093/hmg/11.9.1095. [DOI] [PubMed] [Google Scholar]
- 41.Allikian MJ, Hack AA, Mewborn S, Mayer U, McNally EM. Genetic compensation for sarcoglycan loss by integrin alpha7beta1 in muscle. J Cell Sci. 2004;117:3821–30. doi: 10.1242/jcs.01234. [DOI] [PubMed] [Google Scholar]
- 42.Rooney JE, Welser JV, Dechert MA, Flintoff-Dye NL, Kaufman SJ, Burkin DJ. Severe muscular dystrophy in mice that lack dystrophin and {alpha}7 integrin. J Cell Sci. 2006;119(pt 11):2185–95. doi: 10.1242/jcs.02952. [DOI] [PubMed] [Google Scholar]
- 43.Guo C, Willem M, Werner A, Raivich G, Emerson M, Neyses L, et al. Absence of alpha 7 integrin in dystrophin-deficient mice causes a myopathy similar to Duchenne muscular dystrophy. Hum Mol Genet. 2006;15:989–98. doi: 10.1093/hmg/ddl018. [DOI] [PubMed] [Google Scholar]
- 44.Wang HV, Chang LW, Brixius K, Wickstrom SA, Montanez E, Thievessen I, et al. Integrin-linked kinase stabilizes myotendinous junctions and protects muscle from stress-induced damage. J Cell Biol. 2008;180:1037–49. doi: 10.1083/jcb.200707175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brakebusch C, Fässler R. The integrin-actin connection, an eternal love affair. EMBO J. 2003;22:2324–33. doi: 10.1093/emboj/cdg245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–89. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 47.Colognato H, Winkelmann DA, Yurchenco PD. Laminin polymerization induces a receptor-cytoskeleton network. J Cell Biol. 1999;145:619–31. doi: 10.1083/jcb.145.3.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alcaraz J, Xu R, Mori H, Nelson CM, Mroue R, Spencer VA, et al. Laminin and biomimetic extracellular elasticity enhance functional differentiation in mammary epithelia. EMBO J. 2008;27:2829–38. doi: 10.1038/emboj.2008.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rousselle P, Keene DR, Ruggiero F, Champliaud MF, Rest M, Burgeson RE. Laminin 5 binds the NC-1 domain of type vii collagen. J Cell Biol. 1997;138:719–28. doi: 10.1083/jcb.138.3.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ryan MC, Christiano AM, Engvall E, Wewer UM, Miner JH, Sanes JR, et al. The functions of laminins: Lessons from in vivo studies. Matrix Biol. 1996;15:369–81. doi: 10.1016/s0945-053x(96)90157-2. [DOI] [PubMed] [Google Scholar]
- 51.Nakano A, Chao SC, Pulkkinen L, Murrell D, Bruckner-Tuderman L, Pfendner E, et al. Laminin 5 mutations in junctional epidermolysis bullosa: Molecular basis of Herlitz vs non-herlitz phenotypes. Hum Genet. 2002;110:41–51. doi: 10.1007/s00439-001-0630-1. [DOI] [PubMed] [Google Scholar]
- 52.Ryynanen M, Ryynanen J, Sollberg S, Iozzo RV, Knowlton RG, Uitto J. Genetic linkage of type VII collagen (col7a1) to dominant dystrophic epidermolysis bullosa in families with abnormal anchoring fibrils. J Clin Invest. 1992;89:974–80. doi: 10.1172/JCI115680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Georges-Labouesse E, Messaddeq N, Yehia G, Cadalbert L, Dierich A, Le Meur M. Absence of integrin alpha 6 leads to epidermolysis bullosa and neonatal death in mice. Nat Genet. 1996;13:370–3. doi: 10.1038/ng0796-370. [DOI] [PubMed] [Google Scholar]
- 54.Dowling J, Yu QC, Fuchs E. Beta4 integrin is required for hemidesmosome formation, cell adhesion and cell survival. J Cell Biol. 1996;134:559–72. doi: 10.1083/jcb.134.2.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van der Neut R, Krimpenfort P, Calafat J, Niessen CM, Sonnenberg A. Epithelial detachment due to absence of hemidesmosomes in integrin beta 4 null mice. Nat Genet. 1996;13:366–9. doi: 10.1038/ng0796-366. [DOI] [PubMed] [Google Scholar]
- 56.VanDeVoorde R, Witte D, Kogan J, Goebel J. Pierson syndrome: A novel cause of congenital nephrotic syndrome. Pediatrics. 2006;118:e501–5. doi: 10.1542/peds.2005-3154. [DOI] [PubMed] [Google Scholar]
- 57.Wuhl E, Kogan J, Zurowska A, Matejas V, Vandevoorde RG, Aigner T, et al. Neurodevelopmental deficits in Pierson (microcoria-congenital nephrosis) syndrome. Am J Med Genet A. 2007;143:311–9. doi: 10.1002/ajmg.a.31564. [DOI] [PubMed] [Google Scholar]
- 58.Noakes PG, Miner JH, Gautam M, Cunningham JM, Sanes JR, Merlie JP. The renal glomerulus of mice lacking s-laminin/laminin beta 2: Nephrosis despite molecular compensation by laminin beta 1. Nat Genet. 1995;10:400–6. doi: 10.1038/ng0895-400. [DOI] [PubMed] [Google Scholar]
- 59.Jarad G, Cunningham J, Shaw AS, Miner JH. Proteinuria precedes podocyte abnormalities inlamb2 mice, implicating the glomerular basement membrane as an albumin barrier. J Clin Invest. 2006;116:2272–9. doi: 10.1172/JCI28414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miner JH, Go G, Cunningham J, Patton BL, Jarad G. Transgenic isolation of skeletal muscle and kidney defects in laminin beta2 mutant mice: Implications for pierson syndrome. Development. 2006;133:967–75. doi: 10.1242/dev.02270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jimenez-Mallebrera C, Brown SC, Sewry CA, Muntoni F. Congenital muscular dystrophy: Molecular and cellular aspects. Cell Mol Life Sci. 2005;62:809–23. doi: 10.1007/s00018-004-4510-4. [DOI] [PubMed] [Google Scholar]
- 62.Sunada Y, Bernier SM, Utani A, Yamada Y, Campbell KP. Identification of a novel mutant transcript of laminin alpha 2 chain gene responsible for muscular dystrophy and dysmyelination in dy2j mice. Hum Mol Genet. 1995;4:1055–61. doi: 10.1093/hmg/4.6.1055. [DOI] [PubMed] [Google Scholar]
- 63.Miyagoe Y, Hanaoka K, Nonaka I, Hayasaka M, Nabeshima Y, Arahata K, et al. Laminin alpha2 chain-null mutant mice by targeted disruption of the lama2 gene: A new model of merosin (laminin 2)-deficient congenital muscular dystrophy. FEBS Lett. 1997;415:33–9. doi: 10.1016/s0014-5793(97)01007-7. [DOI] [PubMed] [Google Scholar]
- 64.Kuang W, Xu H, Vilquin JT, Engvall E. Activation of the lama2 gene in muscle regeneration: Abortive regeneration in laminin alpha2-deficiency. Lab Invest. 1999;79:1601–13. [PubMed] [Google Scholar]
- 65.Patton BL, Wang B, Tarumi YS, Seburn KL, Burgess RW. A single point mutation in the ln domain of lama2 causes muscular dystrophy and peripheral amyelination. J Cell Sci. 2008;121(pt 10):1593–604. doi: 10.1242/jcs.015354. [DOI] [PubMed] [Google Scholar]
- 66.Mayer U, Saher G, Fässler R, Bornemann A, Echtermeyer F, von der Mark H, et al. Absence of integrin alpha 7 causes a novel form of muscular dystrophy. Nat Genet. 1997;17:318–23. doi: 10.1038/ng1197-318. [DOI] [PubMed] [Google Scholar]
- 67.Straub V, Duclos F, Venzke DP, Lee JC, Cutshall S, Leveille CJ, et al. Molecular pathogenesis of muscle degeneration in the delta-sarcoglycan-deficient hamster. Am J Pathol. 1998;153:1623–30. doi: 10.1016/s0002-9440(10)65751-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schessl J, Zou Y, Bonnemann CG. Congenital muscular dystrophies and the extracellular matrix. Semin Pediatr Neurol. 2006;13:80–9. doi: 10.1016/j.spen.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 69.Barresi R, Michele DE, Kanagawa M, Harper HA, Dovico SA, Satz JS, et al. LARGE can functionally bypass alpha-dystroglycan glycosylation defects in distinct congenital muscular dystrophies. Nat Med. 2004;10(7):696–703. doi: 10.1038/nm1059. [DOI] [PubMed] [Google Scholar]
- 70.Mayer U. Integrins: Redundant or important players in skeletal muscle? J Biol Chem. 2003;278:14587–90. doi: 10.1074/jbc.R200022200. [DOI] [PubMed] [Google Scholar]
- 71.Bloch RJ, Reed P, O’Neill A, Strong J, Williams M, Porter N, et al. Costameres mediate force transduction in healthy skeletal muscle and are altered in muscular dystrophies. J Muscle Res Cell Motil. 2004;25:590–2. [PubMed] [Google Scholar]
- 72.Ervasti JM, Campbell KP. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol. 1993;122:809–23. doi: 10.1083/jcb.122.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cohn RD, Campbell KP. Molecular basis of muscular dystrophies. Muscle Nerve. 2000;23:1456–71. doi: 10.1002/1097-4598(200010)23:10<1456::aid-mus2>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 74.Michele DE, Campbell KP. Dystrophin-glycoprotein complex: Post-translational processing and dystroglycan function. J Biol Chem. 2003;278:15457–60. doi: 10.1074/jbc.R200031200. [DOI] [PubMed] [Google Scholar]
- 75.Gawlik K, Miyagoe-Suzuki Y, Ekblom P, Takeda S, Durbeej M. Laminin {alpha}1 chain reduces muscular dystrophy in laminin {alpha}2 chain deficient mice. Hum Mol Genet. 2004;13(16):1775–84. doi: 10.1093/hmg/ddh190. [DOI] [PubMed] [Google Scholar]
- 76.Gawlik KI, Li JY, Petersen A, Durbeej M. Laminin alpha1 chain improves laminin alpha2 chain deficient peripheral neuropathy. Hum Mol Genet. 2006;15:2690–700. doi: 10.1093/hmg/ddl201. [DOI] [PubMed] [Google Scholar]
- 77.Meinen S, Barzaghi P, Lin S, Lochmuller H, Ruegg MA. Linker molecules between laminins and dystroglycan ameliorate laminin-alpha2-deficient muscular dystrophy at all disease stages. J Cell Biol. 2007;176:979–93. doi: 10.1083/jcb.200611152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Muntoni F, Brockington M, Brown SC. Glycosylation eases muscular dystrophy. Nat Med. 2004;10:676–7. doi: 10.1038/nm0704-676. [DOI] [PubMed] [Google Scholar]
- 79.Kammerer RA, Schulthess T, Landwehr R, Schumacher B, Lustig A, Yurchenco PD, et al. Interaction of agrin with laminin requires a coiled-coil conformation of the agrin-binding site within the laminin gamma1 chain. EMBO J. 1999;18:6762–70. doi: 10.1093/emboj/18.23.6762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bentzinger CF, Barzaghi P, Lin S, Ruegg MA. Overexpression of mini-agrin in skeletal muscle increases muscle integrity and regenerative capacity in laminin-{alpha}2-deficient mice. FASEB J. 2005;19:934–42. doi: 10.1096/fj.04-3376com. [DOI] [PubMed] [Google Scholar]
- 81.Eusebio A, Oliveri F, Barzaghi P, Ruegg MA. Expression of mouse agrin in normal, denervated and dystrophic muscle. Neuromuscul Disord. 2003;13:408–15. doi: 10.1016/s0960-8966(03)00036-1. [DOI] [PubMed] [Google Scholar]
- 82.Guo LT, Zhang XU, Kuang W, Xu H, Liu LA, Vilquin JT, et al. Laminin alpha2 deficiency and muscular dystrophy; genotype-phenotype correlation in mutant mice. Neuromuscul Disord. 2003;13:207–15. doi: 10.1016/s0960-8966(02)00266-3. [DOI] [PubMed] [Google Scholar]
- 83.Qiao C, Li J, Zhu T, Draviam R, Watkins S, Ye X, et al. Amelioration of laminin-{alpha}2-deficient congenital muscular dystrophy by somatic gene transfer of miniagrin. Proc Natl Acad Sci USA. 2005;102:11999–2004. doi: 10.1073/pnas.0502137102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gilbert JJ, Steinberg MC, Banker BQ. Ultrastructural alterations of the motor end plate in myotonic dystrophy of the mouse (dy2J/dy2J) J Neuropathol Exp Neurol. 1973;32:345–64. doi: 10.1097/00005072-197307000-00001. [DOI] [PubMed] [Google Scholar]
- 85.Law PK, Saito A, Fleischer S. Ultrastructural changes in muscle and motor end-plate of the dystrophic mouse. Exp Neurol. 1983;80:361–82. doi: 10.1016/0014-4886(83)90289-3. [DOI] [PubMed] [Google Scholar]
- 86.Yurchenco PD, Cheng YS, Campbell K, Li S. Loss of basement membrane, receptor and cytoskeletal lattices in a laminin-deficient muscular dystrophy. J Cell Sci. 2004;117:735–42. doi: 10.1242/jcs.00911. [DOI] [PubMed] [Google Scholar]
- 87.Colognato H, Yurchenco PD. The laminin alpha2 expressed by dystrophic dy(2J) mice is defective in its ability to form polymers. Curr Biol. 1999;9:1327–30. doi: 10.1016/s0960-9822(00)80056-1. [DOI] [PubMed] [Google Scholar]
- 88.Chen YY, McDonald D, Cheng C, Magnowski B, Durand J, Zochodne DW. Axon and Schwann cell partnership during nerve re-growth. J Neuropathol Exp Neurol. 2005;64:613–22. doi: 10.1097/01.jnen.0000171650.94341.46. [DOI] [PubMed] [Google Scholar]
- 89.Yang D, Bierman J, Tarumi YS, Zhong YP, Rangwala R, Proctor TM, et al. Coordinate control of axon defasciculation and myelination by laminin-2 and -8. J Cell Biol. 2005;168:655–66. doi: 10.1083/jcb.200411158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bradley WG, Jenkison M. Neural abnormalities in the dystrophic mouse. J Neurol Sci. 1975;25:249–55. doi: 10.1016/0022-510x(75)90144-6. [DOI] [PubMed] [Google Scholar]
- 91.Stirling CA. Abnormalities in Schwann cell sheaths in spinal nerve roots of dystrophic mice. J Anat. 1975;119:169–80. [PMC free article] [PubMed] [Google Scholar]
- 92.Stirling CA. Experimental myelination in dystrophic mice. J Physiol. 1975;245:36P. [PubMed] [Google Scholar]
- 93.Madrid RE, Jaros E, Cullen MJ, Bradley WG. Genetically determined defect of Schwann cell basement membrane in dystrophic mouse. Nature. 1975;257:319–21. doi: 10.1038/257319a0. [DOI] [PubMed] [Google Scholar]
- 94.Patton BL, Miner JH, Chiu AY, Sanes JR. Distribution and function of laminins in the neuromuscular system of developing, adult, and mutant mice. J Cell Biol. 1997;139:1507–21. doi: 10.1083/jcb.139.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Patton BL, Connoll AM, Martin PT, Cunningham JM, Mehta S, Pestronk A, et al. Distribution of ten laminin chains in dystrophic and regenerating muscles. Neuromuscul Disord. 1999;9:423–33. doi: 10.1016/s0960-8966(99)00033-4. [DOI] [PubMed] [Google Scholar]
- 96.Bunge RP, Bunge MB, Eldridge CF. Linkage between axonal ensheathment and basal lamina production by Schwann cells. Annu Rev Neurosci. 1986;9:305–28. doi: 10.1146/annurev.ne.09.030186.001513. [DOI] [PubMed] [Google Scholar]
- 97.Obremski VJ, Johnson MI, Bunge MB. Fibroblasts are required for Schwann cell basal lamina deposition and ensheathment of unmyelinated sympathetic neurites in culture. J Neurocytol. 1993;22:102–17. doi: 10.1007/BF01181574. [DOI] [PubMed] [Google Scholar]
- 98.Obremski VJ, Wood PM, Bunge MB. Fibroblasts promote Schwann cell basal lamina deposition and elongation in the absence of neurons in culture. Dev Biol. 1993;160:119–34. doi: 10.1006/dbio.1993.1291. [DOI] [PubMed] [Google Scholar]
- 99.Obremski VJ, Bunge MB. Addition of purified basal lamina molecules enables Schwann cell ensheathment of sympathetic neurites in culture. Dev Biol. 1995;168:124–37. doi: 10.1006/dbio.1995.1066. [DOI] [PubMed] [Google Scholar]
- 100.Wallquist W, Plantman S, Thams S, Thyboll J, Kortesmaa J, Lannergren J, et al. Impeded interaction between Schwann cells and axons in the absence of laminin alpha4. J Neurosci. 2005;25:3692–700. doi: 10.1523/JNEUROSCI.5225-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Feltri ML, Porta DG, Previtali SC, Nodari A, Migliavacca B, Cassetti A, et al. Conditional disruption of {beta}1 integrin in Schwann cells impedes interactions with axons. J Cell Biol. 2002;156:199–210. doi: 10.1083/jcb.200109021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yu WM, Feltri ML, Wrabetz L, Strickland S, Chen ZL. Schwann cell-specific ablation of laminin gamma1 causes apoptosis and prevents proliferation. J Neurosci. 2005;25:4463–72. doi: 10.1523/JNEUROSCI.5032-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Saito F, Moore SA, Barresi R, Henry MD, Messing A, Ross-Barta SE, et al. Unique role of dystroglycan in peripheral nerve myelination, nodal structure, and sodium channel stabilization. Neuron. 2003;38:747–58. doi: 10.1016/s0896-6273(03)00301-5. [DOI] [PubMed] [Google Scholar]
- 104.Chernousov MA, Kaufman SJ, Stahl RC, Rothblum K, Carey DJ. Alpha7beta1 integrin is a receptor for laminin-2 on Schwann cells. Glia. 2007;55:1134–44. doi: 10.1002/glia.20536. [DOI] [PubMed] [Google Scholar]
- 105.Previtali SC, Dina G, Nodari A, Fasolini M, Wrabetz L, Mayer U, et al. Schwann cells synthesize alpha7beta1 integrin which is dispensable for peripheral nerve development and myelination. Mol Cell Neurosci. 2003;23:210–8. doi: 10.1016/s1044-7431(03)00014-9. [DOI] [PubMed] [Google Scholar]
- 106.Previtali SC, Nodari A, Taveggia C, Pardini C, Dina G, Villa A, et al. Expression of laminin receptors in Schwann cell differentiation: Evidence for distinct roles. J Neurosci. 2003;23:5520–30. doi: 10.1523/JNEUROSCI.23-13-05520.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Timpl R, Wiedemann H, van Delden V, Furthmayr H, Kuhn K. A network model for the organization of type IV collagen molecules in basement membranes. Eur J Biochem. 1981;120:203–11. doi: 10.1111/j.1432-1033.1981.tb05690.x. [DOI] [PubMed] [Google Scholar]
- 108.Oberbaumer I, Wiedemann H, Timpl R, Kuhn K. Shape and assembly of type IV procollagen obtained from cell culture. EMBO J. 1982;1:805–10. doi: 10.1002/j.1460-2075.1982.tb01251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yurchenco PD, Furthmayr H. Self-assembly of basement membrane collagen. Biochemistry. 1984;23:1839–50. doi: 10.1021/bi00303a040. [DOI] [PubMed] [Google Scholar]
- 110.Glanville RW, Qian RQ, Siebold B, Risteli J, Kuhn K. Amino acid sequence of the n-terminal aggregation and cross-linking region (7S domain) of the alpha 1 (IV) chain of human basement membrane collagen. Eur J Biochem. 1985;152:213–9. doi: 10.1111/j.1432-1033.1985.tb09186.x. [DOI] [PubMed] [Google Scholar]
- 111.Yurchenco PD, Furthmayr H. Type IV collagen “7S” Tetramer formation: Aspects of kinetics and thermodynamics. Ann NY Acad Sci. 1986;460:530–3. [Google Scholar]
- 112.Siebold B, Qian RA, Glanville RW, Hofmann H, Deutzmann R, Kuhn K. Construction of a model for the aggregation and cross-linking region (7S domain) of type IV collagen based upon an evaluation of the primary structure of the alpha 1 and alpha 2 chains in this region. Eur J Biochem. 1987;168:569–75. doi: 10.1111/j.1432-1033.1987.tb13455.x. [DOI] [PubMed] [Google Scholar]
- 113.Bachinger HP, Fessler LI, Fessler JH. Mouse procollagen IV. Characterization and supramolecular association. J Biol Chem. 1982;257:9796–803. [PubMed] [Google Scholar]
- 114.Duncan KG, Fessler LI, Bachinger HP, Fessler JH. Procollagen IV. Association to tetramers. J Biol Chem. 1983;258:5869–77. [PubMed] [Google Scholar]
- 115.Weber S, Engel J, Wiedemann H, Glanville RW, Timpl R. Subunit structure and assembly of the globular domain of basement-membrane collagen type IV. Eur J Biochem. 1984;139:401–10. doi: 10.1111/j.1432-1033.1984.tb08019.x. [DOI] [PubMed] [Google Scholar]
- 116.Sundaramoorthy M, Meiyappan M, Todd P, Hudson BG. Crystal structure of NC1 domains: Structural basis for type IV collagen assembly in basement membranes. J Biol Chem. 2002;22:22. doi: 10.1074/jbc.M201740200. [DOI] [PubMed] [Google Scholar]
- 117.Than ME, Henrich S, Huber R, Ries A, Mann K, Kuhn K, et al. The 1.9-Å crystal structure of the noncollagenous (NC1) domain of human placenta collagen IV shows stabilization via a novel type of covalent met-lys cross-link. Proc Natl Acad Sci USA. 2002;99:6607–12. doi: 10.1073/pnas.062183499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Vanacore RM, Shanmugasundararaj S, Friedman DB, Bondar O, Hudson BG, Sundaramoorthy M. The alpha1alpha2 network of collagen IV. Reinforced stabilization of the noncollagenous domain-1 by noncovalent forces and the absence of met-lys cross-links. J Biol Chem. 2004;279:44723–30. doi: 10.1074/jbc.M406344200. [DOI] [PubMed] [Google Scholar]
- 119.Vanacore RM, Friedman DB, Ham AJ, Sundaramoorthy M, Hudson BG. Identification of s-hydroxylysyl-methionine as the covalent cross-link of the noncollagenous (NC1) hexamer of the alpha1alpha1alpha2 collagen IV network: A role for the post-translational modification of lysine 211 to hydroxylysine 211 in hexamer assembly. J Biol Chem. 2005;280:29300–10. doi: 10.1074/jbc.M502752200. [DOI] [PubMed] [Google Scholar]
- 120.Gunwar S, Ballester F, Kalluri R, Timoneda J, Chonko AM, Edwards SJ, et al. Glomerular basement membrane. Identification of dimeric subunits of the noncollagenous domain (hexamer) of collagen IV and the Goodpasture antigen. J Biol Chem. 1991;266:15318–24. [PubMed] [Google Scholar]
- 121.Netzer KO, Suzuki K, Itoh Y, Hudson BG, Khalifah RG. Comparative analysis of the noncollagenous NC1 domain of type IV collagen: Identification of structural features important for assembly, function, and pathogenesis. Protein Sci. 1998;7:1340–51. doi: 10.1002/pro.5560070610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Boutaud A, Borza DB, Bondar O, Gunwar S, Netzer KO, Singh N, et al. Type IV collagen of the glomerular basement membrane: Evidence that the chain specificity of network assembly is encoded by the noncollagenous NC1 domains. J Biol Chem. 2000;275(39):30716–24. doi: 10.1074/jbc.M004569200. [DOI] [PubMed] [Google Scholar]
- 123.Gunwar S, Ballester F, Noelken ME, Sado Y, Ninomiya Y, Hudson BG. Glomerular basement membrane. Identification of a novel disulfide-cross-linked network of alpha3, alpha4, and alpha5 chains of type IV collagen and its implications for the pathogenesis of alport syndrome. J Biol Chem. 1998;273:8767–75. doi: 10.1074/jbc.273.15.8767. [DOI] [PubMed] [Google Scholar]
- 124.Miner JH, Sanes JR. Molecular and functional defects in kidneys of mice lacking collagen alpha 3(IV): Implications for alport syndrome. J Cell Biol. 1996;135:1403–13. doi: 10.1083/jcb.135.5.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Guo XD, Johnson JJ, Kramer JM. Embryonic lethality caused by mutations in basement membrane collagen of C. elegans. Nature. 1991;349:707–9. doi: 10.1038/349707a0. [DOI] [PubMed] [Google Scholar]
- 126.Guo XD, Kramer JM. The two Caenorhabditis elegans basement membrane (type IV) collagen genes are located on separate chromosomes. J Biol Chem. 1989;264:17574–82. [PubMed] [Google Scholar]
- 127.Huang CC, Hall DH, Hedgecock EM, Kao G, Karantza V, Vogel BE, et al. Laminin alpha subunits and their role in c. Elegans development. Development. 2003;130:3343–58. doi: 10.1242/dev.00481. [DOI] [PubMed] [Google Scholar]
- 128.Nagai N, Hosokawa M, Itohara S, Adachi E, Matsushita T, Hosokawa N, et al. Embryonic lethality of molecular chaperone hsp47 knockout mice is associated with defects in collagen biosynthesis. J Cell Biol. 2000;150:1499–506. doi: 10.1083/jcb.150.6.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Koide T, Takahara Y, Asada S, Nagata K. Xaa-arg-gly triplets in the collagen triple helix are dominant binding sites for the molecular chaperone hsp47. J Biol Chem. 2002;277:6178–82. doi: 10.1074/jbc.M106497200. [DOI] [PubMed] [Google Scholar]
- 130.Hudson BG, Tryggvason K, Sundaramoorthy M, Neilson EG. Alport’s syndrome, Goodpasture’s syndrome, and type IV collagen. N Engl J Med. 2003;348:2543–56. doi: 10.1056/NEJMra022296. [DOI] [PubMed] [Google Scholar]
- 131.Gould DB, Phalan FC, Breedveld GJ, van Mil SE, Smith RS, Schimenti JC, et al. Mutations in col4a1 cause perinatal cerebral hemorrhage and porencephaly. Science. 2005;308:1167–71. doi: 10.1126/science.1109418. [DOI] [PubMed] [Google Scholar]
- 132.Gould DB, Phalan FC, van Mil SE, Sundberg JP, Vahedi K, Massin P, et al. Role of col4a1 in small-vessel disease and hemorrhagic stroke. N Engl J Med. 2006;354:1489–96. doi: 10.1056/NEJMoa053727. [DOI] [PubMed] [Google Scholar]
- 133.Durkin ME, Bartos BB, Liu SH, Phillips SL, Chung AE. Primary structure of the mouse laminin b2 chain and comparison with laminin B1. Biochemistry. 1988;27:5198–204. doi: 10.1021/bi00414a038. [DOI] [PubMed] [Google Scholar]
- 134.Fox JW, Mayer U, Nischt R, Aumailley M, Reinhardt D, Wiedemann H, et al. Recombinant nidogen consists of three globular domains and mediates binding of laminin to collagen type IV. EMBO J. 1991;10:3137–46. doi: 10.1002/j.1460-2075.1991.tb04875.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mann K, Deutzmann R, Aumailley M, Timpl R, Raimondi L, Yamada Y, et al. Amino acid sequence of mouse nidogen, a multidomain basement membrane protein with binding activity for laminin, collagen IV and cells. EMBO J. 1989;8:65–72. doi: 10.1002/j.1460-2075.1989.tb03349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kohfeldt E, Sasaki T, Gohring W, Timpl R. Nidogen-2: A new basement membrane protein with diverse binding properties. J Mol Biol. 1998;282:99–109. doi: 10.1006/jmbi.1998.2004. [DOI] [PubMed] [Google Scholar]
- 137.Kimura N, Toyoshima T, Kojima T, Shimane M. Entactin-2: A new member of basement membrane protein with high homology to entactin/nidogen. Exp Cell Res. 1998;241:36–45. doi: 10.1006/excr.1998.4016. [DOI] [PubMed] [Google Scholar]
- 138.Salmivirta K, Talts J, Olsson M, Sasaki T, Timpl R, Ekblom P. Binding of mouse nidogen-2 to basement membrane components and cells and its expression in embryonic and adult tissues suggest complementary functions of the two nidogens. Exp Cell Res. 2002;279:188. doi: 10.1006/excr.2002.5611. [DOI] [PubMed] [Google Scholar]
- 139.Takagi J, Yang Y, Liu JH, Wang JH, Springer TA. Complex between nidogen and laminin fragments reveals a paradigmatic beta-propeller interface. Nature. 2003;424:969–74. doi: 10.1038/nature01873. [DOI] [PubMed] [Google Scholar]
- 140.Ries A, Gohring W, Fox JW, Timpl R, Sasaki T. Recombinant domains of mouse nidogen-1 and their binding to basement membrane proteins and monoclonal antibodies. Eur J Biochem. 2001;268:5119–28. doi: 10.1046/j.0014-2956.2001.02437.x. [DOI] [PubMed] [Google Scholar]
- 141.Brown JC, Sasaki T, Gohring W, Yamada Y, Timpl R. The c-terminal domain v of perlecan promotes beta1 integrin-mediated cell adhesion, binds heparin, nidogen and fibulin-2 and can be modified by glycosaminoglycans. Eur J Biochem. 1997;250:39–46. doi: 10.1111/j.1432-1033.1997.t01-1-00039.x. [DOI] [PubMed] [Google Scholar]
- 142.Hopf M, Gohring W, Kohfeldt E, Yamada Y, Timpl R. Recombinant domain IV of perlecan binds to nidogens, laminin-nidogen complex, fibronectin, fibulin-2 and heparin. Eur J Biochem. 1999;259:917–25. doi: 10.1046/j.1432-1327.1999.00127.x. [DOI] [PubMed] [Google Scholar]
- 143.Ackley BD, Crew JR, Elamaa H, Pihlajaniemi T, Kuo CJ, Kramer JM. The NC1/endostatin domain of Caenorhabditis elegans type xviii collagen affects cell migration and axon guidance. J Cell Biol. 2001;152:1219–32. doi: 10.1083/jcb.152.6.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ackley BD, Kang SH, Crew JR, Suh C, Jin Y, Kramer JM. The basement membrane components nidogen and type XVIII collagen regulate organization of neuromuscular junctions in caenorhabditis elegans. J Neurosci. 2003;23:3577–87. doi: 10.1523/JNEUROSCI.23-09-03577.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kim S, Wadsworth WG. Positioning of longitudinal nerves in C. elegans by nidogen. Science. 2000;288:150–4. doi: 10.1126/science.288.5463.150. [DOI] [PubMed] [Google Scholar]
- 146.Kang SH, Kramer JM. Nidogen is nonessential and not required for normal type IV collagen localization in caenorhabditis elegans. Mol Biol Cell. 2000;11:3911–23. doi: 10.1091/mbc.11.11.3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Bader BL, Smyth N, Nedbal S, Miosge N, Baranowsky A, Mokkapati S, et al. Compound genetic ablation of nidogen 1 and 2 causes basement membrane defects and perinatal lethality in mice. Mol Cell Biol. 2005;25:6846–56. doi: 10.1128/MCB.25.15.6846-6856.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Burgess RW, Skarnes WC, Sanes JR. Agrin isoforms with distinct amino termini: Differential expression, localization, and function. J Cell Biol. 2000;151:41–52. doi: 10.1083/jcb.151.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Yamada H, Denzer AJ, Hori H, Tanaka T, Anderson LV, Fujita S, et al. Dystroglycan is a dual receptor for agrin and laminin-2 in Schwann cell membrane. J Biol Chem. 1996;271:23418–23. doi: 10.1074/jbc.271.38.23418. [DOI] [PubMed] [Google Scholar]
- 150.Denzer AJ, Brandenberger R, Gesemann M, Chiquet M, Ruegg MA. Agrin binds to the nerve-muscle basal lamina via laminin. J Cell Biol. 1997;137:671–83. doi: 10.1083/jcb.137.3.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Denzer AJ, Schulthess T, Fauser C, Schumacher B, Kammerer RA, Engel J, et al. Electron microscopic structure of agrin and mapping of its binding site in laminin-1. EMBO J. 1998;17:335–43. doi: 10.1093/emboj/17.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Gesemann M, Brancaccio A, Schumacher B, Ruegg MA. Agrin is a high-affinity binding protein of dystroglycan in non-muscle tissue. J Biol Chem. 1998;273:600–5. doi: 10.1074/jbc.273.1.600. [DOI] [PubMed] [Google Scholar]
- 153.Noonan DM, Fulle A, Valente P, Cai S, Horigan E, Sasaki M, et al. The complete sequence of perlecan, a basement membrane heparan sulfate proteoglycan, reveals extensive similarity with laminin a chain, low density lipoprotein-receptor, and the neural cell adhesion molecule. J Biol Chem. 1991;266:22939–47. [PubMed] [Google Scholar]
- 154.Murdoch AD, Dodge GR, Cohen I, Tuan RS, Iozzo RV. Primary structure of the human heparan sulfate proteoglycan from basement membrane (HSPG2/perlecan). A chimeric molecule with multiple domains homologous to the low density lipoprotein receptor, laminin, neural cell adhesion molecules, and epidermal growth factor. J Biol Chem. 1992;267:8544–57. [PubMed] [Google Scholar]
- 155.Kallunki P, Tryggvason K. Human basement membrane heparan sulfate proteoglycan core protein: A 467-Kd protein containing multiple domains resembling elements of the low density lipoprotein receptor, laminin, neural cell adhesion molecules, and epidermal growth factor. J Cell Biol. 1992;116:559–71. doi: 10.1083/jcb.116.2.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Bix G, Fu J, Gonzalez EM, Macro L, Barker A, Campbell S, et al. Endorepellin causes endothelial cell disassembly of actin cytoskeleton and focal adhesions through alpha2beta1 integrin. J Cell Biol. 2004;166:97–109. doi: 10.1083/jcb.200401150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Mongiat M, Sweeney SM, San Antonio JD, Fu J, Iozzo RV. Endorepellin, a novel inhibitor of angiogenesis derived from the c terminus of perlecan. J Biol Chem. 2003;278:4238–49. doi: 10.1074/jbc.M210445200. [DOI] [PubMed] [Google Scholar]
- 158.Costell M, Gustafsson E, Aszodi A, Morgelin M, Bloch W, Hunziker E, et al. Perlecan maintains the integrity of cartilage and some basement membranes. J Cell Biol. 1999;147:1109–22. doi: 10.1083/jcb.147.5.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Arikawa-Hirasawa E, Watanabe H, Takami H, Hassell JR, Yamada Y. Perlecan is essential for cartilage and cephalic development. Nat Genet. 1999;23:354–8. doi: 10.1038/15537. [DOI] [PubMed] [Google Scholar]
- 160.Arikawa-Hirasawa E, Le AH, Nishino I, Nonaka I, Ho NC, Francomano CA, et al. Structural and functional mutations of the perlecan gene cause Schwartz-Jampel syndrome, with myotonic myopathy and chondrodysplasia. Am J Hum Genet. 2002;70:5. doi: 10.1086/340390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Nicole S, Davoine CS, Topaloglu H, Cattolico L, Barral D, Beighton P, et al. Perlecan, the major proteoglycan of basement membranes, is altered in patients with Schwartz-Jampel syndrome (chondrodystrophic myotonia) Nat Genet. 2000;26:480–3. doi: 10.1038/82638. [DOI] [PubMed] [Google Scholar]
- 162.Peng HB, Xie H, Rossi SG, Rotundo RL. Acetylcholinesterase clustering at the neuromuscular junction involves perlecan and dystroglycan. J Cell Biol. 1999;145:911–21. doi: 10.1083/jcb.145.4.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kvist AJ, Johnson AE, Morgelin M, Gustafsson E, Bengtsson E, Lindblom K, et al. Chondroitin sulfate perlecan enhances collagen fibril formation. Implications for perlecan chondrodysplasias. J Biol Chem. 2006;281:33127–39. doi: 10.1074/jbc.M607892200. [DOI] [PubMed] [Google Scholar]
- 164.Brittingham R, Uitto J, Fertala A. High-affinity binding of the NC1 domain of collagen vii to laminin 5 and collagen IV. Biochem Biophys Res Commun. 2006;343:692–9. doi: 10.1016/j.bbrc.2006.03.034. [DOI] [PubMed] [Google Scholar]
- 165.Villone D, Fritsch A, Koch M, Bruckner-Tuderman L, Hansen U, Bruckner P. Supramolecular interactions in the dermoepidermal junction zone: Anchoring fibril-collagen VII tightly binds to banded collagen fibrils. J Biol Chem. 2008;283:24506–13. doi: 10.1074/jbc.M802415200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Nakashima Y, Kariya Y, Yasuda C, Miyazaki K. Regulation of cell adhesion and type vii collagen binding by the beta3 chain short arm of laminin-5: Effect of its proteolytic cleavage. J Biochem. 2005;138:539–52. doi: 10.1093/jb/mvi153. [DOI] [PubMed] [Google Scholar]
- 167.Dang N, Murrell DF. Mutation analysis and characterization of col7a1 mutations in dystrophic epidermolysis bullosa. Exp Dermatol. 2008;17:553–68. doi: 10.1111/j.1600-0625.2008.00723.x. [DOI] [PubMed] [Google Scholar]
- 168.Lampe AK, Bushby KM. Collagen VI related muscle disorders. J Med Genet. 2005;42:673–85. doi: 10.1136/jmg.2002.002311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Myers JC, Amenta PS, Dion AS, Sciancalepore JP, Nagaswami C, Weisel JW, et al. The molecular structure of human tissue type XV presents a unique conformation among the collagens. Biochem J. 2007 doi: 10.1042/BJ20070201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Eklund L, Piuhola J, Komulainen J, Sormunen R, Ongvarrasopone C, Fässler R, et al. Lack of type XV collagen causes a skeletal myopathy and cardiovascular defects in mice. Proc Natl Acad Sci USA. 2001;98:1194–9. doi: 10.1073/pnas.031444798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Marneros AG, Olsen BR. Physiological role of collagen XVIII and endostatin. FASEB J. 2005;19:716–28. doi: 10.1096/fj.04-2134rev. [DOI] [PubMed] [Google Scholar]
- 172.Fukai N, Eklund L, Marneros AG, Oh SP, Keene DR, Tamarkin L, et al. Lack of collagen xviii/endostatin results in eye abnormalities. EMBO J. 2002;21:1535–44. doi: 10.1093/emboj/21.7.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Eble JA, Ries A, Lichy A, Mann K, Stanton H, Gavrilovic J, et al. The recognition sites of the integrins alpha1beta1 and alpha2beta1 within collagen IV are protected against gelatinase a attack in the native protein. J Biol Chem. 1996;271:30964–70. doi: 10.1074/jbc.271.48.30964. [DOI] [PubMed] [Google Scholar]
- 174.Colognato-Pyke H, O’Rear JJ, Yamada Y, Carbonetto S, Cheng YS, Yurchenco PD. Mapping of network-forming, heparin-binding, and alpha 1 beta 1 integrin-recognition sites within the alpha-chain short arm of laminin- 1. J Biol Chem. 1995;270:9398–406. doi: 10.1074/jbc.270.16.9398. [DOI] [PubMed] [Google Scholar]
- 175.Colognato H, MacCarrick M, O’Rear JJ, Yurchenco PD. The laminin alpha2-chain short arm mediates cell adhesion through both the alpha1beta1 and alpha2beta1 integrins. J Biol Chem. 1997;272:29330–6. doi: 10.1074/jbc.272.46.29330. [DOI] [PubMed] [Google Scholar]
- 176.Kortesmaa J, Yurchenco P, Tryggvason K. Recombinant laminin-8 (alpha 4beta 1gamma 1). Production, purification, and interactions with integrins. J Biol Chem. 2000;275:14853–9. doi: 10.1074/jbc.275.20.14853. [DOI] [PubMed] [Google Scholar]
- 177.Sasaki T, Timpl R. Domain IVa of laminin alpha5 chain is cell-adhesive and binds beta1 and alphavbeta3 integrins through arg-gly-asp. FEBS Lett. 2001;509:181–5. doi: 10.1016/s0014-5793(01)03167-2. [DOI] [PubMed] [Google Scholar]
- 178.Pall EA, Bolton KM, Ervasti JM. Differential heparin inhibition of skeletal muscle alpha-dystroglycan binding to laminins. J Biol Chem. 1996;271:3817–21. doi: 10.1074/jbc.271.7.3817. [DOI] [PubMed] [Google Scholar]
- 179.Smirnov SP, McDearmon EL, Li S, Ervasti JM, Tryggvason K, Yurchenco PD. Contributions of the lg modules and furin processing to laminin-2 functions. J Biol Chem. 2002;277:18928–37. doi: 10.1074/jbc.M201880200. [DOI] [PubMed] [Google Scholar]
- 180.Ferletta M, Kikkawa Y, Yu H, Talts JF, Durbeej M, Sonnenberg A, et al. Opposing roles of integrin alpha6abeta1 and dystroglycan in laminin-mediated extracellular signal-regulated kinase activation. Mol Biol Cell. 2003;14:2088–103. doi: 10.1091/mbc.E03-01-0852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Friedrich MV, Gohring W, Morgelin M, Brancaccio A, David G, Timpl R. Structural basis of glycosaminoglycan modification and of heterotypic interactions of perlecan domain v. J Mol Biol. 1999;294:259–70. doi: 10.1006/jmbi.1999.3259. [DOI] [PubMed] [Google Scholar]
- 182.Talts JF, Sasaki T, Miosge N, Gohring W, Mann K, Mayne R, et al. Structural and functional analysis of the recombinant g domain of the laminin {alpha}4 chain and its proteolytic processing in tissues. J Biol Chem. 2000;275(45):35192–9. doi: 10.1074/jbc.M003261200. [DOI] [PubMed] [Google Scholar]
- 183.Michele DE, Barresi R, Kanagawa M, Saito F, Cohn RD, Satz JS, et al. Post-translational disruption of dystroglycan ligand interactions in congenital muscular dystrophies. Nature. 2002;418:417–21. doi: 10.1038/nature00837. [DOI] [PubMed] [Google Scholar]
- 184.Yu H, Talts JF. Beta1 integrin and alpha-dystroglycan binding sites are localized to different laminin-G-domain-like (LG) modules within the laminin alpha5 chain G domain. Biochem J. 2003;371:289–99. doi: 10.1042/BJ20021500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Sakashita S, Engvall E, Ruoslahti E. Basement membrane glycoprotein laminin binds to heparin. FEBS Lett. 1980;116:243–46. doi: 10.1016/0014-5793(80)80654-5. [DOI] [PubMed] [Google Scholar]
- 186.Sallum CO, Kammerer RA, Alexandrescu AT. Thermodynamic and structural studies of carbohydrate binding by the agrin-G3 domain. Biochemistry. 2007;44(33):9541–50. doi: 10.1021/bi7006383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Roberts DD, Rao CN, Magnani JL, Spitalnik SL, Liotta LA, Ginsburg V. Laminin binds specifically to sulfated glycolipids. Proc Natl Acad Sci USA. 1985;82:1306–10. doi: 10.1073/pnas.82.5.1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Kalb E, Engel J. Binding and calcium-induced aggregation of laminin onto lipid bilayers. J Biol Chem. 1991;266:19047–52. [PubMed] [Google Scholar]
- 189.Talts JF, Andac Z, Gorhing W, Brancaccio A, Timpl R. Binding of the G domains of laminin alpha1 and alpha2 chains and perlecan to heparin, sulfatides, alpha-dystroglycan and several extracellular matrix proteins. EMBO J. 1999;18:863–70. doi: 10.1093/emboj/18.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Talts JF, Timpl R. Mutation of a basic sequence in the laminin alpha2LG3 module leads to a lack of proteolytic processing and has different effects on beta1 integrin-mediated cell adhesion and alpha-dystroglycan binding. FEBS Lett. 1999;458:319–23. doi: 10.1016/s0014-5793(99)01180-1. [DOI] [PubMed] [Google Scholar]
- 191.Hopf M, Gohring W, Mann K, Timpl R. Mapping of binding sites for nidogens, fibulin-2, fibronectin and heparin to different IG modules of perlecan. J Mol Biol. 2001;311:529–41. doi: 10.1006/jmbi.2001.4878. [DOI] [PubMed] [Google Scholar]
- 192.Hall H, Vorherr T, Schachner M. Characterization of a 21 amino acid peptide sequence of the laminin G2 domain that is involved in hnk-1 carbohydrate binding and cell adhesion. Glycobiology. 1995;5:435–41. doi: 10.1093/glycob/5.4.435. [DOI] [PubMed] [Google Scholar]
- 193.Elola MT, Chiesa ME, Alberti AF, Mordoh J, Fink NE. Galectin-1 receptors in different cell types. J Biomed Sci. 2005;12:13–29. doi: 10.1007/s11373-004-8169-5. [DOI] [PubMed] [Google Scholar]
- 194.Shrivastava A, Radziejewski C, Campbell E, Kovac L, McGlynn M, Ryan TE, et al. An orphan receptor tyrosine kinase family whose members serve as nonintegrin collagen receptors. Mol Cell. 1997;1:25–34. doi: 10.1016/s1097-2765(00)80004-0. [DOI] [PubMed] [Google Scholar]
- 195.Meyer zum Gottesberge AM, Gross O, Becker-Lendzian U, Massing T, Vogel WF. Inner ear defects and hearing loss in mice lacking the collagen receptor DDR1. Lab Invest. 2008;88:27–37. doi: 10.1038/labinvest.3700692. [DOI] [PubMed] [Google Scholar]
- 196.Gross O, Beirowski B, Harvey SJ, McFadden C, Chen D, Tam S, et al. DDR1-deficient mice show localized subepithelial GBM thickening with focal loss of slit diaphragms and proteinuria. Kidney Int. 2004;66:102–11. doi: 10.1111/j.1523-1755.2004.00712.x. [DOI] [PubMed] [Google Scholar]
- 197.Lauer-Fields JL, Malkar NB, Richet G, Drauz K, Fields GB. Melanoma cell cd44 interaction with the alpha 1(IV)1263–1277 region from basement membrane collagen is modulated by ligand glycosylation. J Biol Chem. 2003;278:14321–30. doi: 10.1074/jbc.M212246200. [DOI] [PubMed] [Google Scholar]
- 198.Begovac PC, Hall DE, Shur BD. Laminin fragment E8 mediates PC12 cell neurite outgrowth by binding to cell surface beta 1,4 galactosyltransferase. J Cell Biol. 1991;113:637–44. doi: 10.1083/jcb.113.3.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Yurchenco PD, Smirnov S, Mathus T. In: Methods in cell-matrix adhesion. Adams J, editor. Vol. 69. 2002. pp. 107–40. [Google Scholar]
- 200.Sung U, O’Rear JJ, Yurchenco PD. Cell and heparin binding in the distal long arm of laminin: Identification of active and cryptic sites with recombinant and hybrid glycoprotein. J Cell Biol. 1993;123:1255–68. doi: 10.1083/jcb.123.5.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Borza DB, Bondar O, Ninomiya Y, Sado Y, Naito I, Todd P, et al. The NC1 domain of collagen IV encodes a novel network composed of the alpha 1, alpha 2, alpha 5, and alpha 6 chains in smooth muscle basement membranes. J Biol Chem. 2001;276:28532–40. doi: 10.1074/jbc.M103690200. [DOI] [PubMed] [Google Scholar]