

Abstract
Insensitivity to pain is a rare disorder that is commonly associated with Hereditary and Sensory Autonomic Neuropathies (HSAN I –V) resulting often in autonomic dysfunction and premature death. Very few individuals have been reported with pain insensitivity lacking such autonomic neuropathies. We performed genetic, neurologic, psychological, and psychophysical evaluations in such an individual (OMIM 243000) and her first degree relatives. Sequence analysis of genomic DNA revealed two novel SCN9A mutations in this index case (IC). One was a non-conservative missense mutation (C1719R) in exon 26 present only in the IC and one parent. Further sequence analysis of the child’s DNA revealed a 1-bp splice donor deletion in intron 17 which was also present in the other parent and one sibling. Detailed psychophysical testing was used to phenotypically characterize the IC, her family members, and 10 matched normal controls. Similar to family members and controls the IC showed normal somatosensory functioning for non-nociceptive mechanoreception and warmth. However, she demonstrated diminished ability to detect cool temperatures combined with profound deficits in heat and mechanical nociception. Congenital insensitivity to pain in our IC was associated with two novel SCN9A mutations which most likely resulted in a Nav1.7 channelopathy. However, in contrast to individuals with other SCN9A mutations, the observed pain insensitivity was relative and not absolute, which may be consistent with hypomorphic effects of one or both mutations. The ability to sense at least some danger signals may be advantageous and ameliorate the otherwise increased morbidity and mortality of some individuals with congenital insensitivity to pain.
Keywords: Mutation, SCN9A, Pain Insensitivity
1. Introduction
The experience of “pain” as an unpleasant sensation associated with tissue damage is common to most complex organisms to minimize injuries, and is essential to long-term survival. Exposure to painful stimuli results in adaptive behaviors that can be both reflexive and deliberate to remove the affected body areas from potentially dangerous environments. Pain also provides an important learning opportunity, helping to avoid dangerous situations or behaviors in the future. Pain is a complex sensory and emotional experience that is served by multiple types of primary nociceptive afferents, central pathways, and brain regions (Talbot et al., 1991; Price 1999). Thus, individuals who are congenitally insensitive or indifferent to pain may express deficiencies in different components of pain processing and experience.
Most of these individuals can be diagnosed with one of several Hereditary Sensory and Autonomic Neuropathies (HSAN I to IV) (Klein et al., 2005). The majority of these patients have HSAN I which is autosomal dominant and usually becomes clinically apparent in early adulthood. HSAN II to V are autosomal recessive and usually develop in childhood and can be associated with abnormalities of nociceptive afferents, often in conjunction with autonomic nervous system dysfunction. Developmental abnormalities are rare and are mostly found in HSAN IV. In contrast, individuals of one subgroup (HSAN V) appear to have normal nociceptive afferents without sensory or autonomic deficits.
The first patient with a congenital inability to perceive pain was reported in the early twentieth century (Dearborn 1932) and subsequently only few additional cases have been published. Recently, however, six individuals from three Pakistani families have been reported with congenital inability to perceive any form of pain, in whom all other sensory modalities were intact (Cox et al., 2006). Using positional cloning, genetic analysis of these individuals showed loss of function mutations of the voltage-gated sodium channel gene SCN9A revealing three distinct homozygous nonsense mutations (S459X, I767X and W897X) (Cox et al., 2006). Detailed characterizations showed that these mutations caused loss of function of Nav1.7 resulting in activation currents not greater than background. However, only limited examinations have been made of specific somatosensory deficits in such individuals, including sensitivity to warmth, heat pain, cold or pressure (Nilsen et al., 2009; Goldberg et al., 2007; Ahmad et al., 2007). Here we characterize the somatosensory, psychological, neurological, and genetic abnormalities of a young child described by her parents as insensitive to pain since birth and compare her to her first-degree family member and normal controls (NC). Her condition provided the unique opportunity to apply genetic as well as psychological and standardized psychophysical evaluations (Price 1988; Price and Harkins 1992; Staud et al., 2010; Price et al., 2008) to precisely characterize the abnormalities associated with her clinical condition. Normal pain-free controls were used for comparisons.
2. Methods
2.1 Participants
The IC is a 9 year old female who comes from a non-consanguineous Caucasian family of Northern European descent. Her parents and grandparents are alive and healthy. Her parents as well as her 2 siblings (1 female, 1 male) were also evaluated. The normal pain-free controls (NC) consisted of 9 females and 1 male subject [mean (SD) age: 23.44 (1.6)]. All NC were of Caucasian origin. The study was approved by the Institutional Review Board of the University of Florida and conformed to the ethical guidelines of the 1975 Declaration of Helsinki.
2.2 Definition of pain
The pain definition of the International Association for the Study of Pain (IASP) was used (Merskey and Bogduk 1994) which describes pain as an unpleasant sensory and emotional experience associated with actual or potential tissue damage, or described in terms of such damage. Pain is always subjective. Each individual learns the application of the word through experiences related to injuries in early life. Pain is always unpleasant and therefore also an emotional experience. Experiences which resemble pain but are not unpleasant, e.g., pricking, are not called pain.
2.3 Sensory ratings
A 15 cm mechanical visual analogue scale (M-VAS) (0 – 10) was used for ratings of experimental sensations during mechanical and thermal stimulations (Price and Harkins 1992). The scale was anchored on the left with “no sensation at all” and on the right with “the most intense sensation imaginable”. These endpoints were specifically chosen with the possibility in mind that some individuals might be unable to experience pain with any of the test modalities but could nevertheless perceive increases in intensity within both the innocuous and nociceptive range. For example, someone could indifferent to pain but still notice increases in nociceptive intensity. To further distinguish between indifference and nociceptive insensitivity to pain, every subject was given a two-alternative forced choice at the end of each stimulus to state whether pain was or was not experienced.
2.4 Quantitative Sensory Testing
Instead of pain threshold testing, we used mechanical and thermal stimulus response functions to determine the type of nerve afferent associated with insensitivity to pain in the IC. This approach provides relevant information about the responsiveness of low threshold A-beta and high threshold C-fibers to painful and non-painful mechanical as well as thermal stimuli (Price and Browe 1973)
2. 4.1 Touch Detection Thresholds
Touch detection thresholds were tested at the finger pads of both hands with Semmes-Weinstein monofilaments using a five-alternative forced choice detection procedure. Testing was performed in a quiet room and the subjects sat with their hands positioned on a foam cushion. The subjects could not see their hands to prevent visual cues from the filaments. Stimulation was performed with a series of monofilaments using standardized force (0.02 – 10 g) which had been calibrated using a scale. On each trial, the tip of the monofilament was applied manually to the skin, and pressure was exerted until the filament bent. The nylon filaments were applied to the center of the volar surface of the first segment of each digit on each trial for approximately 1 s. Trials were interrupted by 3 s intervals, after which the stimulus was applied to a different finger.
2.4.2 Mechanical Testing
An electronic algometer (Somedic, Horby, Sweden) was used to obtain pressure pain ratings of all subjects at the thumb nail of either hand. The instrument was placed perpendicular to the nail bed and pressure was slowly increased (50 kPa/s) until pain threshold was reached. During threshold testing the subjects were asked to immediately press a button when pain was felt for the first time.
2.4.3 Heat or Cold Sensory Testing
For heat and cold testing a 3 × 3 cm Peltier thermode (Pathway Neurosensory Analyzer, Medoc Advanced Medical Systems, Ramat Yishai, Israel) was used. The probe was brought into firm contact with the volar surface of each subject’s forearm for 5 s. For heat or cold threshold testing the probe temperature was either increased or decreased from ambient temperature (32°C) to target temperature (35°C – 50°C). Each subject received heat or cold stimuli ranging from 35°C to 50°C (in 3°C steps) or from 35°C to 5°C (in 5°C steps). Each thermal pulse was applied three times in counterbalanced order to either forearm. For sensory ratings the M-VAS was used.
2.5 Questionnaires
To assess the IC’s intelligence and psychosocial functioning a variety of measures were administered, including: (1) the Wechsler Abbreviated Scale of Intelligence (WASI) (Wechsler 2003), (2) the Test of Everyday Attention for Children (TEA-Ch) (Manly et al., 1999; Manly et al., 2001), (3) the Fear Survey Schedule for Children (Gullone and King 1992), the Fear of Pain (FPQIII) (McNeil and Rainwater, III 1998), and (4) the Bryant Empathy Index (Bryant 1982). The IC’s mother completed a number of measures to provide additional assessment of her daughter’s psychosocial functioning, including: (a) the Behavioral Assessment System for Children (Reynolds and Kamphaus 2004), (b) the Adaptive Behavior Scale (Lambert et al., 1993), (c) the Behavior Rating of Executive Functioning (Gioia et al., 2000b; Gioia et al., 2000a), (d) the Griffith Empathy Measure (Dadds et al., 2008), and (e) the Social Skills Rating System (Gresham and Elliott 1990; Demaray et al., 1995). Each of these measures has demonstrated adequate to strong psychometric properties.
2.6 Genetic Testing
2.6.1 DNA and RNA Samples
Blood and buccal swab samples were collected from the IC and her first degree relatives. DNA was extracted from all samples using the PureGene kit (Gentra Systems, Minneapolis, MN). Total cellular RNA was also extracted from IC’s leukocytes (Trizol, Invitrogen), and from lymphocytes that had undergone short-term tissue culture and were harvested after treatment with puromycin using a previously published method (Messiaen et al., 1999). Puromycin was added to inhibit nonsense-mediated decay and to allow better representation of mutant transcripts in the RNA preparation. Puromycin, a translation elongation inhibitor, can stabilize transcripts with nonsense or other premature truncating mutations (which may be unstable due to nonsense-mediated decay), and thus is sometimes used in RNA preparation from cultured cells in case a patient has such a mutation. However, it is not expected to substantially affect the relative amounts of missense transcripts.
2.6.2 DNA Analysis of SCN9A Exons
SCN9A gene numbering is based on NCBI accession NM_02977, gi:25601719, which is the reference sequence in the RefGeneSeq project. SCN9A exons were PCR-amplified from DNA using Hotmaster Taq polymerase with manufacturer’s recommended conditions (Eppendorf, Westbury, NY). The PCR primers used were based on Cox et al. (2006). Exon 26, the last exon of the gene (NCBI consensus reference mRNA NM_002977.2), is very large so we amplified two smaller “A” and “B” overlapping sections (containing coding region) per Cox et al. (2006). Cycling conditions: 1 cycle of 95°C for 5 minutes to activate the polymerase; 35 cycles of 95°C for 15 seconds, 60°C for 30 seconds, 72°C for 1 minute; and a final extension at 72°C for 10 minutes. PCR products were examined on ethidium bromide-stained agarose gels to evaluate quality, specificity, and quantity, purified using the ExoSapit reagent (Amersham Pharmacia, GE Healthcare Life Sciences, Pittsburgh PA), and then sequenced using the ABI Prism BigDye Terminator v.3 cycle sequencing system (ABI, Foster City, CA). PCR primers were used as the sequencing primers. Sequencing reactions were analyzed on an ABI 3130XL Genetics Analyzer at the UF Center for Epigenetics. Data was analyzed using Sequencher software v.4.8 (Gene Codes, Ann Arbor, MI).
2.6.3 Reverse Transcription and Amplification of SCN9A cDNA
To test splicing effects of DNA-based mutations, first strand cDNA was synthesized from 750 ng of total RNA, using the Superscript II kit (Invitrogen). The resulting cDNA was subjected to PCR amplification with two primer-sets designed using the Primer 3 program (http://frodo.wi.mit.edu/): the first had primers in SCN9A exons 16 and 18 (primers SCN16-18F (5′-AGACCCTGATGCAAACAACC) and SCN16-18R (5′-TGAGCAGGATCATGAGGACA) and the second in exons 24 and 26 (primers SCN24-26F (5′-GTCCCTTCCTGCGTTGTTTA) and SCN24-26R (5′-GGTGGAGAGGTGGTGGATGAAGTGG)). The exon 16-18 RT-PCR product was 672 bp in length. The normal exon 24-26 RT-PCR product was 946 bp in length.
2.6.4 Validation of Mutations by Restriction Digest
The putative mutations in exon 26B and exon 17 were confirmed by restriction enzyme digest using enzymes that cut one allele but not the other, due to the mutation. DNA-PCR products from exons 26B (577 bp product) and 17 (292 bp product) were digested with BsaJI and AccI, respectively, per manufacturer’s recommended conditions (New England Biolabs, Ipswich, MA). Digested products were subjected to electrophoresis on an 8% native polyacrylamide gel for 1.5 hours at 200V, and visualized by ethidium bromide staining. BsaJI was predicted to cut at the exon 26B mutation, thereby producing digestion fragments of 236, 142, 136, 51 and 12 bp from the mutant allele, relative to normal (378, 136, 51, and 12 bp fragments). The exon 17 mutation was predicted to create a novel AccI site, thereby producing digestion fragments of 230 and 62 bp from that allele).
3. Results
The index case (IC) was of short stature (2nd percentile) but otherwise normal appearing. She took dexmethylphenidate daily for attention deficit disorder. She was enrolled in 4th grade. The IC, both of her parents, her 12 year old brother, and her 6 year old sister participated in this study. In addition 10 normal pain-free individuals (9 females; 1 male; average age: 22.3 years) served as controls (NC). None of the family and NC was taking any medications at the time of the examination.
The IC’s insensitivity to pain first came to her parents’ attention at age 6 months when she presented with a painless corneal abrasion. The parents also noted that the child did not cry at birth and had never shown any apparent pain behaviors. Subsequently, the girl sustained a second degree hand burn at the age of 2 years without obvious pain behaviors. However, when she acquired otitis media associated with rupture of the tympanic membrane, she briefly complained of pain for the first time in her life. At the age of 4 she underwent tonsillectomy, but did not display any pain behaviors during the post-operative period. Her parents noted that she infrequently held the right side of her head stating that “she hurts”. She was evaluated by a child neurologist in 2006 who described her as insensitive to painful stimuli but otherwise normal. EMG/nerve conduction studies were also normal except absent H-reflexes. A sural nerve biopsy showed normal myelinated and unmyelinated fibers. A chromosome analysis was within normal limits.
3.1 Touch Detection Threshold
Semmes-Weinstein monofilaments from 0.02 to 10 g were used for testing of mechanical sensitivity at the hands. All subjects including the IC were able to identify monofilament pressures of ≥ 1.4 g.
3.2. Psychophysical Testing
3.2.1 Mechanical Pain Thresholds
Pressures of < 450 kPa at the thumbnail were painless for all participants (Figure 1). The IC’s pain-threshold was 711.5 kPa. In contrast, the average (SD) pressure pain threshold of her family members and NC were 422.5 (221.3) and 423.4 (96.7) kPa, respectively.
Figure 1.

Sensory ratings of mechanical stimuli. All subjects received 5 sec pressure stimuli to the thumb nail of either hand with an algometer. The subjects rated the intensity of pressure sensations using a VAS. Additionally, after every stimulus they were asked whether the stimulus was painful. All NC and Family members except the IC and 1 male sibling rated pressure sensations beyond 430 kPa as painful (red symbols). NC=normal control; Family=family except IC; IC=Index Case; VAS=visual analogue scale
3.2.2 Heat Testing
Heat intensity ratings positively accelerated in family members and NC from 0.4 VAS units to 6.0 VAS units (Figure 2). They reported monotonic sensory increases within the warmth (32 – 44°C) and the nociceptive range (44 to 50°C). The IC’s heat ratings, however, increased only within the warm range and failed to accelerate in the nociceptive range (Figure 2). Family members and NC reported heat stimuli of ≥ 47°C as painful, whereas the IC did not report pain with any of the applied heat-stimuli up to 50°C.
Figure 2.

Ratings of warmth and heat stimuli. The study subjects received 3 trains of randomly selected 5 sec warm or heat stimuli (35 – 50°C) to either forearm and rated the stimulus intensity using a VAS. All stimuli were delivered by a 3 × 3 cm thermode (Pathway©). After every rating the subjects were asked to indicate whether the stimulus was painful. Whereas the ratings of NC and Family members positively accelerated with increasing stimulus intensity, reaching pain threshold at ≥ 45°C (red symbols), the ratings of the IC did not seem to increase further with heat stimuli of ≥ 44°C. NC=normal control; Family=family except IC; IC=Index Case
3.2.3 Cold Testing
The cold-intensity ratings of family-members and NC increased with decreasing stimulus temperatures (Figure 3). However, only 50 % of family-members and 10 % of NC perceived 5°C cold stimuli as painful. The IC’s ratings of cold sensations increased with temperatures decreasing to 15°C, but returned to baseline when temperatures reached 5°C (Figure 3).
Figure 3.

Ratings of cold stimuli. All subjects received 3 trains of randomly selected 5 sec cold stimuli (35 – 5°C) to either forearm with a 3 × 3 cm thermode (MEDOC©). The subjects rated the intensity of cold sensations using a VAS. After every rating they were asked to indicate whether the stimulus was painful. Whereas the ratings of NC and Family members seemed to linearly increase with decreasing stimulus temperatures, the ratings of the IC decreased with temperatures of > 15°C. NC=normal control; Family=family except IC; IC=Index Case
3.3 Psychological Testing
The IC’s full scale IQ as measured by the WASI was 106 (66%), which falls in the average range relative to her same-age peers. There was no significant discrepancy between her verbal and performance subscales on the WASI. The female sibling’s full scale IQ on the WASI was 120 (91%), while the male sibling’s full scale IQ on the WASI was 103 (53%). On the Test of Everyday Attention the IC’s subscale scores ranged from low average to average, with no consistent pattern of strengths or weaknesses. Results from the Behavioral Assessment Scale for Children (BASC-II) showed only two at-risk elevations, hyperactivity (t-score = 61) and adaptability (t-score = 38). Her scores on the Adaptive Behavior Scale were in the average range relative to her same-aged peers with no at-risk or clinically significant elevations. On the Revised Fear Survey Schedule for Children (FSSC-R), the IC showed elevations in fear of Failure/Criticism (z-score = 2.05; 97%), Minor Injury/Small Animals (z-score = 1.87; 96%), and The Unknown (z-score = 1.73; 95%); she did not show elevations on the Danger & Death, and Medical Fears subscales on the FSSC-R. On the Behavior Rating of Executive Functioning (BRIEF) the IC displayed at at-risk elevation for overall Behavioral Regulation (t-score = 64; 90%); no other at-risk of clinical elevations were noted on the BRIEF. On the parent-completed Griffith Empathy Scale, the IC’s score of 28 was 10 points below the mean for her same-aged female peers in a previous published paper using this scale. However, on the Bryant Empathy scale, a child-self-report measure, her level of self-reported empathy was in the high-average range. Finally, on the parent-completed Social Skills Rating System (SSRS) the IC’s overall social skills standard score of 71 (3rd percentile) was well below average; her scores were below average on the Cooperation, Assertion, and Responsibility subscales.
3.4 Neurological Examination
The IC’s past history showed no episodes of unexplained vomiting or dysphagia. Her mental status exam was normal. She had normal blood pressure without postural fluctuations. She had normal vision using corrective lenses and normal hearing. Her tear production was normal. Corneal-reflexes and gag-reflex were normal. Her tongue showed normal fungiform papillae;
Sense of smell was tested using the alcohol sniff test (Davidson and Murphy 1997) as well as the three odors: “cloves,” “coffee,” and “rose” (Hummel et al., 2010). These odors were selected because their identification appears to be age independent (Konstantinidis et al., 2006) Using forced choice the IC was unable to identify any of the three odors and therefore was considered anosmic. Peripheral nerves were not palpably enlarged. There was no evidence of motor or sensory neuropathies. She correctly perceived touch, warm and cold, proprioception, tickle and pressure. She was able distinguish soft touch from pin prick. There were no signs of autonomic-nervous-system dysfunction: normal flushing/blushing; no episodes of hyperpyrexia (except during infections); bladder control was normal without history of incontinence or retention. Strength, tone, reflexes including plantar responses, and joint appearance were all normal.
In general, radial and ulnar nerve-conduction studies provided no significant abnormalities except for right ulnar nerve testing showed slowed conduction velocity with normal amplitude and distal latency, which may suggest a demyelinating neuropathy. Sural nerve biopsy showed all nerve-fiber types of normal morphology and distribution by light and electron microscopy. Magnetic-resonance imaging of the brain lacked any abnormalities.
3.5 Genetic Analysis
Sequence analysis of the IC’s genomic DNA did not demonstrate any known mutations of TRKA (NGFR1) (Shatzky et al., 2000; Mardy et al., 1999) or SCN9A (Cox et al., 2006) but revealed heterozygosity for a non-conservative missense mutation of SCN9A in exon 26 in the IC and one of her parents (Figure 4). The mutation (T to C substitution) caused an amino acid substitution of arginine for cysteine (c.5155 T>C; C1719R). Her siblings and the other parent were negative for the C1719R mutation. Subsequent RNA reverse transcription (RT-PCR) and restriction analysis showed that the C1719R mutation was also present in the leukocyte cDNA of the IC and the parent carrying this mutation (data not shown). Further sequence analysis of the child’s DNA revealed heterozygosity for a 1-bp deletion of an A nucleotide in intron 17 which was present in the other parent (c.3467+3 delA, or IVS17+3 delA). This deletion alters the splice donor consensus sequence, which we predicted would cause exon 17 to be skipped at the mRNA level. To test this hypothesis, RT-PCR spanning exons 16–18 was performed using total leukocyte and lymphocyte. RNA from the IC and the carrier parent. Figure 5A (lanes 1 and 3, parent and IC, respectively) shows that, as expected, these two individuals have a normal cDNA product (top fragment) but also a smaller product, whereas the parent not carrying this mutation shows only the normal fragment (lane 2). The lower fragment was isolated and sequenced, confirming that exon 17 was missing from that cDNA fragment. The altered transcript in the IC (lane 3) was at greater levels than the transcript from the normal allele, and compared to the carrier parent. This phenomenon persisted when the assay was repeated with two separate leukocyte RNA samples and the cultured puromycin-treated lymphocytes where nonsense-mediated decay was inhibited. The efficacy of nonsense-mediated decay inhibitors is known to be variable depending on the gene, the mutation, and the genetic background (Zetoune et al., 2008). Therefore, it is not surprising that there was no difference in relative amounts of transcript in puromycin-treated cells compared to untreated cells. The normal-sized fragment thus likely represents the transcript carrying the C1719R mutation, as well as some wild-type transcript that escaped the splicing error (so-called leaky splicing).
Figure 4.

A. Sequencing chromatograms showing the C1719R missense mutation in exon-26B of SCN9A. The first chromatogram shows the results from the IC (note double peak in middle of the sequence below the highlighted base). The second chromatogram shows the same doublet in one parent. The third chromatogram is from the other parent, which has a normal sequence.
B. Ethidium bromide-stained polyacrylamide gel after electrophoresis of restriction enzyme digests of exon PCR products for both mutated SCN9A exons. (I) BsaJI digest of exon-26B confirms the C1719R mutation in the IC (lane 1) and one parent (lane 2) by virtue of the extra fragments of expected size, but not in the other parent (lane 3). Lanes 4 and 5 are unaffected controls. (II). AccI digest of exon-17 PCR products shows that the IC (lane 6) and one parent (lane 8, the parent lacking the exon-26B mutation) have the additional restriction site on one allele consistent with the intronic mutation.
Figure 5.
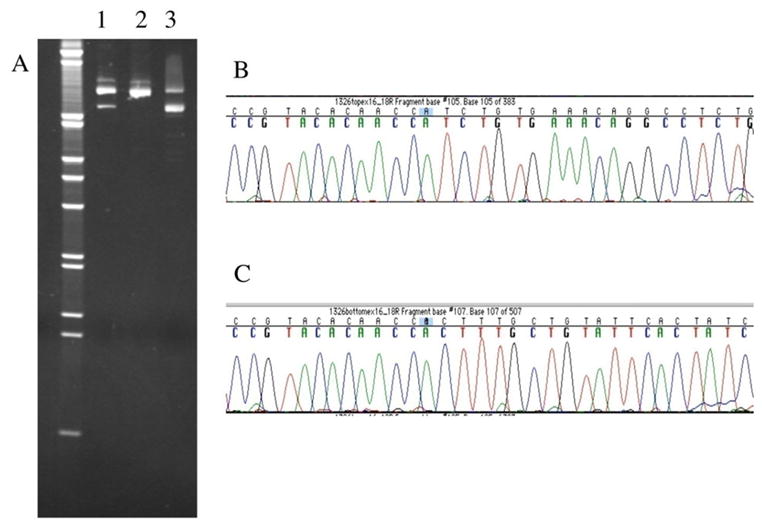
Identification of the exon-17 skip in SCN9A mRNA. (A) This photograph shows an ethidium-bromide stained 8% native polyacrylamide gel separating cDNA RT-PCR products (exon-16 to 18). Samples from the parents are in lanes 1 (carrier) and 2 (non-carries), respectively, and the IC’s sample is in lane 3. (Invitrogen 1 kb molecular weight marker is on the left). Lanes 1 and 3 show two cDNA fragments, while lane 2 shows the one fragment of expected size (672 bp). (B) Sequencing of the cDNA top fragment shows the normal SCN9A sequence at the junction of exon-17 to 18 (shown is non-coding strand, exon-18 is on the left). (C) Sequence chromatogram of the smaller cDNA fragment from the carrier parent shows that the cDNA joins exon 16 directly to exon-18 perfectly, completely skipping the 121 bp exon-17. Identical results were found in the IC for both cDNA fragments, as expected.
Both genetic mutations were confirmed by restriction endonuclease fragment analysis. The C1719R missense mutation causes a gain of a BsaJI site, and the 1bp intron 17 deletion causes a gain of an AccI site. These digests confirmed the heterozygous mutations in the IC and one parent each (Figure 5B). DNA samples from ten unaffected, unrelated Caucasians of northern European descent were screened for the C1719R mutation and were negative for this change. In addition, Singh et al. (2009)(Singh et al., 2009) sequenced this region in 90 unrelated U.S. Caucasians (with febrile seizures, not pain disorders) and did not find this mutation. Together this represents a screening of 200 Caucasian alleles. The failure to detect this change is consistent with C1719R being a pathogenic mutation rather than a neutral polymorphism.
4. Discussion
This study uses detailed psychophysical and psychological tests to characterize possible sensory and psychological abnormalities associated with the presence of two novel mutations of the SCN9A gene. The IC displayed three distinct somatosensory deficits. She was unable to: 1) discriminate heat pain intensity in the nociceptive range of 44 –50°C; 2) discriminate cold sensations from 25 - 5°C, and 3) appreciate normal mechanical pain thresholds. All of these deficits may be relative and not absolute, given the history of this child’s reporting of pain in rather severe medical conditions. Somatosensory and psychological functions were within the normal range on several other tests, confirming the specificity of these three deficits.
The deficit in heat pain is underscored by the fact that almost all mechano-thermal primary nociceptive afferents and the central neurons that they activate show monotonic increases above 44°C in NC (Coghill et al., 1993) and by the fact that psychophysical responses of mammalian organisms, including man, also increase over this range (Almeida et al., 2004). Thus, the IC’s lack of a monotonic sensory increase beyond 44°C is noteworthy and reflects a selective deficit in heat nociception but not warmth appreciation. Similarly, her deficit in mechanical pain sensations occurred without deficits in her ability to detect and normally respond on several other tests of non-painful somatosensory functions. Additionally, her psychological evaluation was found to be mostly normal and therefore not likely to account for the selective deficits. Finally, the combination of her inability to perceive increases in sensations between 44 – 50°C and her lack of categorizing any temperature within this range as painful suggests that her lack of pain report reflects pain insensitivity not indifference. It is unclear whether previously reported cases of SCN9A mutations (Cox et al., 2006; Goldberg et al., 2007; Ahmad et al., 2007) resulting in congenital insensitivity to pain were also associated with deficits in thermal and mechanical pain processing because no extensive quantitative sensory testing was performed. However, one report of a single individual with SCN9A mutations described abnormal warmth and heat pain detection thresholds (Nilsen et al., 2009).
4.1 Genetic Findings
Our genetic analysis confirmed the presence of two novel SCN9A-mutations, most likely responsible for the IC’s pain insensitivity. Although the IC’s parents were heterozygous carriers of these mutations, they were clinically normal and without sensory abnormalities. Consistent with autosomal-recessive transmission, the IC inherited one different SCN9A-mutation from each parent. No other SCN9A-mutations were found and none of her siblings had both mutations. Thus the IC is a compound-heterozygote with two distinct SCN9A-mutations. These mutations have not been previously reported, and are not present in the single nucleotide polymorphism (SNP) databases. The C1719R-mutation is considered to be pathogenic for several reasons: the affected amino acids have substantially different biochemical properties, the cysteine at that position is conserved at least among mammals and birds, and is in an extracellular loop within the fourth transmembrane-domain of Nav1.7, close to two other stop mutations reported in individuals with congenital pain-insensitivity (Drenth and Waxman 2007). The cysteine at position 1719 is conserved among all human and mouse voltage-gated sodium channel alpha subunits, and is also conserved in all SCN9A RefSeq mRNA homologs reported (pig, dog, rhesus monkey, rat, rabbit, cow). No SNPs are reported at this position. There is one variant sequence of SCN3A reported with this substitution (NCBI accession AAK00218, submitted in 2000). However this variant has not been validated, is not cited in any publications, and patient health and tissue source of this mRNA were not indicated; thus the verity of this single report is in question. The lack of this mutation in 200 Caucasian alleles (see Singh et al. (2009) and our results), and highly conserved nature of the residue, suggests that non-pathogenic missense polymorphisms at this codon are exceedingly rare, if they exist at all. Further supporting the notion of C1719 being pathogenic, this conserved cysteine was found mutated (C1756G) in SCN1A in a patient with severe myoclonic epilepsy in infancy (Herini et al., 2010), an autosomal dominant condition (OMIM 607208) that has also been associated with mutations in SCN9A (Singh et al., 2009).
The intron-17 1-bp deletion can be considered pathogenic, since loss of exon-17 from the mRNA causes a frameshift resulting in a truncated (possibly unstable) protein. The deletion of an “A” at the third position of IVS17 (IVS17+3delA) results in the splice donor having the sequence GTAGA, in which the 4th and 5th bases are no longer consensus (GTAAG). However, G in the fourth position is the next most common base (12% vs 70% A) and T occurs at the 5th position 8% of the time. The efficiency of splicing is related to consensus base substitutions in the context of the surrounding sequence, and there are many examples of partially-penetrant splicing errors in situations such as this, particularly where the substitutions are later in the sequence (Roca et al., 2008). Since such splicing-mutations can be leaky, it is possible that this allele could produce minor amounts of full-length Nav1.7,. In addition, the C1719R-mutation might also produce a protein retaining some function, especially since this amino-acid substitution is close to the carboxy-terminus. Thus, one or both mutations could be hypomorphic alleles, possibly allowing low levels of Nav1.7-channel function, which would be novel for SCN9A-related congenital pain insensitivity in which case the IC’s full-length RT-PCR fragment in Figure 5A would represent the C1719 transcript and some wild-type transcript. No amino-acid substitutions have yet been reported in extracellular domains of Nav1.7, including in individuals with known gain-of-function missense mutations associated with two autosomal-dominant congenital pain syndromes [Primary Erythromelalgia (PE) and Paroxysmal Extreme Pain Disorder (PEPD)] (Drenth and Waxman 2007). Prior to this study, only inactivating mutations have been reported in patients with congenital insensitivity to pain, with resulting loss of Nav1.7 function: 10 stop mutations, 4 small frameshift mutations, and one putative splicing mutation (Cox et al., 2006; Ahmad et al., 2007; Goldberg et al., 2007; Nilsen et al., 2009). Most of these were homozygous situations, with three reported compound heterozygotes. Since our IC has no evidence of episodic pain or hyperalgesia, we suggest that the C1719R mutation is not a gain-of-function allele, but rather an inactivating mutation that may have an attenuated effect on channel function. Our IC’s genotype may help explain the unique clinical phenotype of this child compared to other individuals reported with SCN9A mutations.
4.2 Nav1.7 and Pain
SCN9A encodes Nav1.7, the alpha-subunit of a tetrodotoxin-sensitive voltage-gated sodium channel located on chromosome 2q24. This gene is expressed at high levels in peripheral sensory and autonomic neurons (Klugbauer et al., 1995; Toledo-Aral et al., 1997; Sangameswaran et al., 1997). Voltage-gated sodium channels are crucial for the depolarizing phase of action potentials of neurons and seem to determine the excitability and repetitive firing properties of neurons (Rush et al., 2006; Amir et al., 2006). In addition to reduced/loss of function mutations, as in our IC, several gain of function mutation of SCN9A have been reported for two autosomal-dominant pain disorders, PE and PEPD (Fertleman et al., 2006). The former is characterized by severe, episodic burning pain in the extremities in response to warm stimuli or moderate exercise (Yang et al., 2004); the latter disorder demonstrates prominent autonomic manifestations and severe burning pain, most commonly in rectal or facial areas. These SCN9A mutations alter the threshold of activation of the Nav1.7 sodium channel, resulting in hyperexcitability of pain signaling neurons. Mice lacking Nav1.7 in nociceptive neurons show increased mechanical and thermal pain thresholds and decreased inflammatory pain responses (Nassar et al., 2004; Nassar et al., 2005). Global Nav1.7-null mutant mice, however, seem to die shortly after birth (Nassar et al., 2005) due to abnormal feeding behaviors. Different expression patterns of Nav1.7 in rodents and primates may explain the lethal phenotype in Nav1.7-deficient mice. Although Nav1.7 mRNA is expressed in both rat and mouse in the paraventricular hypothalamic and the supraoptic nucleus it appears to be absent in both monkey and human tissue (Ahmad et al., 2007). Similarly, Nav1.7 expression in the pituitary and adrenal glands is detectable in both rodent species but absent in humans. Thus the differential expression and different function of Nav1.7 between rodents and humans may explain the lethal phenotype observed in mice.
4.3 Psychological Consequences of Pain Insensitivity
Because empathy may be strongly influenced by ability to experience pain (Danziger et al., 2006; Singer and Frith 2005) we performed an extensive psychological evaluation of the IC. She appeared to be a bright, pleasant, friendly, and happy young female. She was of average intelligence relative to her peers with no significant pattern of strengths or weakness on the IQ test. Assessment of psychosocial functioning suggested that she did not exhibit any consistent or atypical problems with anxiety, sadness, irritability, oppositional, aggressive or anti-social behaviors. There was some evidence to suggest that she exhibited slightly below average social skills and slightly above average levels of hyperactivity and fears relative to her same aged-peers. The IC’s parents reported that she displays below average levels of empathy compared to other parents’ report of their children, which is consistent with her lower than average social skills. However, it is important to note that the IC self-reported average levels of empathy compared to her same-aged peers, although the validity of self-report of empathy by young children may be questionable.
5. Conclusions
This is the first detailed characterization of two novel SCN9A (Nav1.7) mutations in a female child, including a missense and a splice-donor mutation, resulting in incomplete congenital insensitivity to pain (OMIM 243000). Importantly, these mutations do not seem to be associated with developmental abnormalities typical for some HSAN. Because this child experienced some extreme sensory input as painful, it is likely that her life span will be normal unlike some individuals diagnosed with HSAN.
Acknowledgments
This work was supported by NIH grants NS041670 and AR053541. We would like to acknowledge the Hayward Foundation for financial support for the sequencing core costs. The expert technical assistance of Amber M. Schwier and Michelle Burch is greatly appreciated.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Ahmad S, Dahllund L, Eriksson AB, Hellgren D, Karlsson U, Lund PE, Meijer IA, Meury L, Mills T, Moody A, Morinville A, Morten J, O’Donnell D, Raynoschek C, Salter H, Rouleau GA, Krupp JJ. A stop codon mutation in SCN9A causes lack of pain sensation. Hum Mol Genet. 2007;16(17):2114–2121. doi: 10.1093/hmg/ddm160. [DOI] [PubMed] [Google Scholar]
- Almeida TF, Roizenblatt S, Tufik S. Afferent pain pathways: a neuroanatomical review. Brain Res. 2004;1000(1–2):40–56. doi: 10.1016/j.brainres.2003.10.073. [DOI] [PubMed] [Google Scholar]
- Amir R, Argoff CE, Bennett GJ, Cummins TR. The role of sodium channels in chronic inflammatory and neuropathic pain. J Pain. 2006;7(5):S1–S29. doi: 10.1016/j.jpain.2006.01.444. [DOI] [PubMed] [Google Scholar]
- Bryant BK. An index of empathy for children and adolescents. Child Dev. 1982;53:413–425. [Google Scholar]
- Coghill RC, Mayer DJ, Price DD. The roles of spatial recruitment and discharge frequency in spinal cord coding of pain: a combined electrophysiological and imaging investigation. Pain. 1993;53(3):295–309. doi: 10.1016/0304-3959(93)90226-F. [DOI] [PubMed] [Google Scholar]
- Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, Karbani G, Jafri H, Mannan J, Raashid Y, Al-Gazali L, Hamamy H, Valente EM, Gorman S, Williams R, McHale DP, Wood JN, Gribble FM, Woods CG. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444(7121):894–898. doi: 10.1038/nature05413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dadds MR, Hunter K, Hawes DJ, Frost AD, Vassallo S, Bunn P, Merz S, Masry YE. A measure of cognitive and affective empathy in children using parent ratings. Child Psychiatry Hum Dev. 2008;39(2):111–122. doi: 10.1007/s10578-007-0075-4. [DOI] [PubMed] [Google Scholar]
- Danziger N, Prkachin KM, Willer JC. Is pain the price of empathy? The perception of others’ pain in patients with congenital insensitivity to pain. Brain. 2006;129:2494–2507. doi: 10.1093/brain/awl155. [DOI] [PubMed] [Google Scholar]
- Davidson TM, Murphy C. Rapid clinical evaluation of anosmia. The alcohol sniff test. Arch Otolaryngol Head Neck Surg. 1997;123(6):591–594. doi: 10.1001/archotol.1997.01900060033005. [DOI] [PubMed] [Google Scholar]
- Dearborn GVN. A case of congenital general pure analgesia. J Nerv Ment Dis. 1932;75:612–615. [Google Scholar]
- Demaray MK, Ruffalo SL, Carlson J, Busse RT, Olson AE, McManus SM, Leventhal A. Social skills assessment: A comparative evaluation of six published rating scales. School Psychol Rev. 1995;24:648–671. [Google Scholar]
- Drenth JP, Waxman SG. Mutations in sodium-channel gene SCN9A cause a spectrum of human genetic pain disorders. J Clin Invest. 2007;117(12):3603–3609. doi: 10.1172/JCI33297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fertleman CR, Baker MD, Parker KA, Moffatt S, Elmslie FV, Abrahamsen B, Ostman J, Klugbauer N, Wood JN, Gardiner RM, Rees M. SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron. 2006;52(5):767–774. doi: 10.1016/j.neuron.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Gioia GA, Isquith PK, Guy SC. Construct validity of the Behavior Rating Inventory of Executive Function (BRIEF) J Int Neuropsychol Societ. 2000a;6:139. [Google Scholar]
- Gioia GA, Isquith PK, Guy SC, Kenworthy L. Behavior rating inventory of executive function. Odessa, FL: Psychological Assessment Resources; 2000b. [Google Scholar]
- Goldberg YP, MacFarlane J, MacDonald ML, Thompson J, Dube MP, Mattice M, Fraser R, Young C, Hossain S, Pape T, Payne B, Radomski C, Donaldson G, Ives E, Cox J, Younghusband HB, Green R, Duff A, Boltshauser E, Grinspan GA, Dimon JH, Sibley BG, Andria G, Toscano E, Kerdraon J, Bowsher D, Pimstone SN, Samuels ME, Sherrington R, Hayden MR. Loss-of-function mutations in the Nav1.7 gene underlie congenital indifference to pain in multiple human populations. Clin Genet. 2007;71(4):311–319. doi: 10.1111/j.1399-0004.2007.00790.x. [DOI] [PubMed] [Google Scholar]
- Gresham F, Elliott S. Social Skills Rating Scale. Circle Pines, MN: American Guidance Services; 1990. [Google Scholar]
- Gullone E, King NJ. Psychometric evaluation of a revised fear survey schedule for children and adolescents. J Child Psychol Psychiatry. 1992;33(6):987–998. doi: 10.1111/j.1469-7610.1992.tb00920.x. [DOI] [PubMed] [Google Scholar]
- Herini ES, Gunadi, van Kempen MJ, Yusoff S, Sutaryo Sunartini, Patria SY, Matsuo M, Lindhout D, Nishio H. Novel SCN1A mutations in Indonesian patients with severe myoclonic epilepsy in infancy. Pediatr Int. 2010;52:234–239. doi: 10.1111/j.1442-200X.2009.02916.x. [DOI] [PubMed] [Google Scholar]
- Hummel T, Pfetzing U, Lotsch J. A short olfactory test based on the identification of three odors. J Neurol. 2010 doi: 10.1007/s00415-010-5516-5. in press. [DOI] [PubMed] [Google Scholar]
- Klein CJ, Brown RH, Dyck PJ. Hereditary sensory and autonomic neuropathies: Clinical, pathologic classification, and molecular genetics. In: Dyck PJ, Thomas PK, editors. Peripheral Neuropathy. Vol. 4. Philadelphia: Saunders; 2005. [Google Scholar]
- Klugbauer N, Lacinova L, Flockerzi V, Hofmann F. Structure and functional expression of a new member of the tetrodotoxin-sensitive voltage-activated sodium channel family from human neuroendocrine cells. EMBO J. 1995;14(6):1084–1090. doi: 10.1002/j.1460-2075.1995.tb07091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinidis I, Hummel T, Larsson M. Identification of unpleasant odors is independent of age. Arch Clin Neuropsychol. 2006;21(7):615–621. doi: 10.1016/j.acn.2006.05.006. [DOI] [PubMed] [Google Scholar]
- Lambert N, Nihira K, Leland H. AAMR Adaptive Behavior Scale-School. 2. Austin, TX: PRO-ED; 1993. [Google Scholar]
- Manly T, Anderson V, Nimmo-Smith I, Turner A, Watson P, Robertson IH. The differential assessment of children’s attention: the Test of Everyday Attention for Children (TEA-Ch), normative sample and ADHD performance. J Child Psychol Psychiatry. 2001;42(8):1065–1081. doi: 10.1111/1469-7610.00806. [DOI] [PubMed] [Google Scholar]
- Manly T, Robertson IH, Anderson V, Nimmo-Smith I. The test for everyday attention for children (TEA-CH) Bury St. Edmunds: Thames Valley Test Company; 1999. [Google Scholar]
- Mardy S, Miura Y, Endo F, Matsuda I, Sztriha L, Frossard P, Moosa A, Ismail EA, Macaya A, Andria G, Toscano E, Gibson W, Graham GE, Indo Y. Congenital insensitivity to pain with anhidrosis: novel mutations in the TRKA (NTRK1) gene encoding a high-affinity receptor for nerve growth factor. Am J Hum Genet. 1999;64(6):1570–1579. doi: 10.1086/302422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeil DW, Rainwater AJ., III Development of the Fear of Pain Questionnaire--III. J Behav Med. 1998;21(4):389–410. doi: 10.1023/a:1018782831217. [DOI] [PubMed] [Google Scholar]
- Merskey H, Bogduk N. Classification of chronic pain: Description of chronic pain syndromes and definition of pain terms. 2. Seattle: IASP Press; 1994. [Google Scholar]
- Messiaen LM, Callens T, Roux KJ, Mortier GR, De Paepe A, Abramowicz M, Pericak-Vance MA, Vance JM, Wallace MR. Exon 10b of the NF1 gene represents a mutational hotspot and harbors a recurrent missense mutation Y489C associated with aberrant splicing. Genet Med. 1999;1(6):248–253. doi: 10.1097/00125817-199909000-00002. [DOI] [PubMed] [Google Scholar]
- Nassar MA, Levato A, Stirling LC, Wood JN. Neuropathic pain develops normally in mice lacking both Nav1.7 and Nav1.8. Mol Pain. 2005;1:24. doi: 10.1186/1744-8069-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassar MA, Stirling LC, Forlani G, Baker MD, Matthews EA, Dickenson AH, Wood JN. Nociceptor-specific gene deletion reveals a major role for Na(v)1.7 (PN1) in acute and inflammatory pain. PNAS. 2004;101(34):12706–12711. doi: 10.1073/pnas.0404915101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen KB, Nicholas AK, Woods CG, Mellgren SI, Nebuchennykh M, Aasly J. Two novel SCN9A mutations causing insensitivity to pain. Pain. 2009;143(1–2):155–158. doi: 10.1016/j.pain.2009.02.016. [DOI] [PubMed] [Google Scholar]
- Price DD. Psychologicial and neural mechanisms of pain. New York: Raven Press; 1988. [Google Scholar]
- Price DD. Psychological mechanisms of pain and analgesia: Progress in pain research and management. 15. Seattle: IASP Press; 1999. [Google Scholar]
- Price DD, Browe AC. Responses of spinal cord neurons to graded noxious and non-noxious stimuli. Brain Res. 1973;64:425–429. doi: 10.1016/0006-8993(73)90199-6. [DOI] [PubMed] [Google Scholar]
- Price DD, Harkins SW. Psychophysical approaches to pain measurement and assessment. In: Turk DC, Melzack R, editors. Handbook of Pain Assessment. Vol. 1. New York: Guilford Press; 1992. pp. 114–134. [Google Scholar]
- Price DD, Patel R, Robinson ME, Staud R. Characteristics of electronic visual analogue and numeric scales for ratings of experimental pain in healthy subjects and fibromyalgia patients. Pain. 2008;140:158–166. doi: 10.1016/j.pain.2008.07.028. [DOI] [PubMed] [Google Scholar]
- Reynolds CR, Kamphaus RW. BASC-2 Behavior assessment system for childfen, second edition manual. 2. Circle Pines, MN: American Guidance Service; 2004. [Google Scholar]
- Roca X, Olson AJ, Rao AR, Enerly E, Kristensen VN, Borresen-Dale AL, Andresen BS, Krainer AR, Sachidanandam R. Features of 5′-splice-site efficiency derived from disease-causing mutations and comparative genomics. Genome Res. 2008;18(1):77–87. doi: 10.1101/gr.6859308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush AM, Dib-Hajj SD, Liu SJ, Cummins TR, Black JA, Waxman SG. A single sodium channel mutation produces hyper or hypoexcitability in different types of neurons. PNAS. 2006;103(21):8245–8250. doi: 10.1073/pnas.0602813103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangameswaran L, Fish LM, Koch BD, Rabert DK, Delgado SG, Ilnicka M, Jakeman LB, Novakovic S, Wong K, Sze P, Tzoumaka E, Stewart GR, Herman RC, Chan H, Eglen RM, Hunter JC. A novel tetrodotoxin-sensitive, voltage-gated sodium channel expressed in rat and human dorsal root ganglia. J Biol Chem. 1997;272(23):14805–14809. doi: 10.1074/jbc.272.23.14805. [DOI] [PubMed] [Google Scholar]
- Shatzky S, Moses S, Levy J, Pinsk V, Hershkovitz E, Herzog L, Shorer Z, Luder A, Parvari R. Congenital insensitivity to pain with anhidrosis (CIPA) in Israeli-Bedouins: genetic heterogeneity, novel mutations in the TRKA/NGF receptor gene, clinical findings, and results of nerve conduction studies. Am J Med Genet. 2000;92(5):353–360. doi: 10.1002/1096-8628(20000619)92:5<353::aid-ajmg12>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Singer T, Frith C. The painful side of empathy. Nat Neurosci. 2005;8(7):845–846. doi: 10.1038/nn0705-845. [DOI] [PubMed] [Google Scholar]
- Singh NA, Pappas C, Dahle EJ, Claes LR, Pruess TH, De Jonghe P, Thompson J, Dixon M, Gurnett C, Peiffer A, White HS, Filloux F, Leppert MF. A role of SCN9A in human epilepsies, as a cause of febrile seizures and as a potential modifier of Dravet syndrome. PLoS Genet. 2009;5(9):e1000649. doi: 10.1371/journal.pgen.1000649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staud R, Robinson ME, Price DD. Do past pain events systematically impact pain ratings of healthy subjects or fibromyalgia patients? J Pain. 2010;11(2):142–148. doi: 10.1016/j.jpain.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot JD, Marrett S, Evans AC, Meyer E, Bushnell MC, Duncan GH. Multiple representations of pain in human cerebral cortex. Science. 1991;251(4999):1355–1358. doi: 10.1126/science.2003220. [DOI] [PubMed] [Google Scholar]
- Toledo-Aral JJ, Moss BL, He ZJ, Koszowski AG, Whisenand T, Levinson SR, Wolf JJ, Silos-Santiago I, Halegoua S, Mandel G. Identification of PN1, a predominant voltage-dependent sodium channel expressed principally in peripheral neurons. Proc Natl Acad Sci U S A. 1997;94(4):1527–1532. doi: 10.1073/pnas.94.4.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wechsler D. WISC-IV administrative and scoring manual. San Antonio, TX: The Psychological Corporation; 2003. [Google Scholar]
- Yang Y, Wang Y, Li S, Xu Z, Li H, Ma L, Fan J, Bu D, Liu B, Fan Z, Wu G, Jin J, Ding B, Zhu X, Shen Y. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J Med Genet. 2004;41(3):171–174. doi: 10.1136/jmg.2003.012153. [DOI] [PMC free article] [PubMed] [Google Scholar]