

Abstract
Background
Data regarding possible ion channel mechanisms that predispose to ventricular tachyarrhythmias in patients with phenotype-negative long-QT syndrome (LQTS) are limited.
Methods and Results
We carried out cellular expression studies for the S349W mutation in the KCNQ1 channel, which was identified in 15 patients from the International LQTS Registry who experienced a high rate of cardiac events despite lack of significant QTc prolongation. The clinical outcome of S349W mutation carriers was compared with that of QTc-matched carriers of haploinsufficient missense (n=30) and nonsense (n=45) KCNQ1 mutations. The channels containing the mutant S349W subunit showed a mild reduction in current (<50%), in the haploinsuficient range, with an increase in maximal conductance compared with wild type channels. In contrast, expression of the S349W mutant subunit produced a pronounced effect on both the voltage dependence of activation and the time constant of activation, while haploinsuficient channels showed no effect on either parameter. The cumulative probability of cardiac events from birth through age 20 years was significantly higher among S349W mutation carriers (58%) as compared with carriers of QTc-matched haploinsufficent missense- (21% p=0.004) and nonsense- (25%; p=0.01) mutations.
Conclusions
The S349W mutation in the KCNQ1 potassium channel exerts a relatively mild effect on the ion channel current, whereas an increase in conductance compensates for impaired voltage activation of the channel. The changes observed in voltage activation of the channel may underlie the mechanisms predisposing to arrhythmic risk among LQTS patients with a normal-range QTc.
Keywords: long-QT syndrome, corrected QT interval, potassium ion channel current, ventricular tachycardia, sudden death
INTRODUCTION
The congenital long-QT syndrome (LQTS) is an inherited channelopathy that is characterized by a prolonged corrected QT interval (QTc) and an increased predisposition for polymorphic ventricular arrhythmias and sudden cardiac death (SCD) in young individuals without structural heart disease.1 Genetic mutations leading to LQTS have been identified in 12 susceptibility genes, with the LQT1-3 genotypes comprising more than 95% of genotype positive LQTS.1 Prior studies have shown that prolongation of the corrected QT interval (QTc) is a major risk factor for arrhythmic events in LQTS patients.2-5 Consistently, mutations that cause a ≤50% reduction in ion current have been shown to be associated with both a milder prolongation of QTc and lower risk of cardiac arrhythmias.6 Mild reductions in channel current are typically caused by a decrease in the total level of ion channel protein expressed due to mutations that either forms no or decreased functional protein (haploinsuficiency). However, currently there are limited data regarding possible ion-channel mechanism that may predispose to arrhythmic risk in LQTS patients who do not exhibit the phenotypic QTc prolongation of the disease (i.e., phenotype-negative LQTS). To gain an understanding of the molecular mechanism associated with predisposition for ventricular arrhythmias among patients with phenotype-negative LQTS, we carried out cellular expression studies for channels formed with the KCNQ1 (S349W) mutant subunit, which was identified in the International LQTS Registry as being associated with a high rate of cardiac events and SCD among patients who exhibited a normal-range QTc. Specifically, we hypothesized that since abnormalities in ion-channel current that lead to ventricular arrhythmias are likely associated also with QTc prolongation, additional mechanisms may predispose to arrhythmic risk in LQTS patients with a normal-range QTc.
METHODS
Study Subjects
Study subjects were identified from a population of 731 patients from the International LQTS Registry who were genetically tested and found positive for a known KCNQ1 mutation. Within this population, 15 subjects from 3 families were identified as carriers of the S349W mutation. The clinical course of S349W mutation carriers was compared with QTc-matched LQTS patients who were identified as carriers of mutations previously reported to have a haploinsufficient effect on the KCNQ1 channel (missense: R190Q, R591H, R594Q; nonsense: eA150fs/133, Y171X, L191fs/90, R195fs/40, P400fs/62, P448fs/13, R518X, Q530X, S571fs, A636fs).6-10 Matching of the haploinsufficient mutations was performed on a 2:1 basis for missense mutations and on a 3:1 basis for nonsense mutations. (i.e., 2 [missense] and 3 [nonsense] patients with haploinsufficient mutations were matched by QTc duration [within ± 10 msec] for each patient with the S349W mutation).
The LQTS genotype was determined using standard mutational analytic techniques involving 5 established genetic laboratories associated with the International LQTS Registry. Informed consent for genetic and clinical data was obtained from all patients.
Data Collection and phenotypic definition
Routine clinical and rest ECG parameters were acquired at the time of enrollment in the International LQTS Registry. Measured parameters on the first recorded ECG included QT and R-R intervals in milliseconds, with QT corrected for heart rate by Bazett’s formula.11 A normal-range QTc was defined as ≤440 msec according to accepted criteria for the phenotypic definition of LQTS.12 Clinical data were collected on prospectively designed forms with information on demographic characteristics, personal and family medical history, ECG findings, LQTS-related therapies (including β-blockers, left cardiac sympathetic denervation, and implantation of a pacemaker or a defibrillator), and LQTS-related events during long-term follow-up.
Genotype Characterization
Genetic alterations of the amino acid sequence were characterized by location and by the specific mutation (missense, splice site, in-frame insertions/deletions, nonsense, and frameshift). The transmembrane region of each of the KCNQ1-encoded Kv7.1 channel was defined as amino acid residues from 120 through 355 (S1-6 region).6 The genotype characterization of the 14 KCNQ1 mutations included in the study is presented in Table 1 and the location of the mutations in the KCNQ1 channel is presented in Figure 1.
Table 1.
KCNQ1 mutations by location and coding, type of mutation, and functional Effect.
| LOCATION | NO. OF SUBJECTS | TYPE OF MUTATION | FUNCTIONAL EFFECTS* |
|---|---|---|---|
| Transmembrane | |||
| S349W | 15 | Missense | Unknown |
| R190Q | 3 | Missense | Haploinsufficient |
| R591H | 15 | Missense | Haploinsufficient |
| R594Q | 12 | Missense | Haploinsufficient |
| A150fs/133 | 2 | Nonsense | Haploinsufficient |
| Y171X | 4 | Nonsense | Haploinsufficient |
| L191fs/90 | 5 | Nonsense | Haploinsufficient |
| R195fs/40 | 2 | Nonsense | Haploinsufficient |
| P400fs/62 | 3 | Nonsense | Haploinsufficient |
| P448fs/13 | 5 | Nonsense | Haploinsufficient |
| R518X | 5 | Nonsense | Haploinsufficient |
| Q530X | 15 | Nonsense | Haploinsufficient |
| S571fs | 2 | Nonsense | Haploinsufficient |
| A636fs | 2 | Nonsense | Haploinsufficient |
Functional effects are based on previously reported data (6-10); the functional effect of the S349W mutation was not reported in prior studies.
Figure 1.
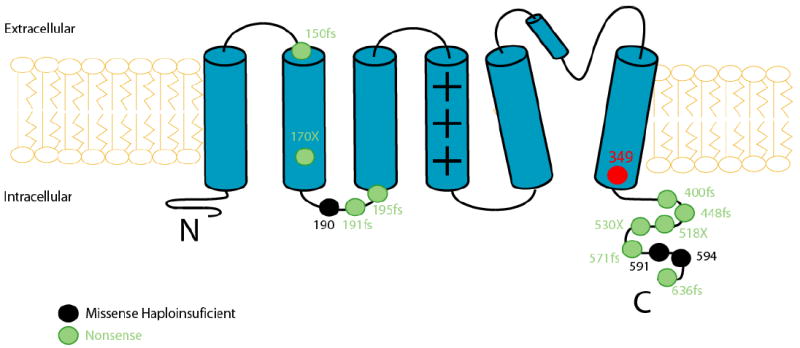
Location of the S349W mutation and the QTc-matched missense and nonsense mutations in the KCNQ1 channel.
Cellular Expression Studies
QT prolongation in LQTS subjects is most commonly produced by delayed repolarization that is associated with reductions in ion channel currents.13 We measured a number of standard functional ion channel characteristics, including changes in activation and deactivation time course, voltage dependence of activation and conductance, to investigate possible pathophysiological disturbances that may predispose LQTS patients with normal-range QTc to ventricular tachyarrthymias. The ion channel characteristics of wild-type channels co-expressed with the S349W mutant subunit (WT+S349W) were compared with those of: 1) wild-type channels; 2) wild-type channels from cells injected with half of their amount of RNA, mimicking a haploinsufficient mutation (WT + H20); and 3) wild-type R591H and R594Q mutant subunits co-expressed (WT+R591H and WT+R594Q, respectively)
Molecular Biology
Human KCNQ1, KCNE1 clones were subcloned in the pGEMsh vector (modified from PGEMHE vector) for oocyte expression.14,15 PCR based site direct mutagenesis was performed using PFU ultra DNA polymerase (Stratagene). Construct sequences were confirmed by DNA sequencing (Cornell, Ithaca). cRNAs were transcribed using the “message machine” kit (Ambion). RNA concentration was estimated using RNA markers (Gibco).
Electrophysiology
Xenopus oocytes were harvested, dissociated and defolliculated by collagenase type I (Sigma) treatment. Channel subunits with the mutations were expressed in combination with wild type subunits to mimic a heterozygous mutation. KCNQ1:KCNE1 cRNA was injected either at a molar ratio of 1:1 (2ng:0.4ng) or 0.5:1 (1ng:0.4ng) in order to mimic the haploinsuficient phenotype. Mutant S349W, R591H, and R594C mutation cRNA was injected at a molar ratio 1:1 (wild-type:mutant), with 1ng wild-type KCNQ1 : 1ng mutant KCNQ1 : 0.4ng KCNE1 per oocyte being injected. Since all mutant subunits were subcloned in the same expression vector, an equivalent amount of protein is expected to be transcribed for each mutant and wild type proteins. Whole-oocyte currents were measured with a GeneClamp 500 amplifier (Axon Instruments). Agarose-cushioned microelectrodes were used with resistances between 0.1 to 1.0 MΩ. Oocytes were constantly superfused with (in mM): 82.5 NaCl, 2 KCl, 1 MgCl2, 1.8 CaCl2, 5 NaOH/HEPES (pH 7.5). Currents were evaluated 1 minute after oocytes impailment. At least 10 oocytes of the same batch and 2-3 oocyte batches were used. One-way ANOVA followed by Dunnett’s Post Hoc test was applied for the assessment of statistical significance when comparing the channel expressing the mutant subunit to wild type channel function. KCNE1 expressed by itself at the concentrations used for these experiments yielded currents at least 10 times smaller than of the S349W mutation currents measured. This current did not significantly affect our analysis of the expressed channels.
Because IKs does not reach a steady level even after long depolarizations at room temperature, we constructed isochronal (t=8s) activation curves to determine the voltage dependence of IKs. Experiments using longer depolarizing pulses (18s compared to 2.7s) showed that the length of the depolarizing pulse affects the voltage dependence of IKs, but relative shifts in the voltage dependence persist independently of the length of the pulse. We measured the IKs tail current at -40 mV after depolarization to a series of voltage steps from -50 to +80 mV every 10 mV. A Boltzmann fit (G = gmax/(1 + exp[-(V - V1/2)/k) of these data was used to determine the voltage that elicits half of the maximal activation (V1/2) of activation. V1/2 values indicate channel sensitivity to activation by voltage. Time constant for activation and deactivation (τ activation: τact and τ deactivation: τdeact) was determined by fitting the activation current with a single exponential. A single exponential fits well the current activation time course at +40mV for human channels (R>0.98). To assess the effect of expression of the S349W mutant channel subunit on the activated current ,we measured the activated current at +40 mV after 4s depolarization (I). All experiments were performed at room temperature.
Western Blotting
Forty-eight hours after RNA injection, oocytes were washed once in oocyte homogenization buffer (80 mM sucrose, 1 mM EDTA, 20 mM Tris/HCl, pH7.4) and homogenized in 20 ul/oocyte of homogenization buffer containing a protease inhibitor cocktail diluted 1:50 (Sigma P2714) by a 20-time passage through a 25 gauge needle. Lysates were centrifuged twice at 200 × g for 5 min at 4° C. The supernatant was collected after the second spin and centrifuged at 14,000 × g for 20 min at 4° C. The pellet was re-suspended in 4 ul/oocyte of 1X SDS Laemmli Buffer (25 mM Tris, 192 mM glycine, 0.1% w/v SDS) containing protease inhibitor cocktail diluted 1:50 (Sigma P2714). Five uL of oocyte lysate mixed with 5 uL 2X Sample Buffer were heated at 60° C for 10 minutes and resolved on a 10% Tris-HCl polyacrylamide gel (BioRad 161-1119). Anti-KCNQ1 antibody was used for immunodetection (Santa Cruz 10646 C-20).
Statistical Analysis
The clinical characteristics of study patients with the S349W mutation were compared with the corresponding characteristics of QTc-matched patients who were carriers of haploinsufficient missense and nonsense KCNQ1 mutations, using the Wilcoxon rank-sum, chi square or Fisher’s exact test, as appropriate. The Kaplan-Meier estimator was used to assess the cumulative event rates, and groups were compared using the log-rank test.
The statistical software used for the analyses was SAS version 9.20 (SAS Institute Inc, Cary, NC). A 2-sided 0.05 significance level was used for hypothesis testing.
RESULTS
Phenotype characterization of S349W mutation carriers
The clinical and ECG characteristics of study patients with the S349W mutation are shown in Table 2. Carriers of the S349W mutation exhibited relatively narrow QTc durations (range: 390 msec to 500 msec; mean [±SD]: 447 ± 28 msec; median: 450 msec [interquartile-range 440 msec to 460 msec). Five patients (33%) with the S349W mutation had a QTc duration of ≤440 msec that is considered to be within the normal-range.12
Table 2.
Baseline and follow-up characteristics of the patients with the S349W mutation
| Characteristics | Patients with S349W mutation (n=15) | Patients with QTc- matched missense mutations (n=30) | patients with QTc- matched nonsense mutations (n=45) | P-value* |
|---|---|---|---|---|
| Female, n (%) | 7 (47) | 24 (60) | 27 (60) | 0.35 |
| Probands, n (%) | 1 (7) | 2 (7) | 2 (4) | 0.87 |
| Family history of SCD | 2 (11) | 4 (8) | 4 (9) | 0.69 |
| QTc (msec) | ||||
| Mean ± SD | 447 ± 28 | 448 ± 32 | 448 ± 32 | NA† |
| Median (IQ range) | 450 (440. 460) | 450 (440. 460) | 450 (440. 460) | NA† |
| ≤ 440 | 5 (33) | 11 (37) | 18 (38) | NA† |
| RR (msec) | ||||
| Mean ±SD | 801 ± 25 | 777 ± 21 | 777 ± 21 | 0.74 |
| Median (IQ range) | 870 (640, 940) | 850 (535, 930) | 850 (535, 930) | 0.92 |
| Therapies | ||||
| β-blockers | 10 (67) | 19 (63) | 28 (62) | 0.56 |
| Pacemaker | 0 (0) | 0 (0) | 0 (0) | - |
| LCSD | 0 (0) | 0 (0) | 0 (0) | - |
| ICD | 0 (0) | 0 (0) | 0 (0) | - |
| Events | ||||
| Syncope | 5 (33) | 5 (17) | 8 (18) | 0.03 |
| ACA | 0 (0) | 0 (0) | 0 (0) | - |
| SCD | 2 (13) | 0 (0) | 0 (0) | 0.12 |
| Cardiac events of any typeठ| 7 (47) | 5 (17) | 8 (18) | 0.02 |
| QTc in patients with events (msec) | ||||
| SCD cases | 440 | NA | NA | - |
| Cardiac events of any type, mean ± SD | 445 ± 10 | 454 ± 12 | 457 ± 11 | 0.27 |
p-value for the comparison among the 3 subgroups.
A statistical comparison is not applicable since groups were matched based on QTc.
During follow-up, 7 patients (47%) experienced cardiac events at a mean age of 11 ± 5 years, despite having a relatively narrow QTc duration (445 ± 10 msec). Furthermore, 2 patients with S349W mutation experienced SCD, despite having a QTc that is within the normal-range and displaying no overt abnormalities on the baseline ECG (Fig. 2).
Figure 2.
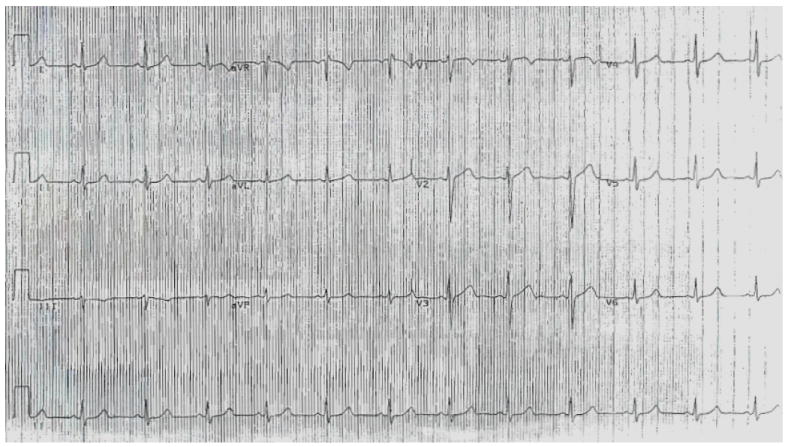
Baseline ECG (obtained at age 9 years) of a patient with the S349W mutation who experienced sudden cardiac death at age 13 years. No repolarization abnormalities are observed, and the corrected QT interval is within the normal-range (approximately 430 msec).
Cellular expression studies
We measured ion channel currents, voltage dependence of activation, and time constant of activation and deactivation associated with the channel expressing wild-type KCNQ1 together with KCNQ1 S349W mutant at a ratio of 1:1 (Fig 3). IKs currents showed a relatively mild decrease for channels expressing KCNQ1 S349W when compared with wild type channels (52±4% of wild type measured at +40 mV after 4 s depolarization, n=25). Typical currents measured through wild-type channels (WT+WT), wild-type channels from cells injected with half of their amount of RNA mimicking a haploinsufficient mutation (WT + H20), and wild-type and S349W and R591H mutant subunits co-expressed (WT+S349 and WT+R591H) are shown in Figure 3A and highlight the relatively mild effect of the S349W mutation on the current (depolarization compared at +40mV highlighted in red). Typical voltage dependence of activation curves are depicted in Figure 3B (from data shown in Fig.3A), showing decreased activation of the S349W containing channels at less depolarized voltages, but increased channel activation for stronger depolarizing voltages when compared with the haploinsuficient channel. These opposing effects underlie the overall mild effect of the mutation on channel current. Summary data are shown in Figure 3C. The S349W containing channels showed an increase in channel maximal conductance, when compared with both the haploinsuficient- and wild type-channels (Fig. 3C right panel). In addition, the S349W containing channels were associated with a right shift in the voltage dependence of activation and slower activation kinetics compared with the wild type channels, whereas the haploinsuficient channel was associated with a decrease in maximal conductance without a significant change in other parameters (Fig. 3C).
Figure 3.
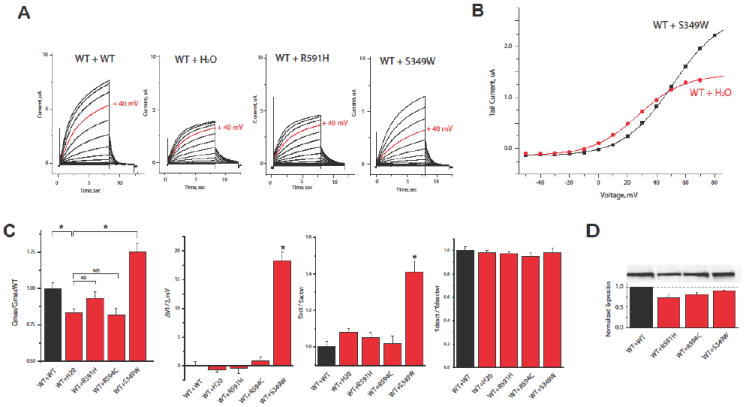
Results from cellular expression studies of the KCNQ1 (S349W) mutation that investigated: (A) Typical ion channel currents associated through wild-type channels (WT+WT), wild-type channels from cells injected with half of their amount of RNA, mimicking a haploinsufficient mutation (WT + H2O), wild-type and S349W mutant subunits co-expressed (WT+S349W); wild-type and R591H mutant subunits co-expressed (WT+R591H) Currents elicited by +40mV depolarization are highlighted in red (B) voltage dependence of activation from data in A; and (C) summary results for changes in maximal conductance voltage dependence of activation, the time constant of activation and time constant of deactivation for WT, WT+ H2O, WT+S349W, WT+R591H and WT+R594C channels (n ≥ 25). (D) Oocytes were homogenized and membrane protein isolated, western-blotted and probed with KCNQ1 antibody. A typical western blot is shown in the top panel showing the relative expression of wild type and mutant protein subunits, as indicated. The band intensities were quantified by Image J software and an average of four blots shown in the bottom panel.
Consistent with the haploinsufficient effect that was previously reported for the R591H and R594Q KCNQ1 missense mutations,9,10 the WT+R591H and WT+R594Q channels showed equivalent currents and activation kinetics to those measured in cells mimicking a haploinsufficient mutation (WT + H20 [Fig. 3]). Cells containing the S349W subunits did not show an increase in expression levels compared to wild type channels (Fig. 3D), suggesting that an increase in expression does not underlie the observed increase in conductance. A mild decrease in protein expression was observed for cells expressing R591H and R594C subunits. Haploinsuficiency occurs when the number of functional channels expressed in the cell is reduced, thus the decrease in protein expression observed for R591H and R594C is consistent with the haploinsuficient functional phenotype observed for these mutants.
Comparison of the clinical course of KCNQ1(S349W) mutation carriers to that of QTc-matched KCNQ1 mutations with a haploinsufficent effect
To further assess the genotype-phenotype correlations of KCNQ1 mutations with a haploinsufficient effect on the ion channel current, we compared the outcome of study patients with the S349W mutation to that of QTc-matched carriers of missense (n=30) and nonsense (n=45) mutations with a haploinsufficient effect on the KCNQ1 channel.6-10 The clinical characteristics of haploinsufficient- mutation carriers were similar to those of patients with the S349W mutation (Table 1). However, during follow-up, S349W mutation carriers had a >2-fold higher frequency of cardiac events (47%) as compared with carriers of haploinsufficient missense- (17%) and nonsense- (18%) mutations. Accordingly, the cumulative probability of a first cardiac event from birth through age 20 years was significantly higher among S349W mutation carriers (58%) as compared with carriers of QTc-matched missense- (21%; p=0.004 [Fig. 5A]) and nonsense- (25%; p=0.01 [Fig. 5B]) mutations. Furthermore, none of the carriers of KNCQ1 haploinsufficent- mutations experienced fatal events during follow-up, whereas 2 patients (13%) with the KNCQ1 S349W mutation experienced SCD prior to age 20 years despite a normal-range QTc on the baseline ECG (Table 2; Fig. 2).
DISCUSSION
The present study is the first to explore possible ion channel mechanisms that may predispose to arrhythmic risk in LQTS patients who do not exhibit the phenotypic QTc prolongation of the disease. We have shown that the S349W mutation exerts a relatively mild effect on the ion channel current, which is within the haploinsufficient range, explaining the lack of a significant prolongation of the corrected QT interval among all S349W mutation carriers. Despite this, however, the S349W mutation was associated with a high rate of cardiac events and SCD during the childhood and adolescence periods. Therefore, it is possible that the pronounced effect of the mutation on the voltage and time dependence of activation may explain the predisposition for arrhythmic events among S349W mutation carriers. These findings stress the importance of mutation-specific functional data for risk assessment in phenotype-negative LQTS.
The clinical course of patients with the congenital long QT syndrome was shown to be variable due to incomplete penetrance. It is influenced by age, genotype, gender, environmental factors, therapy, and possibly other modifier genes.1-6 Recent studies from the International LQTS Registry have assessed the risk for life-threatening events in LQTS patients,3-5 and these studies have consistently demonstrated that ECG and clinical risk factors, including the QTc and age-gender interactions, identify increased risk in the LQTS population. During childhood, affected males and patients with a prolonged QTc were shown to exhibit a higher risk for life threatening events,3 whereas after the onset of adolescence females and patients with a prolonged QTc were shown to exhibit a higher risk.4,5 These studies, however, included mainly phenotype-positive LQTS patients with a QTc ≥450 msec. Thus, it is possible that clinical and ECG factors have more limited applicability for risk assessment among phenotype-negative LQTS patients. Our findings extend prior data, and suggest that genetic information related to the functional effects of the LQTS mutations can be used to identify predisposition to arrhythmic risk in mutations that are associated with a relatively mild effect on the ion channel current and QTc duration.
A recent genotype-phenotype study from the International LQTS Registry has provided important information regarding the effect of the biophysical function of the channel mutations on the phenotypic manifestations and clinical course of LQTS patients.6 The study demonstrated that the biophysical function of KCNQ1 mutations, categorized according to dominant-negative (>50%) or haploinsufficiency (≤50%) reduction in cardiac repolarizing IKs potassium channel current, is an important determinant of outcome. Patients with dominant-negative ion channel dysfunction had more than a 2-fold increase in the risk of cardiac events compared with those who harbored mutations with haploinsufficiency effect.6 The present study consistently shows that the rate of cardiac events among carriers of haploinsufficent- KCNQ1 mutations is relatively low. However, in contrast to the previous report, we have demonstrated that the S349W mutation, which exerts an effect comparable to haploisufficient currents on the IKs channel and on QTc duration, is associated with a high rate of cardiac events. Furthermore, carriers of the S349W mutation were shown to experience SCD despite a normal ECG on baseline risk assessment. These findings suggest that additional ion channel mechanisms may predispose to ventricular tachyarrhythmias in high risk mutations that are associated with mild effects on the ion channel current and QTc duration. The S349W mutation was shown to exert a pronounced effect on the channel activation, including the time constant and the voltage dependence of activation. In contrast, haploinsufficent channels did not affect either parameter of activation. Consistent with these findings, the cumulative probability of cardiac events during childhood and the adolescence period was significantly higher (58%) among carriers of the S349W mutation as compared with QTc-matched KCNQ1 patients with haploinsufficient missense- and nonsense-mutations (21% and 25%, respectively).
Normal cardiac repolarization depends critically on the interplay of multiple ion mechanisms, and these provide some redundancy, or ‘reserve’, to protect against excessive QT prolongation by drugs. Accordingly, a genetic predisposition for reduced repolarization reserve has been suggested to play a role in patients with acquired long-QT syndrome with normal rest QTc durations who experience marked QT prolongation and torsades de pointes following exposure QT-prolonging drugs.16 Similarly, it is possible that patients with phenotype-negative congenital LQTS who harbor ion channel mutations that have a mild effect on current, but also affect channel activation, may be sensitive to drug-induced QT-prolongation despite the fact that the mutation has a relatively mild on ion channel current and QTc duration.
Study limitations
Our findings suggest that changes in the kinetics of activation of the IKs channel may predispose to arrhythmic risk in patients who harbor KCNQ1 mutations that are associated with a relatively mild effect on ion channel current and QTc duration. However, it is possible that additional mechanisms (including environmental, hormonal, the effect of modifier genes, and time-dependent changes in the phenotypic expression of LQTS) that were not explored in the present study may predispose to arrhythmic risk in phenotype-negative LQTS.
Changes in wild type and mutant subunit assembly may contribute to the functional changes caused by mutations, and the stoichiometry of functional channels may depend on the nature of the mutant subunit expressed. Channels formed by different combinations of wild type and mutant subunits co-exist, and contribute to the total macroscopic current. In this study we did not assess the contribution of each channel type to the total current. We assumed that channel assembly and wild-type and mutant subunit stoichiometry in the expression system reflected assembly of the channel in the native environment.
Study patients were not distributed in equivalent proportions among the different mutations (Table 1). Therefore, it is possible the association between mutation and risk may be biased by the inclusion of a higher frequency of symptomatic patients or their family members in the registry. To reduce a possible selection bias, we matched mutation categories by QTc duration. Thus, as seen in Table 2, no statistically significant differences in baseline characteristics (including the frequency of probands and a family history of SCD) were observed among the 3 categories of patients with the S349W mutation, missense mutations, and nonsense mutations, whereas the rate of cardiac events was significantly higher among patients with the S349W mutation.
Conclusions and clinical implications
In recent years there has been a substantial rise in the use of genetic testing for the identification of heritable cardiac arrhythmic disorders. This trend has resulted in a corresponding increase in the diagnosis of mutation-positive asymptomatic young individuals who undergo genetic testing due to a diagnosis of a genetic disorder in a symptomatic family member. Despite this progress, however, a significant barrier remains in the identification of silent carriers of mutations that predispose to ventricular tachyarrhythmias. Our study suggests that genetic data relating to the cellular expression of the ion channel mutation may explain predisposition to arrhythmic risk in LQTS patients who do not exhibit the phenotypic QTc prolongation of the disease. Carriers of the S349W mutation, which was shown to exert a relatively mild effect on the ion channel current but a pronounced effect on channel activation, experienced SCD at an early age despite having a normal ECG during baseline examination. These findings suggest that genetic functional data should be employed for risk assessment in LQTS patients who exhibit a normal-range QTc.
Figure 4.
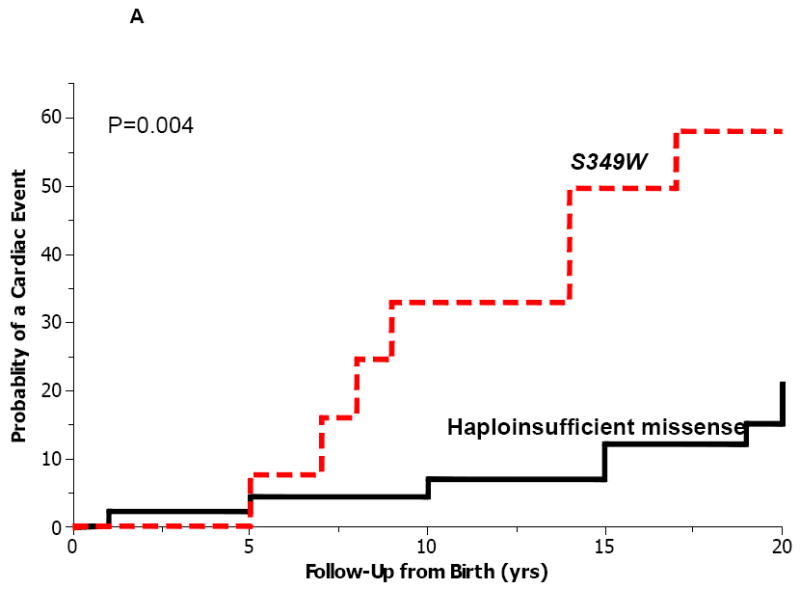
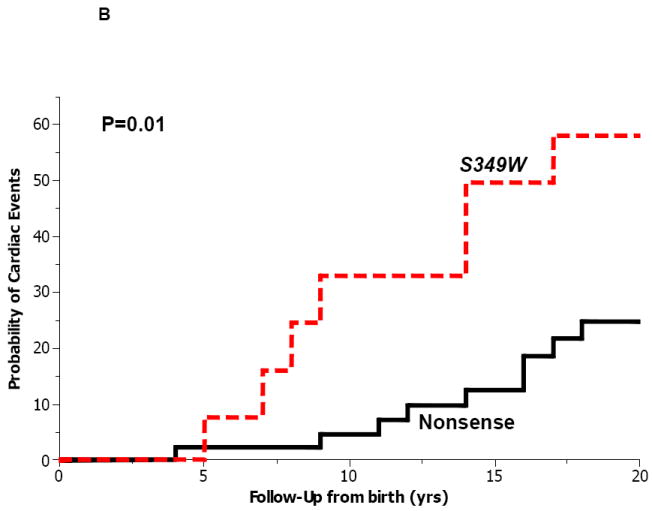
Kaplan-Meier estimates of the probability of a first cardiac event from birth through age 20 years among patients with the S349W mutation (n=15) and patients with QTc-matched haploinsufficient (A) missense (n=30); and (B) nonsense (n=45) mutations.
Acknowledgments
This study was supported in part by research grants HL-33843 and HL 51618 from the National Institutes of Health, Bethesda, Md.
Footnotes
No disclosures.
References
- 1.Goldenberg I, Moss AJ. Long QT Syndrome. J Am Coll Cardiol. 2008;51:2291–2300. doi: 10.1016/j.jacc.2008.02.068. [DOI] [PubMed] [Google Scholar]
- 2.Moss AJ, Schwartz PJ, Crampton RS, Tzivoni D, Locati EH, MacCluer J, Hall WJ, Weitkamp L, Vincent GM, Garson A., Jr The long QT syndrome. Prospective longitudinal study of 328 families. Circulation. 1991;84:1136–1144. doi: 10.1161/01.cir.84.3.1136. [DOI] [PubMed] [Google Scholar]
- 3.Goldenberg I, Moss AJ, Peterson DR, McNitt S, Zareba W, Andrews ML, Robinson JL, Locati EH, Ackerman MJ, Benhorin J, Kaufman ES, Napolitano C, Priori SG, Qi M, Schwartz PJ, Towbin JA, Vincent GM, Zhang L. Risk Factors for aborted cardiac arrest and sudden cardiac death in children with the congenital long-QT syndrome. Circulation. 2008;29(117):2184–2191. doi: 10.1161/CIRCULATIONAHA.107.701243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hobbs JB, Peterson DR, Moss AJ, McNitt S, Zareba W, Goldenberg I, Qi M, Robinson JL, Sauer AJ, Ackerman MJ, Benhorin J, Kaufman ES, Locati EH, Napolitano C, Priori SG, Towbin JA, Vincent GM, Zhang L. Risk of aborted cardiac arrest or sudden cardiac death during adolescence in the long-QT syndrome. JAMA. 2006;296:1249–1254. doi: 10.1001/jama.296.10.1249. [DOI] [PubMed] [Google Scholar]
- 5.Sauer AJ, Moss AJ, McNitt S, Peterson DR, Zareba W, Robinson JL, Qi M, Goldenberg I, Hobbs JB, Ackerman MJ, Benhorin J, Hall WJ, Kaufman ES, Locati EH, Napolitano C, Priori SG, Schwartz PJ, Towbin JA, Vincent GM, Zhang L. Long QT syndrome in adults. J Am Coll Cardiol. 2007;49:329–337. doi: 10.1016/j.jacc.2006.08.057. [DOI] [PubMed] [Google Scholar]
- 6.Moss AJ, Shimizu W, Wilde AA, Towbin JA, Zareba W, Robinson JL, Qi M, Vincent GM, Ackerman MJ, Kaufman ES, Hofman N, Seth R, Kamakura S, Miyamoto Y, Goldenberg I, Andrews ML, McNitt S. Clinical aspects of type-1 long-QT syndrome by location, coding type, and biophysical function of mutations involving the KCNQ1 gene. Circulation. 2007;115:2481–2489. doi: 10.1161/CIRCULATIONAHA.106.665406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Z, Tristani-Firouzi M, Xu Q, Lin M, Keating MT, Sanguinetti MC. Functional effects of mutations in KvLQT1 that cause long QT syndrome. J Cardiovasc Electrophysiol. 1999;10:817–826. doi: 10.1111/j.1540-8167.1999.tb00262.x. [DOI] [PubMed] [Google Scholar]
- 8.Chouabe C, Neyroud N, Richard P, Denjoy I, Hainque B, Romey G, Drici MD, Guicheney P, Barhanin J. Novel mutations in KvLQT1 that affect Iks activation through interactions with Isk. Cardiovasc Res. 2000;45:971–980. doi: 10.1016/s0008-6363(99)00411-3. [DOI] [PubMed] [Google Scholar]
- 9.Grunnet M, Behr ER, Calloe K, Hofman-Bang J, Till J, Christiansen M, McKenna WJ, Olesen SP, Schmitt N. Functional assessment of compound mutations in the KCNQ1 and KCNH2 genes associated with long QT syndrome. Heart Rhythm. 2005;2:1238–1249. doi: 10.1016/j.hrthm.2005.07.025. [DOI] [PubMed] [Google Scholar]
- 10.Huang L, Bitner-Glindzicz M, Tranebjaerg L, Tinker A. A spectrum of functional effects for disease causing mutations in the Jervell and Lange-Nielsen syndrome. Cardiovasc Res. 2001;51:670–680. doi: 10.1016/s0008-6363(01)00350-9. [DOI] [PubMed] [Google Scholar]
- 11.Bazett HC. An analysis of the time relations of electrocardiograms. Heart. 1920;7:353–367. [Google Scholar]
- 12.Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostic criteria for the long QT syndrome: an update. Circulation. 1993;88:782–784. doi: 10.1161/01.cir.88.2.782. [DOI] [PubMed] [Google Scholar]
- 13.Moss AJ, Kass RS. Long QT syndrome: from channels to cardiac arrhythmias. J Clin Invest. 2005;115:2018–3024. doi: 10.1172/JCI25537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matavel A, Lopes CM. PKC activation and PIP(2) depletion underlie biphasic regulation of IKs by Gq-coupled receptors. J Mol Cell Cardiol. 2009;46:704–712. doi: 10.1016/j.yjmcc.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matavel A, Medei E, Lopes CM. PKA and PKC partially rescue long QT type 1 phenotype by restoring channel-PIP(2) interactions. Channels. 2010 Jan 5;4(1) doi: 10.4161/chan.4.1.10227. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 16.Roden DM. Long QT syndrome: reduced repolarization reserve and the genetic link. J Intern Med. 2006;259:59–69. doi: 10.1111/j.1365-2796.2005.01589.x. [DOI] [PubMed] [Google Scholar]