

Abstract
Excessive chloride secretion in sweat (hyperchlorhidrosis), leading to a positive sweat test, is most commonly indicative of cystic fibrosis yet is found also in conjunction with various metabolic, endocrine, and dermatological disorders. There is conflicting evidence regarding the existence of autosomal-recessive hyperchlorhidrosis. We now describe a consanguineous Israeli Bedouin kindred with autosomal-recessive hyperchlohidrosis whose sole symptoms are visible salt precipitates after sweating, a preponderance to hyponatremic dehydration, and poor feeding and slow weight gain at infancy. Through genome-wide linkage analysis, we demonstrate that the phenotype is due to a homozygous mutation in CA12, encoding carbonic anhydrase XII. The mutant (c.427G>A [p.Glu143Lys]) protein showed 71% activity of the wild-type enzyme for catalyzing the CO2 hydration to bicarbonate and H+, and it bound the clinically used sulfonamide inhibitor acetazolamide with high affinity (KI of 10 nM). Unlike the wild-type enzyme, which is not inhibited by chloride, bromide, or iodide (KIs of 73–215 mM), the mutant is inhibited in the submicromolar range by these anions (KIs of 0.37–0.73 mM).
Main Text
Elevated sweat chloride concentrations, as detectable in the sweat test, serve as the classical diagnostic criterion for cystic fibrosis (CF [MIM 219700]).1 Although all CF patients have a positive sweat test (sweat chloride concentrations above 60 mmol/L), not all individuals with a positive sweat test have CF. Abnormal sweat chloride levels have been demonstrated in malnutrition, metabolic disorders (mucopolysacharidoses, type I fucosidosis, and glycogen storage diseases), dermatological diseases (congenital ectodermal dysplasia and atopic dermatitis), and various endocrine disorders, such as panhypopituitarism, Addison disease, isolated adrenocorticoid deficiency, vasopressin-resistant diabetes insipidus, hypothyroidism, and hypoparathyroidism.2 Hyperchlorhidrosis (MIM 143860), autosomal-recessive heredity of isolated excessive chloride exceretion, is an entity whose existence is in dispute. Greenburg et al.3 described 7- and 15-year-old Puerto Rican brothers with elevated sweat chloride. The younger brother was initially studied because of hyponatremic dehydration following sweating. Further analysis of the family identified an abnormal sweat test in the other brother. Both were generally healthy, with no evidence of CF or other disorders causing elevation of sweat chloride. Four other siblings were unaffected. The parents were not related. An unpublished report of the same family by Punnett (OMIM 143860) indicated that the two brothers were sterile (however, one had a 47,XXY karyotype) and suggested that they did have mild CF. Regretfully, the family was not available for molecular genetic testing for resolution of the conflicting claims.
We have identified a large Israeli Bedouin kindred presenting with an apparently autosomal-recessive phenotype of hyperchlorhidrosis (Figure 1). The initial affected individuals (IV:1 and IV:3; Figure 1) had undergone a sweat test as part of routine screening for CF when they presented with hyponatremic dehydration with coincidental gastroenteritis at infancy. The quantitative pilocarpine iontophoresis test (QPIT) was performed for the collection of sweat and a chemical analysis of its chloride content, per the standard guidelines of the Cystic Fibrosis Foundation.1 In some detail, at least 50–100 mg of sweat was collected. When a macroduct coil was used for collection, sweat was stimulated with a disposable Pilogel electrode using the Webster Sweat Inducer for 5 min. After 30 min, 15 μl was considered to be the minimum acceptable sample. Sweat chloride values of more than 60 mmol/L of chloride were considered to be abnormal. Screening of the entire kindred (Figure 1) identified seven individuals with sweat chloride levels substantially above 60 mmol/L (Table 1). The medical records of all seven affected individuals were reviewed, and all family members underwent careful clinical evaluation by a pediatric pulmonologist, a pediatric gastroenterologist, and a clinical geneticist. The study was approved by the institutional review board of Soroka Medical Center, and informed consent was obtained from all participants or their legal guardians. All seven affected individuals (and not their healthy siblings) reported dramatically excessive amounts of visible salt precipitates in sweat (especially in the nape). Five of the seven individuals (yet none of their nonaffected siblings) were hospitalized once before the age of one year for hyponatremic dehydration, with reports of poor feeding and slow weight gain. Sodium supplementation was effective in the treatment of hyponatremia in all cases. By one year of age, all affected individuals achieved normal height and weight and had no further feeding problems. Psychomotor development and intelligence of affected individuals was normal, and physical examination of all affected individuals demonstrated no further abnormal findings or dysmorphism. All affected individuals were too young to allow investigation of fertility. As mentioned above, medical records of five of the seven affected individuals demonstrated that they had a single episode of hyponatremic dehydration during infancy (mostly following mild gastroenteritis), with steatorrhea recorded in two of the cases. However, aside from these single episodes, repeat tests of serum and urinary electrolytes (including sodium and potassium) were normal in all affected individuals, as were serum amylase and fecal elastase. Renal function tests, renin-aldosterone axis function tests, urinalysis, and blood and urinary pH tests were normal. None of the affected individuals had any abnormal respiratory symptoms or signs, and none had chronic rhinitis or nasal polyps. All affected family members had normal pulmonary function tests (FEV1). Nasal potential difference (NPD) measurement, an electrophysiological test that assesses CF transmembrane conductance regulator (CFTR) activity and is a recognized diagnostic tool in CF, was tested in six affected individuals as previously described4 and was normal (data not shown).
Figure 1.
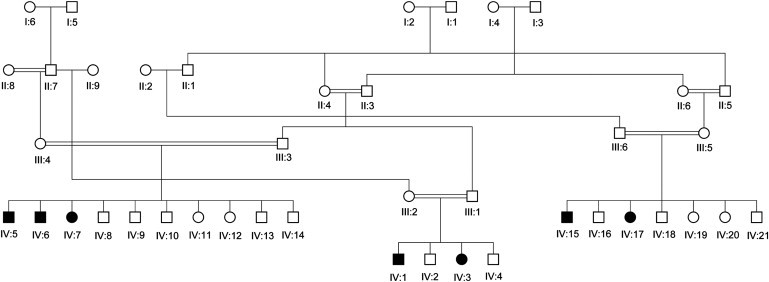
Pedigree of the Affected Israeli-Bedouin Kindred
The pedigree is compatible with autosomal-recessive heredity.
Table 1.
Sweat Test Measurements of Individuals in the Israeli Bedouin Kindred Studied
| Individual No. | Sweat Test (mmol/L) |
|---|---|
| III:1 | 41 |
| III:2 | 34 |
| IV:1 | 110 |
| IV:2 | ND |
| IV:3 | 156 |
| IV:4 | 25 |
| III:3 | 56 |
| III:4 | 54 |
| IV:5 | 95 |
| IV:6 | 128 |
| IV:7 | 116 |
| IV:8 | 35 |
| IV:9 | ND |
| IV:10 | 34 |
| IV:11 | 58 |
| IV:12 | 56 |
| IV:13 | 59 |
| IV:14 | 52 |
| III:5 | 29 |
| IV:15 | 155 |
| IV:16 | ND |
| IV:17 | 85 |
| IV:18 | 21 |
| IV:19 | 9.5 |
| IV:20 | 19 |
| IV:21 | 30 |
ND, not done.
Assuming autosomal-recessive heredity due to a founder effect, homozygosity mapping was performed to elucidate the molecular basis of the clinical phenotype. DNA samples of the seven affected individuals and 19 nonaffected family members were obtained with informed consent after approval of the Soroka Medical Center institutional review board. DNA was isolated from whole blood of affected individuals with the use of the FlexiGene DNA Extraction Kit per the manufacturer's instructions (QIAGEN) as previously described.5 Lymphoblast cell lines were established by Epstein-Barr virus (EBV) transformation via standard methods.6 We first tested the affected individuals for homozygosity at the locus of CFTR by using microsatellite markers as previously described (data not shown).7 In brief, microsatellite markers were derived from Marshfield maps. Intronic primer pairs were designed with the Primer 3 (version 0.4.0) software, based on DNA sequences obtained from the UCSC Genome Browser (sequences available on request). PCR products were separated on polyacrylamide gel with the use of silver staining for detection.7 Affected individuals did not share homozygosity at the CFTR locus (data not shown).
Next, we performed genome-wide linkage analysis testing seven affected individuals (Figure 1; individuals IV:1, IV:3, IV:5, IV:6, IV:7, IV:15, and IV:17), four obligate carrier parents (Figure 1; individuals III:1, III:2, III:3, and III:4), and one healthy sibling (Figure 1; individual IV:16). A genome-wide scan was performed at the microarray facility at the Biological Services Department of Ben Gurion University, with the use of the high-density-genotyping GeneChip Human Mapping 250K Array Nsp (Affymetrix) according to manufacturer's guidelines as previously described.8 Homozygosity-by-descent analysis was carried out with the use of an in-house-generated tool for homozygosity mapping (B.M., unpublished data). The analysis identified a segment of homozygosity on chromosome 15 that was common to all seven patients. Few additional significantly smaller regions of shared homozygosity, shorter than 1 Mb, were spotted. However, because none of these were contained within larger stretches of individual homozygosity, we focused on the chromosome 15 locus. The interval of homozygosity was confirmed, and fine mapping was performed by testing DNA samples of all seven affected individuals and 19 nonaffected members of the extended family, then genotyping with additional microsatellite markers derived from Marshfield maps or with ones designed with the Tandem Repeats Finder (TRF) program and the UCSC Genome Browser. Analysis was performed with the use of markers within the interval between D15S155 and D15S984: chr15:61090417-61090468, D15S970, D15S1036, D15S987, D15S1507, D15S153, D15S988, D15S983, D15S650, D15S131, D15S124, and D15S818. Haplotypes for the 26 family members were manually constructed and analyzed with GENEHUNTER.
All of the obligatory carriers were shown to be heterozygous for the haplotype. Furthermore, most unaffected family members were heterozygous for this haplotype. As seen in Figure 2, an 18.4 cM interval of homozygosity between markers chr15:61090417-61090468 and D15S650 (according to the Marshfield map), corresponding to ∼9.17 Mb, was common to all affected individuals in the family studied. To determine the exact borders of the homozygosity region shared by all of the affected individuals, we integrated the data from the polymorphic markers studies with the data of the SNP arrays. The phenotype-associated homozygosity locus was shown to lie within 11 cM (corresponding to ∼5.6 Mb) between chr15:61090417-61090468 and rs16949924 at physical positions 61090421 and 66727347 (UCSC Genome Browser, February 2009 assembly), respectively.
Figure 2.
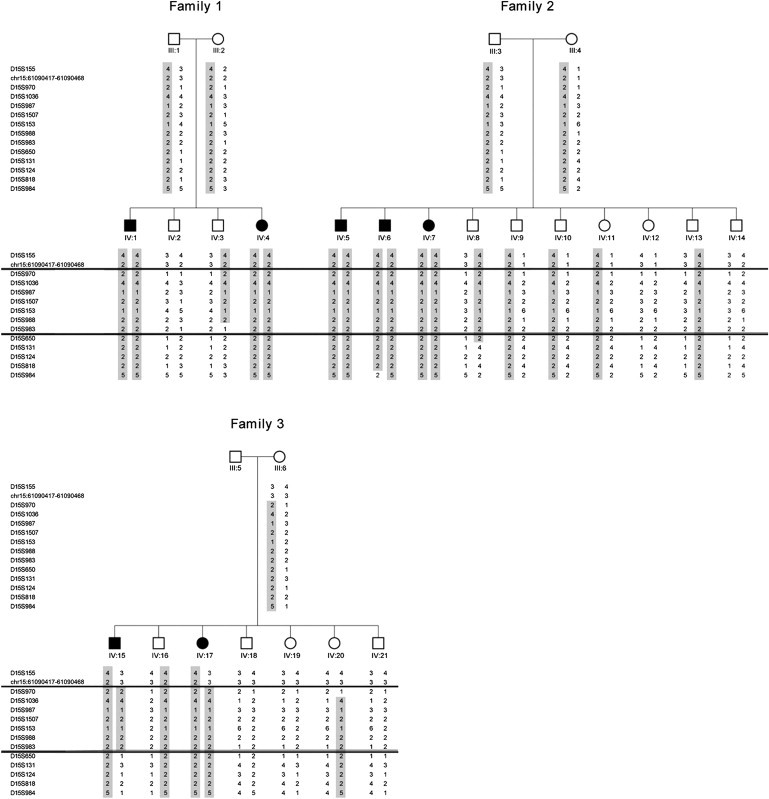
Fine Mapping of the Chromosome 15 Locus
Partial pedigree of the large Israeli-Bedouin kindered and disease-haplotype segregation. The two lines indicate a crossing over event. Disease associated haplotype shown in boxes. Markers chr15:61090417-61090468 and rs16949924 define the minimal homozygosity locus associated with the disease (Individual IV:15). The affected haplotype is shaded. Numbers below symbols correspond to the position of individuals within the pedigree (see Figure 1). Blank spaces indicate non-genotyped markers.
Two-point LOD-score calculation was performed with the use of SUPERLINK,9 assuming an autosomal-recessive mode of inheritance with a penetrance of 0.99, a disease mutant gene frequency of 0.0001, and a uniform distribution of allele frequencies. For LOD-score calculations, the number of alleles was set as the number observed in the pedigree, rather than as the number observed elsewhere, to provide a conservative estimate of the LOD score. Through testing of the entire 26 samples of the kindred, the maximum two point LOD score calculated was [Zmax] = 6.65 (θ = 0.0) at D15S153.
We then proceeded to sequence candidate genes within the identified locus. To obtain a prioritized list of the 56 candidate genes within the locus, we used our Syndrome to Gene (S2G) software,10 using CFTR as a reference gene. The software selects for any syndrome a prioritized list of candidate genes, on the basis of their known molecular interactions with genes associated with phenotypically similar syndromes. Candidate genes were sequenced from lymphoblastoid cDNA of patients: RNA was extracted from cultured cells of EBV-transformed lymphoblastoid cell lines with the use of the RNeasy Mini Kit (QIAGEN), and cDNA was reverse transcribed by the Verso RT-PCR Kits (TAMAR) as previously described.6 Primer pairs for PCR amplification from cDNA and/or exons of genomic DNA (including flanking intron sequences) of the genes in the putative chromosome 15 locus were designed on the basis of the known mRNA and genomic sequences with the use of Primer 3. Primer sequences and PCR conditions are available upon request. PCR products were directly sequenced with the ABI PRISM 3730 DNA Analyzer according to the manufacturer's protocols (Applied Biosystems, Foster City, CA, USA). Sequence variations were confirmed by bidirectional sequencing. Of the top candidate genes suggested by S2G, we sequenced SLC24A1 (MIM 603617) (encoding solute carrier family 24, a potassium-dependent sodium/calcium exchanger)11 and CA12 (MIM 603263). Testing cDNA and genomic DNA from lymphoblastoid cells of an affected individual, the entire coding regions and flanking ∼50 bp intron sequences of the two genes were sequenced. No mutation was found in SLC24A. A missense mutation (c.427G>A) was identified in exon 4 of CA12 (GenBank accession number NM_001218.3), predicted to generate p.Glu143Lys mutant carbonic anhydrase 12 protein (Figure 3). Testing for the CA12 mutation in the entire family and controls was performed via restriction analysis, based on the fact that the mutation creates an MnlI restriction site that does not exist in the wild-type sequence. PCR amplification of genomic DNA was performed with the use of the following primers: F-5′-TCCAGTCTCGCTACAGTGC-3′ and R-3′-AGAGCAGCCACTGCCAGACT-5′. MnlI restriction analysis of the PCR amplicons generated differential cleavage products of the wild-type (uncut, 139 bp) versus mutant (95 bp and 44 bp fragments) allele. Fragments were separated by electrophoresis on 3% agarose gel. Restriction analysis showed complete segregation (and thus full penetrance) of the mutation with the disease-associated phenotype in all investigated family members, heterozygosity in all obligatory carrier parents, and no homozygosity in nonaffected individuals. Of 300 healthy Bedouin controls, the mutation was found in only one individual in a heterozygous state and in none in a homozygous state.
Figure 3.
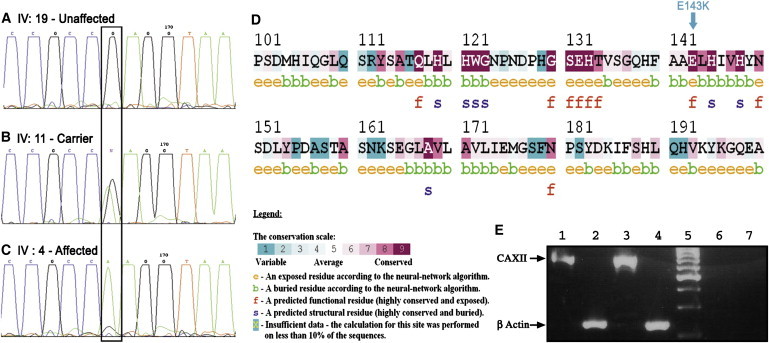
The c.427G>A (p.Glu143Lys) Mutation in Exon 4 of CA12, and CA12 Expression
Sequence analysis is shown for an unaffected individual (A), an obligatory carrier (B), and an affected individual (C). (D) ConSeq analysis for the p.Glu143Lys residue (marked with an arrow), demonstrating conservation grade 9 (highly conserved). (E) RT-PCR demonstrating that CA12 (as well as actin control) are transcribed in normal human foreskin (1,2) and arm skin (3,4). PCR primer sequences available upon request. Lane 5 = size marker. Lanes 6,7 represent negative controls for CA12 and actin PCR reactions, with no cDNA template.
To test whether the CA12 missense mutation alters the enzymatic activity of the encoded carbonic anhydrase (CA [EC 4.2.1.1]), the mutant p.Glu143Lys CA XII was cloned to generate GST-tagged mutant protein as previously reported by this group for the wild-type enzyme.12,13 The enzyme was purified in two steps as described earlier for the wild-type CA XII,12,13 and its activity was assayed by a stopped-flow method for the physiological reaction.14 An Applied Photophysics stopped-flow instrument was used for assaying the CA-catalyzed CO2 hydration activity.14 Phenol red (at a concentration of 0.2 mM) was used as an indicator, working at the absorbance maximum of 557 nm, with 20 mM HEPES (pH 7.4) and 20 mM NaBF4 (for maintaining constant ionic strength), following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor, at least six traces of the initial 5%–10% of the reaction were used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (10 mM) were prepared in distilled-deionized water, and dilutions up to 0.01 μM were performed thereafter with distilled-deionized water. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to the assay, in order to allow for the formation of the E-I complex. The inhibition constants were obtained by nonlinear least-squares methods with the use of PRISM 3, whereas the kinetic parameters for the uninhibited enzymes were from Lineweaver-Burk plots, as reported earlier,12,13 and represent the mean from at least three different determinations. The catalytic activity of p.Glu143Lys CA XII compared to that of wild-type CA XII and other members of the α-CA family, such as CA I, II, IV, VI, and IX, are shown in Table 2. Inhibition data with acetazolamide, a clinically used sulfonamide inhibitor [4] are also shown in Table 2. Table 3 and Figure 4 present the inhibition data of the mutant and wild-type CA XII with anions, compared to those of the physiologically relevant enzymes CA I and II.13,15
Table 2.
Kinetic Parameters and Inhibitory Properties of Acetazolamide for Different CA Isozymes, Including the Mutant CA XII15
| Isoenzyme | kcat (s−1) | KM (mM) | kcat/KM (M−1s−1) | KI (Acetazolamidea) (nM) | Subcellular Localization |
|---|---|---|---|---|---|
| hCA I | 2.0 × 105 | 4.0 | 5.0 × 107 | 250 | cytosol |
| hCA II | 1.4 × 106 | 9.3 | 1.5 × 108 | 12 | cytosol |
| hCA IV | 1.1 × 106 | 21.5 | 5.1 × 107 | 74 | GPI-anchored |
| hCA VI | 3.4 × 105 | 6.9 | 4.9 × 107 | 11 | secreted |
| hCA IX (catalytic domain) | 3.8 × 105 | 6.9 | 5.5 × 107 | 25 | transmembrane |
| hCA IX (full length) | 1.1 × 106 | 7.5 | 1.5 × 108 | 16 | transmembrane |
| hCA XII | 4.2 × 105 | 12.0 | 3.5 × 107 | 5.7 | transmembrane |
| hCA XII mutant | 3.0 × 105 | 11.9 | 2.5 × 107 | 9.9 | transmembrane |
5-acetamido-1,3,4-thiadiazole-2-sulfonamide.
Table 3.
Inhibition Constants for Anionic Inhibitors of Human Isozymes hCA I, hCAII, and hCAXII and Mutant hCA XII for the CO2 Hydration Reaction at 20°C14
| Inhibitor |
KIa |
[mM] |
||
|---|---|---|---|---|
| hCA I | hCA II | hCA XII | Mutant hCA XII | |
| F− | > 300 | > 300 | 0.56 | 0.59 |
| Cl− | 6 | 200 | 73 | 0.49 |
| Br− | 4 | 63 | 82 | 0.37 |
| I− | 0.3 | 26 | 215 | 0.73 |
| CNO− | 0.0007 | 0.03 | 0.73 | 0.58 |
| SCN− | 0.2 | 1.6 | 0.80 | 0.52 |
| CN− | 0.0005 | 0.02 | 0.001 | 0.004 |
| N3− | 0.0012 | 1.5 | 0.08 | 0.005 |
| HCO3− | 12 | 85 | 0.75 | 0.67 |
| CO32− | 15 | 73 | 0.64 | 0.47 |
| NO3− | 7 | 35 | 79 | 0.58 |
| NO2− | 8.4 | 63 | 94 | 0.64 |
| HS− | 0.0006 | 0.04 | 4.85 | 0.54 |
| HSO3− | 18 | 89 | 0.84 | 0.92 |
| SO42− | 63 | > 200 | 0.77 | 0.74 |
| BF4− | > 200 | > 200 | > 200 | > 200 |
| H2NSO3Hb> | 0.021 | 0.39 | 0.70 | 7.6 μM |
| H2NSO2NH2 | 0.31 | 1.13 | 0.83 | 6.5 μM |
| PhB(OH)2 | 58.6 | 23.1 | 0.80 | 6.1 μM |
| PhAsO3H2b | 31.7 | 49.2 | 0.84 | 5.2 μM |
Errors were in the range of 3%–5% of the reported values, from three different assays.
As sodium salt.
Figure 4.
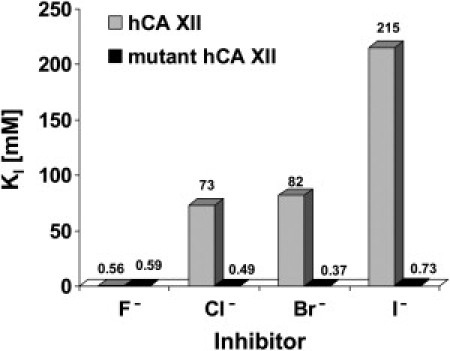
Inhibition Constants for Anionic Inhibitors of Human Wild-Type and Mutant hCA XII, for the CO2 Hydration Reaction, at 20°C14
The mutant CA XII investigated in this study has a glutamic acid mutated to a lysine at position 143. This amino acid residue is found near the third Zn(II) ligand (His145), being in a highly conserved protein region in all mammalian CA isozymes.15 Indeed, the residues in the neighborhood of the three histidine residues acting as Zn(II) ligands are conserved in all CAs, which is probably due to the fact that the tetrahedral geometry of Zn(II) is essential for the proper catalytic activity of the enzyme. In fact, in addition to the three His ligands mentioned above, the metal ion is also coordinated by a water molecule and a hydroxide ion that acts as nucleophile in the reactions catalyzed by these enzymes. This nonprotein zinc ligand is also involved in extensive hydrogen bonding with amino acid residues in its neighborhood (primarily conserved Thr and Glu residues), which assures both its proper orientation and its nucleophility needed to act on the substrates of the enzyme. Normally, perturbations of such hydrogen bonds will lead to a drastic diminution of the catalytic activity.15,16 However, unexpectedly, the mutant investigated here, in which a negatively charged amino acid residue (Glu143) near the third Zn(II) ligand is replaced by a positively charged one (Lys), still maintains a high enough catalytic activity for the physiologic reaction. Indeed, the mutant enzyme showed around 70% activity, as compared to the wild-type enzyme, for the physiologic CO2 hydration reaction, with a kcat of 3.0 × 105 s−1 and a kcat/KM of 2.5 × 107 M−1s−1 (Table 2). Furthermore, this mutant enzyme was highly inhibited by acetazolamide, a clinically used sulfonamide,15,16 with an inhibition constant of 10 nM (Table 2), in the same order of magnitude as the wild-type CA XII (which is anyhow better inhibited by acetzolamide, with a KI of 5.7 nM).
We have also investigated the inhibition of the mutant CA XII with a series of anions, some of which are physiologically relevant species,15 such as chloride, sulfate, bicarbonate, etc. (Table 3). Data in Table 3 and Figure 4 show that the mutant CA XII investigated here is much more sensitive to inhibition by anions, unlike the wild-type CA XII, which was generally weakly inhibited by halides, except fluoride.13 Thus, the mutant CA XII had an inhibition constant for halides of 0.49 mM for chloride, 0.37 mM for bromide, and 0.73 mM for iodide, being thus 148.9 times more sensitive to chloride inhibition than the wild-type enzyme, 221.6 times more sensitive to bromide, and 294.5 times more sensitive to iodide, as compared to the wild-type CA XII. Bicarbonate, carbonate, and pseudohalides generally showed similar inhibitory activity against the two enzymes (in the millimolar range), although the mutant was usually slightly better inhibited as compared to the wild-type CA XII. The only anion that was not at all inhibitory against both proteins was tetrafluroborate. Cyanide and azide were quite potent micromolar inhibitors of the mutant CA XII (inhibition constants of 4–5 μM). Sulfamic acid, sulfamide, phenylboronic acid, and phenylarsonic acid were also low micromolar inhibitors of the mutant CA XII (inhibition constants in the range of 5.2–7.6 μM), whereas they were submillimolar ones against wild-type CA XII. Thus, the p.Glu143Lys CA XII shows an inhibition profile with this class of inhibitors very different from that of the wild-type enzyme and the cytosolic, ubiquitous CA I and II (Table 3). It is highly unusual that a point mutation in a CA isoform leads to such a drastic change in the affinity for anion inhibitors.15,16 It is tempting to speculate that the positively charged amino acid residue (Lys143) near the third Zn(II) ligand present in this mutant is the main reason for this enhanced affinity for anion inhibitors, but it is probable that the hydrogen bonding network in the neighborhood of the catalytic center is completely changed as a result of this mutation.
The CA metalloenzymes catalyze a very simple but essential physiological reaction, carbon dioxide hydration to bicarbonate and protons.15 This reaction also occurs without a catalyst, but it is too slow. Given that CO2, bicarbonate, and protons are essential molecules and ions in many important physiologic processes in all life kingdoms (Bacteria, Archaea, and Eukarya), throughout the phylogenetic tree, and relatively high amounts of them are present in different tissues or cell compartments of all such organisms, it is no wonder that CAs evolved independently at least five times, with five genetically distinct enzyme families known to date: the α-, β-, γ-, δ- and ζ-CAs.15
In many organisms these enzymes are involved in crucial physiological processes connected with respiration and transport of CO2 and bicarbonate, pH and CO2 homeostasis, electrolyte secretion in a variety of tissues and organs, biosynthetic reactions (such as gluconeogenesis, lipogenesis, and ureagenesis), bone resorption, calcification, tumorigenicity, and many other physiologic or pathologic processes (thoroughly studied in vertebrates),15,16 whereas in algae, plants, and some bacteria they play an important role in photosynthesis and other biosynthetic reactions.15 In diatoms δ- and ζ-CAs play a crucial role in CO2 fixation.15,16
So far, 16 different α-CA isoforms have been isolated and characterized in mammals, where they play important physiological roles, as briefly outlined above. Some of them are cytosolic (CA I, CA II, CA III, CA VII, and CA XIII), others are membrane-bound (CA IV, CA IX, CA XII, CA XIV, and CA XV), CA VA and CA VB are mitochondrial, and CA VI is secreted in saliva and milk. Three acatalytic forms are also known: the CA-related proteins (CARP), CARP VIII, CARP X, and CARP XI, which seem to be cytosolic proteins as well.15 The mammalian CAs were the first such enzymes isolated and studied in detail,15 and many of them are established therapeutic targets with the potential to be inhibited or activated to treat a wide range of disorders.16
Isozymes CA IX and CA XII are predominantly found in tumor cells and show a restricted expression in normal tissues.15 It has been recently proven that by efficiently hydrating carbon dioxide to protons and bicarbonate, these CAs contribute significantly to the extracellular acidification of solid tumors, whereas their inhibition reverts this phenomenon to a certain extent.15 CA IX and CA XII are overexpressed in many such tumors in response to the hypoxia inducible factor (HIF) pathway, and research on the involvement of these isozymes in cancer has progressed significantly in recent years. Interestingly, an alternatively spliced isoform of CA XII was demonstrated in diffusely infiltrating astrocytic gliomas.17 There are recent efforts to design inhibitors against these antitumor targets.16 In relevance to the present report, it is of interest to note that CA II has been shown to be expressed in sweat glands18 and that its inhibitor, topiramate, has been demonstrated to induce oligohydrosis in humans and in mice.18,19
CA XII transcripts were previously demonstrated in normal human kidney, colon, prostate, pancreas, ovary, testis, lung, eye, and brain.20–22 CA XII is also overexpressed in the eyes of glaucoamtouos patients, and its inhibition may lead to a strong antiglaucoma effect.15,22 Using RT-PCR, we have now demonstrated that CA XII is also transcribed in human skin (Figure 3E). This finding is in line with previous studies, showing that different proteins that participate in the regulation of electrolyte transport as well as in acid base balance in the kidney and the colon are expressed in the sweat glands. For example, in isolated sweat glands, bumetanide (an inhibitor of the sodium-potassium and chloride cotransportes NKCC1 and NKCC2) inhibits sweat secretion. Immunoelectron microscopy demonstrated abundant NKCC1 and NHE1 labeling of the basolateral plasma membrane of mouse sweat glands.14 The presence of an Na+/H+ exchanger of isoform 1 (NHE1) in the human eccrine sweat duct has also been shown with the use of an NHE1 antibody.15 In addition, normal human epidermis stained positively for CFTR, a cAMP-dependent chloride channel protein, as densely as did the eccrine sweat gland.16
We have thus shown that a consistent autosomal-recessive phenotype of hyperchlorhidrosis does exist and should be considered in the differential diagnosis of a positive sweat test, especially in the context of hyponatremic dehydration during infancy. We have further demonstrated that the phenotype can be caused by a homozygous missense mutation in CA12. The mutant protein demonstrated 70% CA activity as compared to the wild-type enzyme. This might explain why the phenotype is mild, with no significant gastrointestinal or renal involvement—despite the fact that CA12 is expressed in those tissues. It is also feasible that in the kidney and colon (yet not in the skin) CA12 activity is complemented by that of other enzymes, making the 30% reduction in activity insignificant in those tissues. Unlike the wild-type enzyme, which is not inhibited by chloride, bromide, or iodide (KIs of 73–215 mM), the mutant is inhibited in the submicromolar range by these anions (KIs of 0.37–0.73 mM). Thus, we demonstrate that the CA12 mutation reduces the CA activity and causes a dramatic change in chloride-mediated feedback regulation of this enzyme, leading to excessive chloride secretion in sweat.
Acknowledgments
The authors deeply thank the United States Cystic Fibrosis Foundation (CFF) and the Morris Kahn Family Foundation for making this study possible.
Web Resources
The URLs for data presented herein are as follows:
HaploPainter, http://haplopainter.sourceforge.net/index.html
Marshfield Clinic Comprehensive Human Genetic Maps, http://research.marshfieldclinic.org/genetics/GeneticResearch/compMaps.asp
NEBcutter V2.0, http://tools.neb.com/NEBcutter2/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Primer3, http://frodo.wi.mit.edu/primer3/
Simple Modular Architecture Research Tool (SMART), http://smart.embl-heidelberg.de/
Superlink online version 1.5, http://bioinfo.cs.technion.ac.il/superlink-online/
Syndrome to Gene (S2G), http://fohs.bgu.ac.il/s2g
UCSC Genome Browser website, http://genome.ucsc.edu/
References
- 1.LeGrys V.A., Yankaskas J.R., Quittell L.M., Marshall B.C., Mogayzel P.J., Jr., Cystic Fibrosis Foundation Diagnostic sweat testing: the Cystic Fibrosis Foundation guidelines. J. Pediatr. 2007;151:85–89. doi: 10.1016/j.jpeds.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 2.Casaulta C., Stirnimann A., Schoeni M.H., Barben J. Sweat test in patients with glucose-6-phosphate-1-dehydrogenase deficiency. Arch. Dis. Child. 2008;93:878–879. doi: 10.1136/adc.2007.132688. [DOI] [PubMed] [Google Scholar]
- 3.Greenburg F., Schidlow D., Palmer N., Huang N. Isolated hyperchlorhidrosis without evidence of cystic fibrosis in two brothers, a possible autosomal recessive trait. Am. J. Hum. Genet. 1979;31:73A. [Google Scholar]
- 4.Jaron R., Yaakov Y., Rivlin J., Blau H., Bentur L., Yahav Y., Kerem E., Bibi H., Picard E., Wilschanski M. Nasal potential difference in non-classic cystic fibrosis-long term follow up. Pediatr. Pulmonol. 2008;43:545–549. doi: 10.1002/ppul.20807. [DOI] [PubMed] [Google Scholar]
- 5.Narkis G., Ofir R., Manor E., Landau D., Elbedour K., Birk O.S. Lethal congenital contractural syndrome type 2 (LCCS2) is caused by a mutation in ERBB3 (Her3), a modulator of the phosphatidylinositol-3-kinase/Akt pathway. Am. J. Hum. Genet. 2007;81:589–595. doi: 10.1086/520770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Narkis G., Ofir R., Landau D., Manor E., Volokita M., Hershkowitz R., Elbedour K., Birk O.S. Lethal contractural syndrome type 3 (LCCS3) is caused by a mutation in PIP5K1C, which encodes PIPKI gamma of the phophatidylinsitol pathway. Am. J. Hum. Genet. 2007;81:530–539. doi: 10.1086/520771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Birnbaum R.Y., Zvulunov A., Hallel-Halevy D., Cagnano E., Finer G., Ofir R., Geiger D., Silberstein E., Feferman Y., Birk O.S. Seborrhea-like dermatitis with psoriasiform elements caused by a mutation in ZNF750, encoding a putative C2H2 zinc finger protein. Nat. Genet. 2006;38:749–751. doi: 10.1038/ng1813. [DOI] [PubMed] [Google Scholar]
- 8.Khateeb S., Flusser H., Ofir R., Shelef I., Narkis G., Vardi G., Shorer Z., Levy R., Galil A., Elbedour K., Birk O.S. PLA2G6 mutation underlies infantile neuroaxonal dystrophy. Am. J. Hum. Genet. 2006;79:942–948. doi: 10.1086/508572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Silberstein M., Tzemach A., Dovgolevsky N., Fishelson M., Schuster A., Geiger D. Online system for faster multipoint linkage analysis via parallel execution on thousands of personal computers. Am. J. Hum. Genet. 2006;78:922–935. doi: 10.1086/504158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gefen A., Cohen R., Birk O.S. Syndrome to gene (S2G): in-silico identification of candidate genes for human diseases. Hum. Mutat. 2010;31:229–236. doi: 10.1002/humu.21171. [DOI] [PubMed] [Google Scholar]
- 11.Schnetkamp P.P. The SLC24 Na+/Ca2+-K+ exchanger family: vision and beyond. Pflugers Arch. 2004;447:683–688. doi: 10.1007/s00424-003-1069-0. [DOI] [PubMed] [Google Scholar]
- 12.Vullo D., Innocenti A., Nishimori I., Pastorek J., Scozzafava A., Pastoreková S., Supuran C.T. Carbonic anhydrase inhibitors. Inhibition of the transmembrane isozyme XII with sulfonamides-a new target for the design of antitumor and antiglaucoma drugs? Bioorg. Med. Chem. Lett. 2005;15:963–969. doi: 10.1016/j.bmcl.2004.12.053. [DOI] [PubMed] [Google Scholar]
- 13.Innocenti A., Vullo D., Pastorek J., Scozzafava A., Pastorekova S., Nishimori I., Supuran C.T. Carbonic anhydrase inhibitors. Inhibition of transmembrane isozymes XII (cancer-associated) and XIV with anions. Bioorg. Med. Chem. Lett. 2007;17:1532–1537. doi: 10.1016/j.bmcl.2006.12.113. [DOI] [PubMed] [Google Scholar]
- 14.Khalifah R.G. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J. Biol. Chem. 1971;246:2561–2573. [PubMed] [Google Scholar]
- 15.Supuran C.T. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008;7:168–181. doi: 10.1038/nrd2467. [DOI] [PubMed] [Google Scholar]
- 16.Supuran C.T., Scozzafava A., Casini A. Carbonic anhydrase inhibitors. Med. Res. Rev. 2003;23:146–189. doi: 10.1002/med.10025. [DOI] [PubMed] [Google Scholar]
- 17.Haapasalo J., Hilvo M., Nordfors K., Haapasalo H., Parkkila S., Hyrskyluoto A., Rantala I., Waheed A., Sly W.S., Pastorekova S. Identification of an alternatively spliced isoform of carbonic anhydrase XII in diffusely infiltrating astrocytic gliomas. Neuro-oncol. 2008;10:131–138. doi: 10.1215/15228517-2007-065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma L., Huang Y.G., Deng Y.C., Tian J.Y., Rao Z.R., Che H.L., Zhang H.F., Zhao G. Topiramate reduced sweat secretion and aquaporin-5 expression in sweat glands of mice. Life Sci. 2007;80:2461–2468. doi: 10.1016/j.lfs.2007.04.018. [DOI] [PubMed] [Google Scholar]
- 19.Ben-Zeev B., Watemberg N., Augarten A., Brand N., Yahav Y., Efrati O., Topper L., Blatt I. Oligohydrosis and hyperthermia: pilot study of a novel topiramate adverse effect. J. Child Neurol. 2003;18:254–257. doi: 10.1177/08830738030180041001. [DOI] [PubMed] [Google Scholar]
- 20.Türeci O., Sahin U., Vollmar E., Siemer S., Göttert E., Seitz G., Parkkila A.K., Shah G.N., Grubb J.H., Pfreundschuh M., Sly W.S. Human carbonic anhydrase XII: cDNA cloning, expression, and chromosomal localization of a carbonic anhydrase gene that is overexpressed in some renal cell cancers. Proc. Natl. Acad. Sci. USA. 1998;95:7608–7613. doi: 10.1073/pnas.95.13.7608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kallio H., Pastorekova S., Pastorek J., Waheed A., Sly W.S., Mannisto S., Heikinheimo M., Parkkila S. Expression of carbonic anhydrases IX and XII during mouse embryonic development. BMC Dev. Biol. 2006;6:22. doi: 10.1186/1471-213X-6-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liao S.Y., Ivanov S., Ivanova A., Ghosh S., Cote M.A., Keefe K., Coca-Prados M., Stanbridge E.J., Lerman M.I. Expression of cell surface transmembrane carbonic anhydrase genes CA9 and CA12 in the human eye: overexpression of CA12 (CAXII) in glaucoma. J. Med. Genet. 2003;40:257–261. doi: 10.1136/jmg.40.4.257. [DOI] [PMC free article] [PubMed] [Google Scholar]