

Abstract
Fibrochondrogenesis is a severe, autosomal-recessive, short-limbed skeletal dysplasia. In a single case of fibrochondrogenesis, whole-genome SNP genotyping identified unknown ancestral consanguinity by detecting three autozygous regions. Because of the predominantly skeletal nature of the phenotype, the 389 genes localized to the autozygous intervals were prioritized for mutation analysis by correlation of their expression with known cartilage-selective genes via the UCLA Gene Expression Tool, UGET. The gene encoding the α1 chain of type XI collagen (COL11A1) was the only cartilage-selective gene among the three candidate intervals. Sequence analysis of COL11A1 in two genetically independent fibrochondrogenesis cases demonstrated that each was a compound heterozygote for a loss-of-function mutation on one allele and a mutation predicting substitution for a conserved triple-helical glycine residue on the other. The parents who were carriers of missense mutations had myopia. Early-onset hearing loss was noted in both parents who carried a loss-of-function allele, suggesting COL11A1 as a locus for mild, dominantly inherited hearing loss. These findings identify COL11A1 as a locus for fibrochondrogenesis and indicate that there might be phenotypic manifestations among carriers.
Main Text
Fibrochondrogenesis (MIM 228520) is a severe, autosomal-recessive, short-limbed skeletal dysplasia first described in 1978.1 The disease is clinically characterized by a flat midface with a small nose and anteverted nares, significant shortening of all limb segments but relatively normal hands and feet, and a small bell-shaped thorax with a protuberant abdomen.2–8 Radiographically, the long bones are short and have broad metaphyseal ends, giving them a dumb-bell shape. The vertebral bodies are flat and, on lateral view, have a distinctive pinched appearance, with a hypoplastic posterior end and a rounded anterior end. The ribs are typically short and wide and have metaphyseal cupping at both ends. The phenotype was named on the basis of the abnormal morphology of the growth plate, in which the chondrocytes had a fibroblastic appearance and there were regions of fibrous cartilage extracellular matrix.2–5,7,8
The index case reported here was born at term to parents of Mexican origin. The couple already had five unaffected children. Although consanguinity was denied, to determine for counseling purposes whether the phenotype might be recessive, we performed whole-genome SNP genotyping on a clinical basis. Three large regions of homozygosity were identified: 12.8 Mb at 91,985,291–104,782,487 bp on chromosome 1p21.1-p22.2; 5.5 Mb at 140,785,938–146,264,218 bp on chromosome 8q24.3; and 15.3 Mb at 17,935,721–33,266,609 bp on chromosome 13q11-q13.2 (NCBI build 36/hg18). These data indicated that the parents had an unknown ancestral genetic relationship and implied that the phenotype was likely to be recessively inherited and produced by homozygosity for a mutation in a gene in one of the three intervals. However, after the initial clinical study, the family declined to participate further, so additional experiments could not be carried out in the index case.
The three intervals on chromosomes 1, 8, and 13 contained 389 genes in total. To prioritize these genes for mutation screening, we hypothesized that the exclusively skeletal phenotype could be consistent with a mutation in a gene selectively expressed in cartilage. Gene expression data for all of the genes was correlated with the expression patterns of known cartilage-selective genes via the UCLA Gene Expression Tool (UGET).9 The gene expression data in UGET are derived from Celsius,10 a large conormalized microarray dataset of Affymetrix-based gene expression. In this study, the UGET tool was seeded with the known cartilage-selective genes,11 C10orf49, COL2A1 (MIM 120140), MATN1 (MIM 115437), LECT1 (MIM 605147), ACAN (MIM 155760), CSPG4 (MIM 601172), MMP13 (MIM 600108), COL9A3 (MIM 120270), TRPV4 (MIM 605427), and COL11A2 (MIM 120290), and expression data for the Affymetrix HG-U133 Plus 2 platform were used for identifying genes that were within the candidate intervals and whose expression patterns were correlated with those of the seed genes. The highest correlation was observed for the gene encoding the α1 chain of collagen XI (COL11A1; MIM 120280), which was the only known cartilage-selective gene identified by UGET.
Mutation analysis of COL11A1 was carried out commercially (Connective Tissue Gene Tests, Allentown, PA) by amplification and sequence analysis of all of the 68 coding exons and splice junction consensus sequences in two genetically independent cases of fibrochondrogenesis. The studies were carried out under a protocol approved by an institutional review board, and informed consent was obtained from all participants. The proband from family 1 (International Skeletal Dysplasia Registry reference number R00-394A) was the first child of parents of European descent and was stillborn at 32 weeks of gestation. Subsequently, the couple had two unaffected children. The diagnosis was based on the radiographic presentation (Figure 1), cartilage histology demonstrating a fibroblastic appearance of the chondrocytes (Figure 2A), and transmission electron microscopy of cartilage showing frayed and irregular collagen fibrils (Figure 2C). Sequence analysis of COL11A1 (NM_001854.3) revealed compound heterozygosity for an exon 18 c.1786dupG duplication that produced a frameshift and subsequent premature termination codon (p.Ala596GlyfsX8), and an exon 42 c.3124G>A transition that predicts a triple-helical glycine-to-arginine (p.Gly1042Arg) substitution (Table 1). The mother had had myopia from about 10 years of age, was of average stature, had normal hearing, and was heterozygous for the missense mutation. This mutation was absent from 214 control chromosomes of ethnically matched individuals. The father reported that he had hearing loss at age 7 and wore glasses from 6 years of age. He was of average stature but had experienced joint pain since childhood. A DNA sample with which to determine whether he was a carrier for the frameshift mutation was not available.
Figure 1.
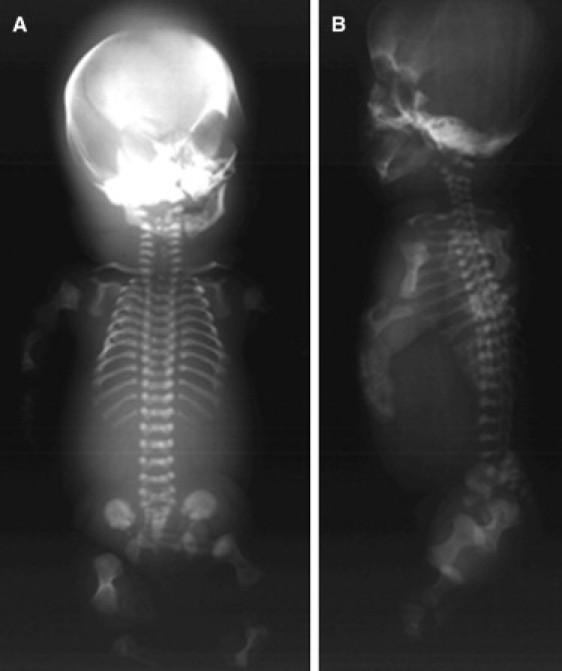
Radiographs of Fibrochondrogenesis Case R00-394 at 32 Weeks Gestation
(A) Anterior-posterior view.
(B) Lateral view. Note the short long bones with broad metaphyses, short ribs with metaphyseal cupping, and flat vertebral bodies with hypoplastic posterior and rounded anterior ends.
Figure 2.
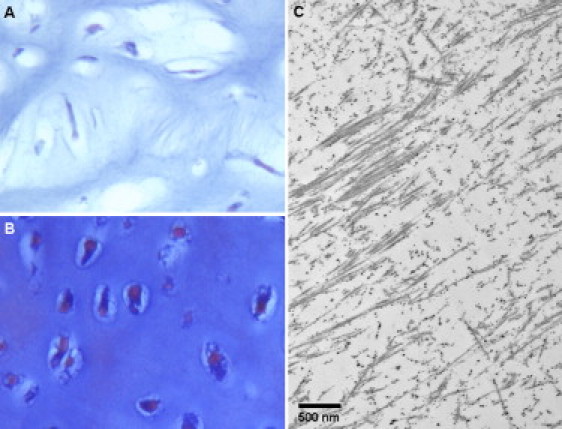
Histologic Analysis of Distal Femur Cartilage from Case R00-394 and Control
(A and B) Light microscopy. Cartilage was decalcified, embedded in paraffin, and stained with von Kossa trichrome. The fibrochondrogenesis case (A, 32 weeks gestation) exhibits fibroblastic chondrocytes and a fibrous intercellular matrix not present in the 28-weeks-gestation control (B). Magnification is 40×.
(C) Transmission electron microscopy showing frayed and irregular collagen fibrils.
Table 1.
COL11A1 Mutations in Fibrochondrogenesis Cases
| Patient ID | Exon | Nucleotide Change | Amino Acid Change |
|---|---|---|---|
| R06-573C | 30 | c.2386G>C | p.Gly796Arg |
| 53 | c.3943G>T | p.Gly1315X | |
| R00-394 | 18 | c.1786dupG | p.Ala596GlyfsX8 |
| 42 | c.3124G>A | p.Gly1042Arg |
The nucleotide changes are shown with respect to COL11A1 mRNA sequence NM_001854.3, and the corresponding predicted amino acid change is numbered from the initiating methionine residue, where applicable. Exons are numbered sequentially 1–68, and exons 6 and 7 represent the alternatively spliced exons 6A and 6B, respectively.
To determine whether the c.1786dupG change was effectively a null mutation or could lead to the production of a truncated protein, we examined allele-specific expression based on the c.3124G>A (p.Gly1042Arg) missense mutation (Figure 3). Total RNA was isolated (RNeasy Mini Kit, QIAGEN) from P3 cultured chondrocytes derived from the proband, cDNA was synthesized (High-Capacity cDNA Reverse Transcription Kit, Applied Biosystems), and a fragment containing the c.3124G>A (p.Gly1042Arg) mutation was amplified by PCR. Sequence analysis revealed that all of the amplified products were derived from the missense allele, consistent with nonsense-mediated decay of the transcript derived from the allele with the insertion mutation.
Figure 3.
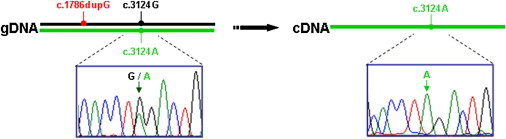
Allele-Specific Expression Based on the c.3124G>A Missense Mutation in Case R00-394
On the left, the sequence trace of an amplified genomic DNA fragment containing the c.3124A missense mutation is shown. On the right, the sequence trace of an amplified cDNA fragment containing the same region is shown, demonstrating the absence of the c.3124G nucleotide derived from the c.1786dupG allele.
The proband from family 2 (R06-573C; Figure 4) was the second affected child born to a father of European descent and an African-American mother. The pregnancy for the first affected child was terminated at 24 weeks of gestation (Figure 4), whereas the second affected child was born at term. Although the radiographs supported a diagnosis of fibrochondrogenesis in both affected offspring, the clinical phenotype of the second child was milder than previously reported for fibrochondrogenesis, and he is currently alive at 3 years of age. He has a flat midface, prominent eyes, short stature, and short limbs, and his hands exhibit brachyclinodactyly and have some soft-tissue syndactyly in the web spaces. His trunk is short and narrow, he has a pectus carinatum, and he breathes without assistance. He has high myopia and had a left cataract, and there is mild-to-moderate hearing loss. Sequence analysis of COL11A1 revealed compound heterozygosity for an exon 30 c.2386G>C change that predicts a glycine-to-arginine (p.Gly796Arg) substitution and an exon 53 c.3943G>T change that predicts a change from a glycine to a premature termination codon (p.Gly1315X; Table 1). The latter change was expected to be functionally null, likely via nonsense-mediated decay of the transcript. Analysis of DNA from the parents showed that the father was heterozygous for the null mutation and that the mother was heterozygous for the missense mutation. The missense mutation was absent from 216 ethnically matched control chromosomes. The father is of average stature and has normal vision and mild hearing loss. He reports that his father and paternal uncle had hearing loss that began in their 20s and that they required the use of hearing aids. The mother is of mild short stature for her family and has normal hearing and mild myopia.
Figure 4.
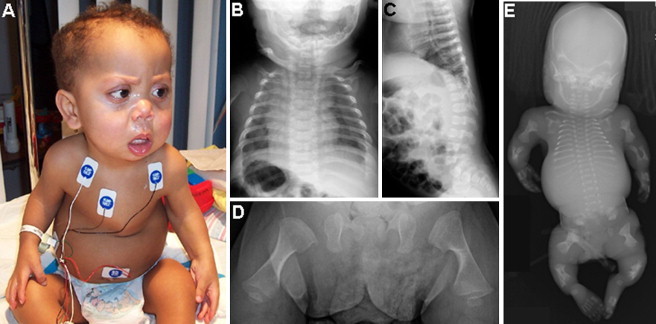
Clinical Photograph and Radiographs of Fibrochondrogenesis Cases from Family R06-573
(A) The surviving boy (R06-573C) at 19 months of age, presenting with a flat midface, small nose with anteverted nares, and a small bell-shaped thorax.
(B) An anterior-posterior chest radiograph of the boy at 2 months of age shows short and wide ribs with metaphyseal cupping.
(C) A lateral-view radiograph of the spine shows flat vertebral bodies.
(D) An anterior-posterior radiograph of the femurs shows short long bones with broad metaphyses.
(E) Anterior-posterior radiograph of the first affected fetus (R06-573A) in the family at 24 weeks gestation.
This study demonstrates that mutations in the gene encoding the α1 chain of type XI procollagen produce the recessively inherited fibrochondrogenesis phenotype. Type XI procollagen is a heterotrimeric protein assembled from the products of three genes, COL11A1, COL11A2 (MIM 120290), and COL2A1 (MIM 120140). Like all fibrillar collagens, the structure of the protein is defined by a long, uninterrupted triple-helical domain specified by the repeating amino acid sequence Gly-Xaa-Yaa. Glycine residues at every third position are strictly required for proper folding of the triple helix, and the helix is stabilized by hydroxylated proline residues in the Yaa position. Mature type XI collagen, which accounts for 3%–10% of the collagenous protein content of cartilage,12,13 forms heterotypic fibrils with type II collagen, and the ratio between the molecules appears in part to determine fibril diameter.14 Molecules composed of different hetero-combinations of type V and XI collagen chains are also expressed in a tissue-dependent manner, supporting a role for the type V/XI collagen polymer as a filamentous template for type I and II collagen fibrils.15
The fibrochondrogenesis cases studied resulted from compound heterozygosity for two types of COL11A1 mutations that imply two mechanisms that together contribute to development of the disease. First, the null allele predicts reduced synthesis of the proα1(XI) chain. Although this might predict reduced synthesis of type XI procollagen, in mice heterozygous for the chondrodysplasia (cho/+) Col11a1 null mutation,16 there is some incorporation of proα1(V) chains into type XI procollagen molecules, and this partially compensates for the reduced synthesis of the proα1(XI) chain.14 Second, mutant proα1(XI) chains, encoded by the allele with the mutation leading to the glycine substitution, would be expected to disrupt the triple-helical structure of molecules into which they were incorporated. Thus, the combined effects of these two mechanisms could lead to the disruption of the structure of the type II/type XI collagen heterotypic fibril observed in fibrochondrogenesis. However, it was not possible to define the consequences of the mutations on type XI collagen synthesis because cartilage for biochemical analysis was not available for these two cases.
Mutations in COL11A1 have previously been reported to cause autosomal-dominant forms of Stickler (MIM 604841) and Marshall (MIM 154780) syndromes, which are characterized by mild facial dysmorphism, sensorineural hearing loss, and myopia.17–21 In eight of the reported cases, the phenotype results from heterozygosity for mutations leading to glycine substitutions in the proα1(XI) chain. These data suggest that the phenotypic abnormalities in the fibrochondrogenesis carrier parents, who were not identified as having Stickler/Marshall syndrome, could result from their mutant COL11A1 alleles. For the null alleles, hearing loss appears to be the major manifestation, suggesting COL11A1 as a locus for mild, early-onset hearing loss. Mutations in the paralogous type XI procollagen gene, COL11A2, are known to produce both autosomal-dominant and recessive forms of hearing loss.22,23 For the glycine substitutions, the parental phenotypes included mild short stature and myopia. However, it will be necessary to study additional carriers to determine how consistent the observed pattern is, as well as to document in detail the extent of phenotypic abnormalities observed in the hearing and vision of carriers. Furthermore, for glycine substitutions in fibrillar collagen molecules, phenotypic consequences are known to vary with the specific amino acid and substituting residue involved, as most extensively documented for osteogenesis imperfecta.24 It will therefore be important to study additional cases to develop a clearer understanding of the range of expression of the carrier phenotype among parents and unaffected siblings of fibrochondrogenesis cases.
The chondrodysplasia (cho/cho) mouse,16 which results from homozygosity for a functional null allele of Col11a1, might represent an appropriate animal model for fibrochondrogenesis. Homozygous mutant animals have a short snout, their long bones and ribs are short, wide, and have flared metaphyses, their spine is short with irregular vertebrae, and their thoracic cage is small, likely explaining the neonatal lethality of the phenotype.25 At the ultrastructural level, collagen fibrils in the cartilage extracellular matrix are sparsely distributed and abnormally thick, and they display a repeating transverse banding pattern14,25 not unlike that seen in fibrochondrogenesis. Thus, it is possible that, in addition to the mechanisms described here, absence of the COL11A1 gene product could also produce fibrochondrogenesis.
Fibrochondrogenesis thus defines the most severe end of a spectrum of disorders involving the type XI procollagen genes characterized to date. Especially given the nonlethal case described here, which bears some similarity to a case of recessive spondyloepimetaphyseal dysplasia,26 we suspect that additional mutations in COL11A1 will be found to cause dominant or recessive disorders of intermediate severity between fibrochondrogenesis and Stickler/Marshall syndrome. Identification of mutations in fibrochondrogenesis will facilitate diagnostic testing for families and, for the carrier parents, siblings, and other family members, early surveillance for abnormalities in vision and hearing.
Acknowledgments
We thank the families who participated in this work. This work was supported in part by grants from the National Institutes of Health (DE019567, HD22657, and M01-RR00425).
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
UCLA Gene Expression tool (UGET), https://secure.genome.ucla.edu/index.php/UGET_HOWTO
References
- 1.Lazzaroni-Fossati F., Stanescu V., Stanescu R., Serra G., Magliano P., Maroteaux P. Fibrochondrogenesis. Arch. Fr. Pediatr. 1978;35:1096–1104. [PubMed] [Google Scholar]
- 2.Whitley C.B., Langer L.O., Jr., Ophoven J., Gilbert E.F., Gonzalez C.H., Mammel M., Coleman M., Rosemberg S., Rodriques C.J., Sibley R. Fibrochondrogenesis: Lethal, autosomal recessive chondrodysplasia with distinctive cartilage histopathology. Am. J. Med. Genet. 1984;19:265–275. doi: 10.1002/ajmg.1320190209. [DOI] [PubMed] [Google Scholar]
- 3.Eteson D.J., Adomian G.E., Ornoy A., Koide T., Sugiura Y., Calabro A., Lungarotti S., Mastroiacovo P., Lachman R.S., Rimoin D.L. Fibrochondrogenesis: Radiologic and histologic studies. Am. J. Med. Genet. 1984;19:277–290. doi: 10.1002/ajmg.1320190210. [DOI] [PubMed] [Google Scholar]
- 4.Bankier A., Fortune D., Duke J., Sillence D.O. Fibrochondrogenesis in male twins at 24 weeks gestation. Am. J. Med. Genet. 1991;38:95–98. doi: 10.1002/ajmg.1320380121. [DOI] [PubMed] [Google Scholar]
- 5.Martínez-Frías M.L., García A., Cuevas J., Rodríguez J.I., Urioste M. A new case of fibrochondrogenesis from Spain. J. Med. Genet. 1996;33:429–431. doi: 10.1136/jmg.33.5.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.al-Gazali L.I., Bakir M., Dawodu A., Haas D. Recurrence of fibrochondrogenesis in a consanguineous family. Clin. Dysmorphol. 1999;8:59–61. [PubMed] [Google Scholar]
- 7.Randrianaivo H., Haddad G., Roman H., Delezoide A.L., Toutain A., Le Merrer M., Moraine C., Lise A. Fetal fibrochondrogenesis at 26 weeks' gestation. Prenat. Diagn. 2002;22:806–810. doi: 10.1002/pd.423. [DOI] [PubMed] [Google Scholar]
- 8.Leeners B., Funk A., Cotarelo C.L., Sauer I. Two sibs with fibrochondrogenesis. Am. J. Med. Genet. 2004;127:318–320. doi: 10.1002/ajmg.a.20620. [DOI] [PubMed] [Google Scholar]
- 9.Day A., Dong J., Funari V.A., Harry B., Strom S.P., Cohn D.H., Nelson S.F. Disease gene characterization through large-scale co-expression analysis. PLoS ONE. 2009;31:e8491. doi: 10.1371/journal.pone.0008491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Day A., Carlson M.R., Dong J., O'Connor B.D., Nelson S.F. Celsius: A community resource for Affymetrix microarray data. Genome Biol. 2007;8:R112. doi: 10.1186/gb-2007-8-6-r112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Funari V.A., Day A., Krakow D., Cohn Z.A., Chen Z., Nelson S.F., Cohn D.H. Cartilage-selective genes identified in genome-scale analysis of non-cartilage and cartilage gene expression. BMC Genomics. 2007;12:165. doi: 10.1186/1471-2164-8-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eyre D.R. The collagens of articular cartilage. Semin. Arthritis Rheum. 1991;2:2–11. doi: 10.1016/0049-0172(91)90035-x. [DOI] [PubMed] [Google Scholar]
- 13.Eyre D.R., Wu J.J., Fernandes R.J., Pietka T.A., Weis M.A. Recent developments in cartilage research: Matrix biology of the collagen II/IX/XI heterofibril network. Biochem. Soc. Trans. 2002;30:893–899. doi: 10.1042/bst0300893. [DOI] [PubMed] [Google Scholar]
- 14.Fernandes R.J., Weis M., Scott M.A., Seegmiller R.E., Eyre D.R. Collagen XI chain misassembly in cartilage of the chondrodysplasia (cho) mouse. Matrix Biol. 2007;26:597–603. doi: 10.1016/j.matbio.2007.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu J.J., Weis M.A., Kim L.S., Carter B.G., Eyre D.R. Differences in chain usage and cross-linking specificities of cartilage type V/XI collagen isoforms with age and tissue. J. Biol. Chem. 2009;284:5539–5545. doi: 10.1074/jbc.M806369200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Y., Lacerda D.A., Warman M.L., Beier D.R., Yoshioka H., Ninomiya Y., Oxford J.T., Morris N.P., Andrikopoulos K., Ramirez F. A fibrillar collagen gene, Col11a1, is essential for skeletal morphogenesis. Cell. 1995;80:423–430. doi: 10.1016/0092-8674(95)90492-1. [DOI] [PubMed] [Google Scholar]
- 17.Majava M., Hoornaert K.P., Bartholdi D., Bouma M.C., Bouman K., Carrera M., Devriendt K., Hurst J., Kitsos G., Niedrist D. A report on 10 new patients with heterozygous mutations in the COL11A1 gene and a review of genotype-phenotype correlations in type XI collagenopathies. Am. J. Med. Genet. A. 2007;143:258–264. doi: 10.1002/ajmg.a.31586. [DOI] [PubMed] [Google Scholar]
- 18.Annunen S., Körkkö J., Czarny M., Warman M.L., Brunner H.G., Kääriäinen H., Mulliken J.B., Tranebjaerg L., Brooks D.G., Cox G.F. Splicing mutations of 54-bp exons in the COL11A1 gene cause Marshall syndrome, but other mutations cause overlapping Marshall/Stickler phenotypes. Am. J. Hum. Genet. 1999;65:974–983. doi: 10.1086/302585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin S., Richards A.J., Yates J.R., Scott J.D., Pope M., Snead M.P. Stickler syndrome: further mutations in COL11A1 and evidence for additional locus heterogeneity. Eur. J. Hum. Genet. 1999;7:807–814. doi: 10.1038/sj.ejhg.5200377. [DOI] [PubMed] [Google Scholar]
- 20.Richards A.J., Yates J.R., Williams R., Payne S.J., Pope F.M., Scott J.D., Snead M.P. A family with Stickler syndrome type 2 has a mutation in the COL11A1 gene resulting in the substitution of glycine 97 by valine in alpha 1 (XI) collagen. Hum. Mol. Genet. 1996;5:1339–1343. doi: 10.1093/hmg/5.9.1339. [DOI] [PubMed] [Google Scholar]
- 21.Richards A.J., McNinch A., Martin H., Oakhill K., Rai H., Waller S., Treacy B., Whittaker J., Meredith S., Poulson A., Snead M.P. Stickler syndrome and the vitreous phenotype: Mutations in COL2A1 and COL11A1. Hum. Mutat. 2010;31:E1461–E1471. doi: 10.1002/humu.21257. [DOI] [PubMed] [Google Scholar]
- 22.McGuirt W.T., Prasad S.D., Griffith A.J., Kunst H.P., Green G.E., Shpargel K.B., Runge C., Huybrechts C., Mueller R.F., Lynch E. Mutations in COL11A2 cause non-syndromic hearing loss (DFNA13) Nat. Genet. 1999;23:413–419. doi: 10.1038/70516. [DOI] [PubMed] [Google Scholar]
- 23.Chen W., Kahrizi K., Meyer N.C., Riazalhosseini Y., Van Camp G., Najmabadi H., Smith R.J. Mutation of COL11A2 causes autosomal recessive non-syndromic hearing loss at the DFNB53 locus. J. Med. Genet. 2005;42:e61. doi: 10.1136/jmg.2005.032615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marini J.C., Forlino A., Cabral W.A., Barnes A.M., San Antonio J.D., Milgrom S., Hyland J.C., Körkkö J., Prockop D.J., De Paepe A. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: Regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum. Mutat. 2007;28:209–221. doi: 10.1002/humu.20429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seegmiller R., Fraser F.C., Sheldon H. A new chondrodystrophic mutant in mice. Electron microscopy of normal and abnormal chondrogenesis. J. Cell Biol. 1971;48:580–593. doi: 10.1083/jcb.48.3.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Farag T.I., Al-Awadi S.A., Hunt M.C., Satyanath S., Zahran M., Usha R., Uma R. A family with spondyloepimetaphyseal dwarfism: a ‘new’ dysplasia or Kniest disease with autosomal recessive inheritance? J. Med. Genet. 1987;24:597–601. doi: 10.1136/jmg.24.10.597. [DOI] [PMC free article] [PubMed] [Google Scholar]