

Abstract
Spinocerebellar ataxias (SCAs) are dominantly inherited neurodegenerative disorders characterized by progressive cerebellar ataxia and dysarthria. We have identified missense mutations in prodynorphin (PDYN) that cause SCA23 in four Dutch families displaying progressive gait and limb ataxia. PDYN is the precursor protein for the opioid neuropeptides, α-neoendorphin, and dynorphins A and B (Dyn A and B). Dynorphins regulate pain processing and modulate the rewarding effects of addictive substances. Three mutations were located in Dyn A, a peptide with both opioid activities and nonopioid neurodegenerative actions. Two of these mutations resulted in excessive generation of Dyn A in a cellular model system. In addition, two of the mutant Dyn A peptides induced toxicity above that of wild-type Dyn A in cultured striatal neurons. The fourth mutation was located in the nonopioid PDYN domain and was associated with altered expression of components of the opioid and glutamate system, as evident from analysis of SCA23 autopsy tissue. Thus, alterations in Dyn A activities and/or impairment of secretory pathways by mutant PDYN may lead to glutamate neurotoxicity, which underlies Purkinje cell degeneration and ataxia. PDYN mutations are identified in a small subset of ataxia families, indicating that SCA23 is an infrequent SCA type (∼0.5%) in the Netherlands and suggesting further genetic SCA heterogeneity.
Introduction
The dominant spinocerebellar ataxias (SCAs) are a genetically heterogeneous group of neurodegenerative disorders characterized by progressive cerebellar ataxia, dysarthria, oculomotor abnormalities, and many additional symptoms.1 The ataxia results from selective atrophy of the Purkinje cells in the cerebellum. To date, 31 different SCA loci have been described, but the causative mutation and corresponding gene has only been identified in 19 SCA types.2,3 Nine of the 19 SCAs are caused by coding CAG repeat (SCA1–3, SCA6, SCA7, and SCA17; MIM #164400, MIM #183090, MIM #109150, MIM #183086, MIM #164500, and MIM #607136) or noncoding CAG, CTG, or ATTCT repeat expansions. These coding CAG (glutamine) repeat expansions lead to elongated polyglutamine tracts that subsequently cause misfolding of the corresponding proteins. The functional implication of the noncoding repeat expansions is suggested to induce RNA-mediated gain-of-function mechanisms, leading to neurotoxicity. However, the remainders of the known SCA types are due to missense mutations or chromosomal rearrangements (SCA5, SCA11, SCA13–15, SCA20, SCA27, SCA28, and SCA31; MIM #600224, MIM #604432, MIM #605259, MIM #605361, MIM #606658, MIM #608687, MIM #609307, MIM #610246, and MIM #117210).2 The SCA genes involved play a role in a wide range of biological processes. Recent studies into the function of the disease proteins revealed the existence of common pathways leading to ataxia consisting of changes in gene transcription and RNA processing or synaptic transmission via calcium and glutamate signaling.4,5
Currently, only 70% of the Dutch SCA families can be diagnosed by mutation analysis of seven of the known SCA genes (SCA1–3, SCA6, SCA7, SCA12, and SCA14). The identification of causal mutations and novel genes in the group of genetically undiagnosed families will provide additional insights into the underlying pathological pathways leading to cerebellar neurodegeneration and will perhaps indicate therapeutic approaches.
We previously mapped the SCA23 locus (MIM #610245) to a 6 Mb region located on chromosomal region 20p12.3-p13 in a single, large Dutch ataxia family without mutations in the SCA genes that were recognized at the time.6 MRI and neuropathological examination of SCA23 autopsy tissue revealed neuronal loss in the Purkinje cell layer, dentate nuclei, and inferior olivary nuclei. The patients showed a relatively slowly progressive, isolated, cerebellar ataxia. Additional neurological examination revealed dysarthria, oculomotor problems such as slowing saccades, and ocular dysmetria. Decreased vibration sense below the knee was observed in three affected individuals, hyper-reflexia was observed in four patients, and two patients displayed Babinski's signs. Because no affected individuals of the first generation were still alive at the time of the assessment, clinical anticipation could not be confirmed. The SCA23 locus comprises 97 genes and transcripts, of which at least 54 are expressed in the cerebellum. Previous attempts to identify one or more mutations causing the disease did not yield any results.6,7
In the present study, we identified four missense mutations in prodynorphin, PDYN (MIM #131340), in the originally reported SCA23 family and in three families from a Dutch ataxia cohort. PDYN is the precursor protein for the opioid neuropeptides α-neoendorphin, Dyn A, and Dyn B, ligands for the κ-opioid receptor (OPRK1; MIM #165196).8 Neuropeptides constitute a large family of signaling molecules that mediate and modulate neuronal communication by acting on cell surface receptors and thus regulate diverse physiological functions and behavior.9,10 Neuropeptides and their precursor molecules have not yet been identified as causative factors for neurodegenerative disorders.
Subjects and Methods
Human Subjects
The participants from the SCA23 family all gave informed consent, as approved by the Medical Ethical Committee of the University Medical Centre Utrecht. The 1100 ataxia patients (single index cases) screened in this study were obtained from the genetic diagnostic centers in Utrecht and Groningen in The Netherlands. All extended DNA analyses were performed in a diagnostic setting (accredited diagnostic DNA laboratory). Only DNA samples from patients who were referred for genetic testing for SCA were used and had no mutations in seven known SCA (SCA1–3, SCA6, SCA7, SCA12, and SCA14) genes. This cohort is comprised of 10% familial cases (8% dominant and 2% recessive) and 90% seemingly sporadic cases. The additional tests were thus performed in line with the original diagnostic request. Moreover, when the blood samples were taken, patients were asked whether they had objections to the use of their DNA for future (anonymous) studies to help develop or improve techniques. If they had objections, this was indicated on the original “Request for DNA Test” form, and the DNA was not used. The unrelated 500 control individuals were obtained from the Dutch blood bank.
Postmortem Human Specimens
SCA23 cerebellar tissues from the subject with a p.R138S mutation and from a control subject were obtained from the University Medical Center Nijmegen, Nijmegen, The Netherlands. Samples of the cerebellum (Cb), dorsolateral prefrontal cortex (PFC; Brodmann area 9), and nucleus accumbens (NAc) from three control subjects, all females of European descent (for demographic data, see Table S6 available online), were collected at the Karolinska Institute, Stockholm, Sweden, by qualified pathologists. The collection was performed under full ethical clearance from the University Medical Center Nijmegen and the Stockholm Ethical Review Board. Informed written consent from the next of kin was also obtained.
PDYN Sequencing
DNA was extracted from peripheral blood by a routine salting-out procedure. The genomic DNA of all participating subjects in this study was used to amplify the PDYN coding exons 3 and 4 (GenBank accession number: NM 024411.3) by PCR. The resulting amplicons, including the intron-exon boundaries, were screened for mutations via Sanger sequencing on an ABI 3700 (Applied Biosystems). The PCR, primer sequences, and conditions are shown in Table S7.
Dynorphin Radioimmunoassay
The radioimmunoassay (RIA) procedure was described elsewhere.11,12 Briefly, cells were extracted in 1 M acetic acid, extracts were run through an SP-Sephadex ion exchange C-25 column, and peptides were eluted and analyzed by RIA.
Peptide Synthesis
Dyn A peptides were synthesized at the Leiden University Medical Center, Leiden, The Netherlands, purified by reverse-phase chromatography and Superdex column, and analyzed by reverse-phase chromatography and MALDI-TOF MS. The purity of all peptides was ∼98%.
Neuron-Enriched Cultures, Peptide Treatment, and Assessment of Neuron Viability
Neurons were isolated from E15–E16 imprinting control region CD-1 mouse striatum as published.13 Briefly, the tissue was enzymatically and mechanically dissociated and filtered through a 70 μm pore nylon mesh. Neurons were plated onto poly-L-lysine (Sigma-Aldrich) coated glass coverslips inserted into a 24-well multiwell plate (5 × 105 neurons suspended per well). Neurons were maintained in Neurobasal media with added B27 supplement (Invitrogen), antibiotics, 0.5 mM L-glutamine, and 0.025 μM glutamate (both Invitrogen) at 37°C in 5% CO2 at ∼95% humidity. Cultures matured for 1 week prior to the start of experiments and were almost exclusively neurons when assessed by immunostaining for either NeuN or MAP2a/b (glial contamination < 1%). Neurons were treated with wild-type or mutant Dyn A peptides (p.L211S, p.R212W, or p.R215C) at 100 nM for 60 hr.
Time-lapse digital images of the same neuron were recorded at 20 min intervals for 60 hr with a Zeiss AxioObserver Z.1 microscope equipped with an automated, computer-controlled stage encoder, digital camera (Zeiss MRm), and environmental control chamber (PeCon Instruments) at 37°C with 95% humidity and 5% CO2. In each experiment, cells from 2–3 pups were pooled and distributed across treatments such that the neurotoxic effects of each peptide variant were directly compared in the same population of cells. Approximately 50 healthy neurons with well-defined dendritic and axonal arbors were identified within ≥8 overlapping fields (40× magnification; Mark&Find and Time Lapse modules Zeiss AxioVision 4.6) in each individual culture well prior to treatment (0 hr). Individual neurons were tracked throughout the experiment, and death was defined by the culmination of a series of events resulting in nuclear fragmentation and destruction of the cell body. Events preceding neuronal death included dendritic and axonal pruning, the dissolution of the Nissl substance, appearance of cytoplasmic swelling and vacuolization, nuclear damage and pyknosis, and eventual destruction of the cell body.13,14 Death was confirmed by viability markers such as ethidium homodimer, ethidium monoazide, or trypan blue (data not shown). These markers all possess inherent cytotoxicity and cannot be used to monitor cells during the entire course of the experiment. The effect of each treatment on neuron survival (percent pretreatment value) was analyzed statistically at 4 hr intervals via one-way repeated-measures analysis of variance (ANOVA; Graphpad Prism software) and was reported as mean neuron survival ± standard error of the mean (SEM) from n = 2–4 separate experiments (100–200 total neurons per treatment).
mRNA Analysis By Quantitative Real-Time PCR with TaqMan Low-Density Arrays
RNA preparation was performed with RNeasy Lipid Tissue Mini Kit (QIAGEN). RNA was quantified with microspectrophotometry by Nanodrop. RNA Quality Indicator (RQI) was measured with Bio-Rad Experion. RNA samples with RQI values above 5.0 are generally considered to be suitable for qRT-PCR.15–17 Average RQI was 7.28 ± 1.55 (mean, standard deviation [SD]), demonstrating high quality of isolated RNA. cDNA was synthesized with the High-Capacity cDNA Archive Kit (Applied Biosystems). mRNA levels were quantified by TaqMan low-density arrays (Applied Biosystems). For each gene, every sample was run in triplicate on the same array. To measure the quantity of a given RNA species, we monitored the threshold cycles (Ct) by the Applied Biosystems 7900HT Fast Real-Time PCR System. mRNA levels were calculated by relative quantification by using a normalization factor (geometric mean of four reference genes: beta-actin, ACTB; ribosomal large P0, RPLP0; polymerase (RNA) II [DNA-directed) polypeptide A, POLR2A; and ubiquitin C, UBC) and the qBASE program for internal and external calibration and for easy care of large RT-PCR data sets.
Immunohistochemistry
The cerebellar tissue was obtained from an SCA23 patient with a p.R138S6 and from a control subject via a rapid autopsy protocol with a warm postmortem interval of 4 or 5 hr (Table S4). The control subject was without cerebellar ataxia or other neurological disorders. Fresh-frozen cerebellar tissue samples were fixed in 4% phosphate-buffered paraformaldehyde, frozen, cut in 5 μm thick sections, and processed as described previously, with modifications.18,19 Briefly, the sections were incubated with PBS/0.2% triton and 0.1 M sodium citrate buffer (pH = 4.5) exposed to microwave irradiation and incubated with rabbit polyclonal anti-PDYN, Dyn A, or Dyn B antibodies (IgG fraction)12,19 or mouse monoclonal anti-SLC1A6 (alias EAAT4; MIM #600637) antibodies (Abcam). After incubation with peroxidase-labeled secondary antibodies (Santa Cruz Biotechnology), the staining was visualized by 3-amino-9-ethylcarbazole (AEC; Sigma-Aldrich) and counterstained with hematoxylin. The sections were analyzed with a bright-field microscope (Leica; DM3000).
Cell Culture, Transfection, and Plasmids
RINm-5F cells were grown in RPMI-1960 (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen) in a 37°C incubator with 5% CO2. Transfections were carried out with Lipofectamine (Invitrogen). pCMV4 empty plasmid was used for mock transfection. The wild-type pCMV4-fl-h-PDYN construct was described previously.19 The p.R138S, p.L211S, p.R212W, and p.R215C mutations were introduced into the PDYN cDNA with the Quickchange II Site-Directed Mutagenesis kit (Stratagene). The mutagenesis primers that were used to generate the mutant PDYN constructs are listed in Table S8. Sequencing was performed to verify the presence of the mutations and to check PDYN cDNA sequence.
Western Blotting
To enrich PDYN in extracts of the cerebellum characterized by low PDYN expression levels, we extracted the tissues with 1 M acetic acid followed by SEP-PAC C18 reverse-phase chromatography or with Buffer C, as described before.12,19 Both procedures gave essentially the same results. Tissues for analysis of EAAT4 and calbindin (MIM #114050) and cell pellets for analysis of PDYN were extracted with 4% SDS buffer supplemented with DTT and Compete Inhibitor Cocktail (Roche), as described previously.20 Protein concentration was determined with DC protein assay (Bio-Rad Laboratory). Proteins were resolved by SDS-PAGE on 10% Tricine gels, transferred onto nitrocellulose membranes (Schleicher and Schuell), and stained with MemCode reversible Protein Stain Kit (Pierce), as described previously.12,19 Membranes blocked with 5% nonfat dry milk were probed with anti-PDYN, anti-Dyn B, and anti-EAAT4 antibodies (Santa Cruz Biotechnology) or anti-calbindin antibody (Sigma-Aldrich) and were incubated with peroxidase-conjugated anti-rabbit, anti-mouse (Bio-Rad Laboratory), and anti-goat secondary antibodies (Sigma-Aldrich). Densitometric analysis was performed with Image Gauge V3.12 (Fujifilm). Protein levels were calculated as the ratio of optical density (OD) of protein immunoreactivity to MemCode OD. Measurements of all proteins were performed within the linear range of detection. The correlation coefficient between protein immunoreactivity measured as OD and protein load measured as MemCode OD was 0.98 or higher.
Sample Preparation for Mass Spectrometry Analysis
Powdered brain tissue samples were sonicated in the extraction buffer (6 M urea, 2 M thiourea, and 1% octyl-β-D-glucopyranoside [all Sigma-Aldrich] in 10 mM Tris-HCl) and further exchanged to 50 mM NH4HCO3 (Sigma-Aldrich) with PD SpinTrap G-25 columns (GE Healthcare). The protein was measured via the microBCA assay (Pierce). The samples were processed via a modification of protocol described elsewhere.21 The samples were reduced with 15 mM 1,4-dithiothreitol (Roche), alkylated by incubation with 30 mM iodoacetamide (Sigma-Aldrich), and then processed on the 10 kDa cutoff filters (Millipore). Stepwise washing with 2% acetonitrile (Sigma-Aldrich) in 50 mM NH4HCO3, 50% acetonitrile in 50 mM NH4HCO3 and 50 mM NH4HCO3 was followed by incubation with trypsin (1 μg per 50 μg protein; Roche) for 20 hr at 37°C. Peptides were eluted from the filters with 0.1% formic acid (Sigma-Aldrich) and were dried on SpeedVac. The aliquots of each sample containing the same amount of protein (3 μg) were prepared by dilution in 0.1% trifluoroacetic acid.
High-Performance Liquid Chromatography/Tandem Mass Spectrometry and Key-Node Analysis
Experiments were performed on an LTQ-FT Ultra mass spectrometer (ThermoFisher Scientific) used online with an Agilent 1100 nanoflow high-performance liquid chromatography and a nano electrospray ion source (Proxeon Biosystems). A 15 cm C18 nanocolumn (75 μM inner diameter, 375 μm outer diameter) was used for peptide elution at a flow of 200 nL/min with a gradient from 2% to 50% acetonitrile for 100 min. Mass spectrometric analysis was performed by recording of high-resolution (100,000) survey mass spectrum in Fourier transform mass spectrometry and consecutive low-resolution, collision-induced dissociation of up to five of the most abundant ions in the LTQ. Acquired data (.RAW files) were converted to the Mascot search engine (version 2.1.3, Matrix Science). Monoisotopic mass tolerance was set to 10 ppm, and for fragment ions it was ±0.9 Da. A protein was considered to be positively matched by using the stringent threshold of 27 for the Mowse score for all its peptides (p ≤ 0.05). The Mascot htm output and .RAW files of each sample were used as input files for quantification with an in-house-written program package (C++). The area of the chromatographic peak was taken as the peptide abundance. Sum of the abundances of all “bold red” nonidentical peptides was attributed to the protein expression level (≥1 peptide per protein). Protein expression data are shown in Table S4. Protein IDs and protein abundances were loaded into ExPlain 2.4.1 tool (BIOBASE GmbH), where protein IDs were substituted by respective gene IDs. Then we searched upstream for the key nodes most relevant for the input gene products.22 Key nodes are signaling molecules found on pathway intersections in the upstream vicinity of the genes from the input list. Each key node found was given a score reflecting its connectivity, i.e., how many input-list genes are reached and the proximities to those genes. The score calculation also included the abundances of the downstream proteins. Changes in the key node scores thus reflected the changes in the activation levels of the corresponding signaling networks.22
Results
We previously reported the mapping of the SCA23 locus to chromosomal region 20p12.3-p13.6 After excluding 32 prioritized genes via sequencing,7 we identified a c.414G>T (p.R138S) transition resulting in a missense mutation in exon 4 of PDYN in all affected family members (n = 10), but not in unaffected relatives (n = 4) of the originally reported SCA23 family (Figures 1A and 1B). To confirm a possible role of PDYN in cerebellar degeneration, we screened the coding region of PDYN in a panel of Dutch ataxia patients (n = 1100). The panel consists of ataxia patients who applied for regular DNA diagnostics and had no mutations in seven known SCA genes (SCA1–3, SCA6, SCA7, SCA12, and SCA14). Only in 10% of these cases was a clear familial history of the disease described (8% dominant and 2% recessive), and 90% seemed apparently sporadic.
Figure 1.
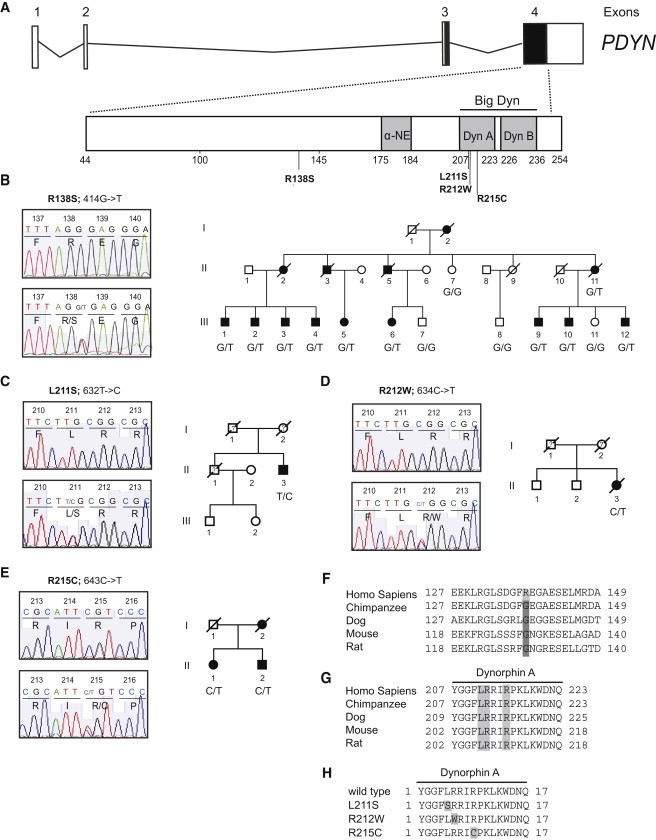
PDYN Mutations Causing SCA23
(A) PDYN exons 3 and 4 encode PDYN, which gives rise to the opioid peptides α-neoendorphin (α-NE), dynorphin A (Dyn A), dynorphin B (Dyn B), and big dynorphin (Big Dyn), which encompasses Dyn A and Dyn B. DNA sequence analysis identified four missense mutations (c.414G>T, c.632T>C, c.634C>T, and c.643C>T) in SCA23 subjects.
(B) Sequence electropherogram and pedigree of original Dutch SCA23 family. The c.414G>T; p.R138S mutation was identified in 10 affected individuals, but not in the four unaffected family members.
(C–E) Sequence electropherograms and pedigrees of the patients with Dyn A mutations: c.632T>C; p.L211S, c.634C>T; p.R212W, and c.643C>T; p.R215C. At the moment, no DNA material of additional family members was available for mutation screening in the p.L211S and p.R212W families. The p.R215C mutation was identified in two affected siblings. Closed symbols denote affected; open symbols denote unaffected; ? denotes disease status unknown; / denotes deceased.
(F and G) Conservation of the mutated amino acids. Arginine 138 is human specific; other species analyzed have glycine 138. Leucine 211, Arginine 212, and Arginine 215 are conserved across species.
(H) Localization of mutations in Dyn A.
We identified three additional missense mutations, c.632T>C (p.L211S), c.634C>T (p.R212W), and c.643C>T (p.R215C), among the 1100 screened Dutch ataxia patients (one familial case with dominant inheritance of the disease and two apparently sporadic cases with an unknown family history; Figures 1C–1E). We were able to show segregation of the p.R215C substitution in two affected siblings (patients II-1 and II-2 in Figure 1E). For the other two families (Figures 1C and 1D), no additional family members were available. The man with the p.L211S mutation (Figure 1C, patient II:3) suffered from progressive gait and upper limb ataxia, oculomotor abnormalities, distal sensory neuropathy, and pyramidal signs of the legs. In addition, subtle parkinsonian features were noted. The disease symptoms started at age 73. The family history was incomplete, and ataxia was not mentioned. The woman with the p.R212W mutation (Figure 1D, patient II:3) displayed a slowly progressive cerebellar ataxia of the legs and arms, as well as a progressive mixed axonal polyneuropathy. The onset of the symptoms started at age 54, and the family history was negative for ataxia. An MRI scan of the brain, performed 4 months prior to death, revealed generalized cerebral cortical and subcortical atrophy, agenesis of the corpus callosum, and prominent atrophy of the cerebellar vermis, the pons, and the inferior olivary nucleus. The woman carrying the p.R215C mutation (Figure 1E, patient II:1) was initially diagnosed with an essential tremor of head and postural arms. From about age 50 onward, memory and word finding deteriorated. A neurological exam at age 53 revealed slowed mental processes, slight dysarthria, mild ataxia of upper and lower limbs, postural arm tremor and a tremor of the head, proximal paresis of the legs with mild signs of sensory neuropathy, and a bilateral pes cavus. Her brother (Figure 1E, patient II:2) was already considered clumsy at school age. Around age 54, mild gait impairment and slowing were noted. The neurological examination at age 56 revealed head tremor, fixation instability at neutral gaze, very mild dysarthria, action tremor of the hands, and a subtle ataxia of the hands and arms. The mother of these two patients (not examined by us) suffered from mild late-onset ataxia and head and limb tremor since primary school, and her mother likely suffered from dementia. The clinical details are summarized in Table S1. All four mutations were absent in 1000 Dutch blood bank control chromosomes. In addition, three known SNPs (rs77155664, rs45469293, and rs6045819) were identified in exon 4, and no difference in their distribution was observed between ataxia cases and controls (data not shown).
Of the four altered amino acids, Arg138 is confined to humans (Figure 1F), whereas Lys211, Arg212, and Arg215 were conserved across the vertebrate species examined (Figures 1G and 1H). In silico analysis with four different bioinformatics programs (polyPhen, SIFT, SWAP, and Align GVD) predicted three (p.L211S, p.R212W, and p.R215C) of the four missense mutations to have damaging effects on protein structure and function, whereas the p.R138S mutation was predicted to be benign (Table S2). In addition, the p.R138S and p.L211S mutations were also predicted to increase PDYN phosphorylation levels by generating potential phosphorylation sites (Table S3).
Proprotein convertases 1 and 2 cleave PDYN at both paired and single basic residues, resulting in defined sets of neuropeptides.8,23 The human-specific Arg138 is located in the nonopioid domain of PDYN and may affect N-terminal trimming of the 8 kDa and 10 kDa PDYN processing intermediates. The p.L211S, p.R212W, and p.R215C mutations in PDYN substitute amino acid residues Leu5, Arg6, and Arg9 to Ser5, Trp6, and Cys9, respectively, in the 17 amino acid Dyn A sequence (Figure 1H). These changes may abrogate Dyn A conversion to Dyn A(1–8), Leu-enkephalin-Arg, or Leu-enkephalin; Dyn A is cleaved between the 8th and 9th, 6th and 7th, and 5th and 6th amino acid residues, and both Arg6 and Arg9 are critical for Dyn A processing.23,24
To examine whether the mutations affect PDYN synthesis and processing, we transfected the cDNA wild-type (WT) or mutant PDYN cDNA into rat insulinoma RINm-5F cells. Protein and peptide products were analyzed by immunoblotting with anti-PDYN and Dyn B antibodies and by RIA for Dyn A, Dyn B, and the dynorphin conversion product Leu-enkephalin-Arg (Figures 2A–2D).19 RINm-5F cells do not produce endogenous PDYN and efficiently process ectopic PDYN into opioid peptides.19 Analysis of cells transfected with PDYN plasmids demonstrated that WT and all mutants produced the 28 kDa PDYNs at similar levels (Figure 2A). However, the efficiency of processing of these proteins to opioid peptides was dramatically affected by the mutations (Figures 2B–2D). The levels of Dyn A produced by PDYN p.L211S and p.R212W were 10- to 18-fold elevated compared to WT, whereas the levels of Dyn B produced by PDYN p.R138S, p.R212W, and p.R215C, as well as the levels of Leu-enkephalin-Arg produced by PDYN p.R138S and p.R215C, were decreased approximately 2-fold. Thus, all three Dyn A mutants resulted in a 2- to 35-fold higher Dyn A production compared to Dyn B.
Figure 2.
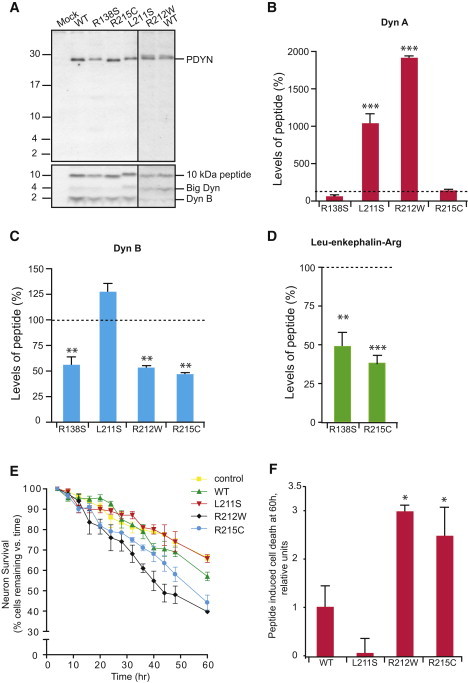
SCA23 Mutations Affect PDYN Processing and Enhance Dyn A Toxicity
(A) Expression and processing of WT and mutant PDYNs in RINm-5F cells. Immunoblotting was performed with anti-PDYN (against C-terminal fragment; top) or anti-Dyn B (bottom) antibodies. Synthetic big dynorphin (a 32 amino acid peptide consisting of Dyn A and Dyn B) and Dyn B were loaded on the same gel as peptide markers and produced the 4 and 2 kDa bands identified by anti-Dyn B antibodies (data not shown; however, their positions are shown on the right). The 10, 4, and 2 kDa PDYN processing products were detected in RINm-5F cells transfected with PDYN expression plasmids.
(B–D) Peptide levels in cells expressing WT PDYN were taken as 100%. One-way ANOVA followed by Dunnett's multiple comparison test; ∗∗p < 0.01, ∗∗∗p < 0.001; mutant versus wild-type. Data are shown as means ± SEM.
(B) RINm-5F cells expressing PDYN p.L211S and p.R212W showed significant elevation in levels of Dyn A compared to cells expressing WT PDYN (ANOVA; p < 0.001). The Dyn A RIA was not hindered by the mutations because they did not affect the binding of the mutant peptides to the Dyn A antibodies. The antibodies were generated against the C-terminal Dyn A fragment.12,19
(C) Dyn B levels showed significant decrease in cells expressing PDYN p.R138S, p.R212W, and p.R215C (ANOVA; p < 0.001).
(D) PDYN p.R138S and p.R215C produced lower levels of Leu-enkephalin-Arg compared to WT PDYN (ANOVA; p < 0.005). Leu-enkephalin-Arg peptides derived from PDYN p.L211S and p.R212W were not analyzed by RIA because these two mutations are located within the Leu-enkephalin-Arg antigenic epitope and may hinder the binding of these mutant peptides to anti-Leu-enkephalin-Arg antibodies.12,19
(E and F) Time-lapse imaging of striatial neurons treated with Dyn A peptides (100 nM) revealed marked loss of neuronal viability induced by Dyn A p.R212W and Dyn A p.R215C peptides compared to vehicle-treated control or WT Dyn A (one-way repeated-measures ANOVA; p < 0.005). Data are represented as the means ± SEM.
(F) Neuronal death at 60 hr induced by mutant Dyn A peptides. The level of neuronal death as a result of excess WT Dyn A was considered as 1. Bonferroni's multiple correction test, ∗p < 0.05. Data are represented as the means ± SD.
Previous studies have shown that Dyn A can induce toxicity and neuronal cell death.25–27 To examine whether mutant Dyn A would be more neurotoxic, we analyzed effects of WT and mutant Dyn A peptides on the viability of striatal neurons via environmentally controlled time-lapse imaging. Although mild toxic effects were induced by WT Dyn A, we observed marked increases in neuronal losses following exposure to p.R212W and p.R215C mutant Dyn A peptides compared to vehicle-treated controls or the WT peptide after 60 hr of exposure (Figures 2E and 2F). No significant differences in cell viability were observed between control and the p.L211S Dyn A-treated neurons. Thus, two mutations in Dyn A may enhance the intrinsic neurotoxicity of WT Dyn A.
The bias, based almost exclusively on data in mature rodents, is that the endogenous opioid peptides and their receptors are not produced and do not regulate neuronal activity in the cerebellum.28 To gain insight into pathophysiological mechanisms of PDYN actions, we characterized these systems in three controls and SCA23 cerebella. Only a single brain of a SCA23 PDYN p.R138S subject was available for postmortem analysis.6 Our results showed that PDYN was expressed in the human cerebellum, although at low levels compared to the nucleus accumbens (NAc) and prefrontal cortex (PFC; Figure 3A). In contrast to animals, the κ- and μ- (OPRM1; MIM #600018) opioid receptors and proenkephalin (PENK; MIM #131330) were expressed in human cerebellum at levels that were comparable with those in the NAc and PFC, whereas the δ-opioid receptor (OPRD1) was not expressed (Figure 3A).29 In SCA23 tissue, cerebellar OPRK1, OPRM1, and PENK were 2- to 3.5-fold upregulated, whereas the level of PDYN mRNA was slightly lower than in controls (Figure 3B). In contrast to mRNA, cerebellar PDYN levels were intermediate between those in PFC and NAc and were similar in controls and SCA23 (Figures 3C and 3D). The high protein levels in relation to PDYN expression in cerebellum in comparison to NAc and PFC suggest slower processing of PDYN to mature peptides in this area.
Figure 3.
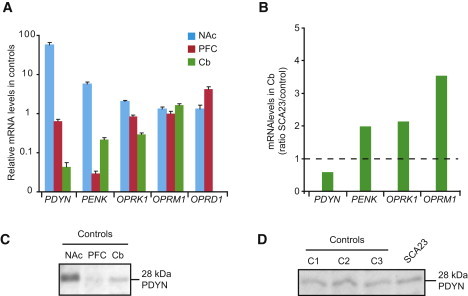
Impact of the PDYN p.R138S Mutation on Expression of the Opioid Genes and PDYN Levels in Human Brain
(A) Levels of opioid mRNA in the nucleus accumbens (NAc), prefrontal cortex (PFC), and cerebellum (Cb) were quantified by TaqMan low-density arrays in postmortem samples from three control subjects. PDYN expression was substantially lower in Cb compared to NAc and PFC, the two brain areas with high and moderate gene expression. In the Cb, PENK showed high expression levels compared to the PFC but lower expression levels compared to the NAc. Both OPRK1 and OPRM1 are expressed in Cb, NAc, and PFC at similar levels, whereas OPRD1 expression was not detectable in the Cb. Data bars represent the means ± SEM.
(B) Comparison of mRNA levels in the Cb between three control and SCA23 cerebella. The data is shown as the ratio of levels in SCA23 to those in controls. Mean levels in controls were set to 1, as indicated by the dashed line. PENK, OPRK1, and OPRM1 expression was 2- and 3.5-fold increased, whereas PDYN expression was slightly decreased in the SCA23 Cb compared to controls.
(C) Analysis of PDYN in the human NAc, PFC, and Cb by immunoblot analysis. Tissue samples were pooled from three control subjects and purified on SEP-PAC reverse-phase columns prior to SDS-PAGE. PDYN levels in cerebellum are intermediate between those in NAc and PFC.
(D) No differences were evident between PDYN levels in control (C1-3) and SCA23 cerebella.
To determine in which cell types PDYN, Dyn A, and Dyn B are localized in cerebellum, we performed immunohistochemistry. This analysis demonstrated that PDYN and its major peptide products are mainly located in the Purkinje cells in both control and SCA23 cerebellum (Figure 4A). The SCA23 cerebellum showed significant loss of Purkinje cells and atrophic dendrites, as was described previously.6 No marked differences in the localization and generation of PDYN and the Dyn A and B peptides were observed between control and SCA23 cerebella.
Figure 4.
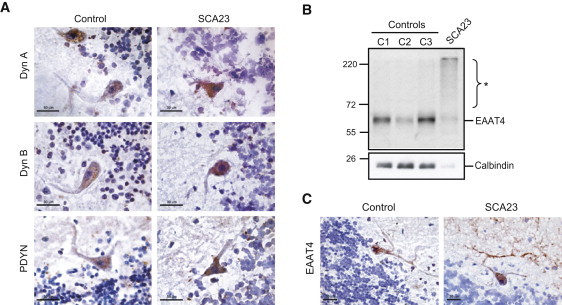
Localization of PDYN and Dynorphins, and Analysis of the Purkinje Cell-Specific Excitatory Amino Acid Transporter, in SCA23 Cerebellum
(A) Immunohistochemistry of cerebellum from the PDYN p.R138S subject and a control individual without neurological disease. Immunoperoxidase labeling with anti-Dyn A, anti-Dyn B, or anti-PDYN antibodies was visualized by 3-Amino-9-ethylcarbazole as the chromogen and was counterstained with hematoxylin.
(B) Immunoblot analysis of excitatory amino acid transporter (EAAT4) and calbindin, both Purkinje cell markers. High molecular weight species were detected in the SCA23 cerebellum (∗).
(C) Immunoperoxidase labeling of EAAT4 in SCA23 and control cerebellum.
Scale bars represent 30 μm (A and C).
Dyn A has been shown to induce neurodegeneration through a nonopioid mechanism that may involve glutamate receptors and acid-sensing ion channels.9,30 To unravel cellular pathways affected by PDYN p.R138S, we performed shotgun proteomic analysis of control and SCA23 cerebella. Proteins involved in cell adhesion, synaptogenesis, secretory pathways, and signal transduction were up- or downregulated in SCA23 (Figure S1 and Table S4). Key node molecules in cellular pathways affected in SCA23 cerebellum identified by pathway analysis include enzymes regulating glutamate cycling and proteins involved in neuroprotection, apoptosis, and signal transduction (Table S5). The solute carrier, SLC1A6 (alias EAAT4; excitatory amino acid transporter), a marker of Purkinje cells that has been previously found to be downregulated or aggregated in SCA1 and SCA5,31,32 was decreased in the proteome of SCA23 PDYN p.R138S (Table S4). Immunoblotting confirmed a reduction in the 65 kDa EAAT4 in the SCA23 cerebellum by 3-fold (Figure 4B). About 70% of EAAT4 was accumulated as aggregated or polyubiquitinated protein in SCA23 (Figure 4C). EAAT4 showed strong staining in perikarya and dendrites of the Purkinje cells in control and SCA23 cerebella (Figure 4C). Calbindin is another protein marker of the Purkinje cells and was also found to be downregulated in SCA1 and SCA5 cerebella.31,32 Although the proteomic analysis showed only marginal 1.6-fold decrease in calbindin levels in SCA23, further immunoblot analysis demonstrated substantial 7-fold downregulation of this protein in the pathological cerebellum (Figure 4B).
Discussion
We have shown that missense mutations in PDYN cause a relatively slowly progressive, cerebellar ataxia with some distinctive features such as distal sensory neuropathy and pyramidal signs, proximal paresis of the legs, and tremors of the head and upper limbs. SCA23 was assumed to be a relatively pure cerebellar ataxia. However, our findings showed that the clinical phenotype of SCA23 can be highly variable and that no common starting symptom was present in our four different SCA23 families. Mutations in PDYN are identified here in a small subset of Dutch ataxia families (4 of 1100), indicating that SCA23 seems an infrequent SCA type (±0.5%) in the Netherlands and suggesting further genetic heterogeneity. However, the syndromes present in our ataxia panel are phenotypically not well defined, which is reflected in the low success rate (∼20%) to identify mutations in known SCA genes via diagnostic screening. This may explain why we have identified only 4 ataxia families exhibiting PDYN mutations in 1100 cases. Although we can only speculate about the frequency of SCA23 in a phenotypically better-defined cohort, we assume that SCA23 is a rare cause of ataxia and will not account for more than ∼0.5% of the Dutch ataxia cases. To exclude the possibility that the PDYN p.R138S mutation would be a random sequence variation that cosegregates with the true disease mutation within the linkage interval, we sequenced 1000 unrelated blood bank control chromosomes in which this variation was not observed, and we did not identify any other additional coding sequence variation in 31 previously sequenced candidate genes that perfectly cosegregated with the disease phenotype in this family.7 In two families (PDYN p.R138S and p.R215C), we could confirm that the mutations segregated with the disease.
The in silico analysis predicts damaging effects of three Dyn A mutations (p.L211S, p.R212W, and p.R215C) on protein structure and possibly function, whereas the p.R138S mutation was assessed as benign. The negative prediction may be due to the insufficient sensitivity of the programs used in analysis, because they are based on similar algorithms and may have the same limitation. The analysis may also be hindered by the absence of any structural information for this class of proteins. Formation of an additional phosphorylation site in PDYN by the p.R138S mutation may affect processing or trafficking of mutant protein, thus representing an alternative mechanism of the pathogenic activity of PDYN.
Model cell experiments provided evidence that the PDYN mutations p.L211S (Leu5) and p.R212W (Arg6) result in enhanced Dyn A levels. This may be due to slow conversion of mutant Dyn A to short enkephalins. Indeed, conversion of Dyn A to short fragments generally occurs at di- and monobasic amino acid residues, including Leu5 and Arg9, that are substituted by Ser and Cys in mutant PDYN.23 In addition, analysis of the effect of mutant Dyn A peptides on striatal neurons demonstrated high toxicity of Dyn A p.R212W and Dyn A p.R215C. The elevated neurotoxicity of Dyn A mutants compared to WT Dyn A suggests a dominant-negative effect of these mutations rather than a loss of function. The increased stabilization of the mutant Dyn A peptides may not be the only factor underlying neurotoxicity, because Dyn A p.L211S, which is apparently resistant for conversion to a short fragment, does not induce any toxic response.
Immunohistochemical experiments revealed that PDYN, Dyn A, and Dyn B are mainly located in the Purkinje cells in both control and SCA23 cerebella. The absence of significant differences in PDYN and dynorphin localization and PDYN levels between control and PDYN p.R138S cerebella, observed by both the cell staining and immunoblotting, further argues against a possible loss of function and suggests that PDYN is strongly elevated in the Purkinje cells remaining in SCA23.
Further analysis of the endogenous opioid systems and the proteome in cerebellar autopsy tissue of the PDYN p.R138S subject revealed marked alterations in expression of crucial components of the opioid and glutamate systems. These alterations were evident for OPRM1, OPRK1, enzymes regulating glutamate cycling, and the excitatory amino acid transporter, EAAT4. These findings are consistent with previous observations demonstrating alterations in expression and localization of EAAT4 in SCA1 and SCA5 cerebella and provide additional evidence for the hypothesis that the dysfunction of glutamate signaling is a common downstream mechanism for SCA.31,32
Two putative mechanisms may be envisaged to explain the pathogenic effects of mutant PDYNs. First, modifications of nonopioid neurodegenerative and/or opioid activities of Dyn A or its longer pathological form, such as the 32 amino acid big dynorphin, may lead to alterations in glutamate signaling and excitotoxicity.9,26,30,33 Several lines of evidence support this notion. Thus, Dyn A, but not other dynorphins, elicits various pathological effects, including neurological dysfunction and cell death. Intracerebroventricular injection of this peptide induces abnormal motor effects such as wild running, barrel rolling, and ataxia in mice, whereas intrathecal infusion produces allodynia, paralysis, and neuronal loss.9,34–39 Consistently, increased production of Dyn A following trauma contributes to spinal cord injury and neuropathic pain.9,37–39 Dyn A is also upregulated in aging rodent brain, contributing to age-related impairment of spatial learning and memory.40 In the prefrontal cortex of Alzheimer disease patients, the elevated Dyn A levels correlate with neuritic plaque density.41 Second, synthesis of mutant PDYNs may affect secretory pathways, inducing endoplasmic reticulum stress and impairing maturation and trafficking of glutamate transporters and other signaling proteins. Actions of mutant PDYNs accumulating over the years may ultimately lead to Purkinje cell degeneration. The p.R138S mutation located in the nonopioid PDYN domain supports the second hypothesis.
Neuropeptides are a large group of signaling molecules that regulate a wide variety of physiological and pathological processes.9,10 A mutation that causes early-onset obesity and adrenal insufficiency had been found in proopiomelanocortin, the protein precursor to adrenocorticotropin, α-melanocyte-stimulating hormone, and β-endorphin.42 Our findings demonstrate that mutations in a neuropeptide gene may cause an inherited neurodegenerative disorder. Neuropeptides usually exert their actions as ligands of G protein-coupled receptors. In the case of Dyn A, although physiological effects are largely mediated through the κ-opioid receptors, pathological effects may be generally mediated through direct action of this peptide on the plasma membrane, possibly by inducing pore formation and increased calcium influx in neurons.9,43–45 Remarkably, three mutations identified in SCA23 families alter the amino acid sequence of Dyn A in a domain associated with non-receptor-mediated, neurodegenerative actions. Two peptides with these mutations cause significant toxicity in cultured neurons, raising the possibility that some SCA23 degenerative processes are due to direct effects of mutant peptides on neurons. We may speculate that mutations in other neuropeptide precursor genes are involved in the etiology of cerebellar ataxia and that their identification will provide further insight into mechanisms of synaptic neurotransmission and neurodegeneration.
Acknowledgments
We thank the participating family members for their cooperation and all the members of the SpinoCerebellaire Ataxie Nederland working group for their input and help regarding this study. We acknowledge Henk Moorlag for preparing the cerebellar slides, Alexander Zubarev for protein quantification software, Henrik Druid, Kanar Alkass, and Pieter Wesseling for human brain samples, and Jackie Senior for improving the manuscript. This work was supported in part by a Rosalind Franklin Fellowship from the University of Groningen (D.S.V.) and a National Institute on Drug Abuse Independent Scientist Award (DA027374; K.F.H.), as well as by the Swedish Science Council, Swedish Institute, AFA Forsäkring and Swedish Council for Working Life and Social Research (G.B.), and an Uppsala University Fellowship (H.W.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Align GVGD, http://agvgd.iarc.fr/agvgd_input.php
BIOBASE Biological Databases, http://www.biobase-international.com
GenBank, http://www.ncbi.nlm.nih.gov/genbank/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
PolyPhen, http://genetics.bwh.harvard.edu/pph/
SIFT, http://sift.jcvi.org/
References
- 1.Schöls L., Amoiridis G., Büttner T., Przuntek H., Epplen J.T., Riess O. Autosomal dominant cerebellar ataxia: Phenotypic differences in genetically defined subtypes? Ann. Neurol. 1997;42:924–932. doi: 10.1002/ana.410420615. [DOI] [PubMed] [Google Scholar]
- 2.Matilla-Dueñas A., Sánchez I., Corral-Juan M., Dávalos A., Alvarez R., Latorre P. Cellular and molecular pathways triggering neurodegeneration in the spinocerebellar ataxias. Cerebellum. 2010;9:148–166. doi: 10.1007/s12311-009-0144-2. [DOI] [PubMed] [Google Scholar]
- 3.Sato N., Amino T., Kobayashi K., Asakawa S., Ishiguro T., Tsunemi T., Takahashi M., Matsuura T., Flanigan K.M., Iwasaki S. Spinocerebellar ataxia type 31 is associated with “inserted” penta-nucleotide repeats containing (TGGAA)n. Am. J. Hum. Genet. 2009;85:544–557. doi: 10.1016/j.ajhg.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schorge S., van de Leemput J., Singleton A., Houlden H., Hardy J. Human ataxias: A genetic dissection of inositol triphosphate receptor (ITPR1)-dependent signaling. Trends Neurosci. 2010;33:211–219. doi: 10.1016/j.tins.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carlson K.M., Andresen J.M., Orr H.T. Emerging pathogenic pathways in the spinocerebellar ataxias. Curr. Opin. Genet. Dev. 2009;19:247–253. doi: 10.1016/j.gde.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Verbeek D.S., van de Warrenburg B.P., Wesseling P., Pearson P.L., Kremer H.P., Sinke R.J. Mapping of the SCA23 locus involved in autosomal dominant cerebellar ataxia to chromosome region 20p13-12.3. Brain. 2004;127:2551–2557. doi: 10.1093/brain/awh276. [DOI] [PubMed] [Google Scholar]
- 7.Verbeek D.S. Spinocerebellar ataxia type 23: A genetic update. Cerebellum. 2009;8:104–107. doi: 10.1007/s12311-008-0085-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akil H., Watson S.J., Young E., Lewis M.E., Khachaturian H., Walker J.M. Endogenous opioids: Biology and function. Annu. Rev. Neurosci. 1984;7:223–255. doi: 10.1146/annurev.ne.07.030184.001255. [DOI] [PubMed] [Google Scholar]
- 9.Hauser K.F., Aldrich J.V., Anderson K.J., Bakalkin G., Christie M.J., Hall E.D., Knapp P.E., Scheff S.W., Singh I.N., Vissel B. Pathobiology of dynorphins in trauma and disease. Front. Biosci. 2005;10:216–235. doi: 10.2741/1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ludwig M., Leng G. Dendritic peptide release and peptide-dependent behaviours. Nat. Rev. Neurosci. 2006;7:126–136. doi: 10.1038/nrn1845. [DOI] [PubMed] [Google Scholar]
- 11.Christensson-Nylander I., Nyberg F., Ragnarsson U., Terenius L. A general procedure for analysis of proenkephalin B derived opioid peptides. Regul. Pept. 1985;11:65–76. doi: 10.1016/0167-0115(85)90032-1. [DOI] [PubMed] [Google Scholar]
- 12.Yakovleva T., Bazov I., Cebers G., Marinova Z., Hara Y., Ahmed A., Vlaskovska M., Johansson B., Hochgeschwender U., Singh I.N. Prodynorphin storage and processing in axon terminals and dendrites. FASEB J. 2006;20:2124–2126. doi: 10.1096/fj.06-6174fje. [DOI] [PubMed] [Google Scholar]
- 13.Singh I.N., Goody R.J., Dean C., Ahmad N.M., Lutz S.E., Knapp P.E., Nath A., Hauser K.F. Apoptotic death of striatal neurons induced by human immunodeficiency virus-1 Tat and gp120: Differential involvement of caspase-3 and endonuclease G. J. Neurovirol. 2004;10:141–151. doi: 10.1080/13550280490441103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh I.N., El-Hage N., Campbell M.E., Lutz S.E., Knapp P.E., Nath A., Hauser K.F. Differential involvement of p38 and JNK MAP kinases in HIV-1 Tat and gp120-induced apoptosis and neurite degeneration in striatal neurons. Neuroscience. 2005;135:781–790. doi: 10.1016/j.neuroscience.2005.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fleige S., Pfaffl M.W. RNA integrity and the effect on the real-time qRT-PCR performance. Mol. Aspects Med. 2006;27:126–139. doi: 10.1016/j.mam.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 16.Fleige S., Walf V., Huch S., Prgomet C., Sehm J., Pfaffl M.W. Comparison of relative mRNA quantification models and the impact of RNA integrity in quantitative real-time RT-PCR. Biotechnol. Lett. 2006;28:1601–1613. doi: 10.1007/s10529-006-9127-2. [DOI] [PubMed] [Google Scholar]
- 17.Stadler F., Kolb G., Rubusch L., Baker S.P., Jones E.G., Akbarian S. Histone methylation at gene promoters is associated with developmental regulation and region-specific expression of ionotropic and metabotropic glutamate receptors in human brain. J. Neurochem. 2005;94:324–336. doi: 10.1111/j.1471-4159.2005.03190.x. [DOI] [PubMed] [Google Scholar]
- 18.Waldvogel H.J., Curtis M.A., Baer K., Rees M.I., Faull R.L. Immunohistochemical staining of post-mortem adult human brain sections. Nat. Protoc. 2006;1:2719–2732. doi: 10.1038/nprot.2006.354. [DOI] [PubMed] [Google Scholar]
- 19.Nikoshkov A., Hurd Y.L., Yakovleva T., Bazov I., Marinova Z., Cebers G., Pasikova N., Gharibyan A., Terenius L., Bakalkin G. Prodynorphin transcripts and proteins differentially expressed and regulated in the adult human brain. FASEB J. 2005;19:1543–1545. doi: 10.1096/fj.05-3743fje. [DOI] [PubMed] [Google Scholar]
- 20.Okvist A., Johansson S., Kuzmin A., Bazov I., Merino-Martinez R., Ponomarev I., Mayfield R.D., Harris R.A., Sheedy D., Garrick T. Neuroadaptations in human chronic alcoholics: Dysregulation of the NF-kappaB system. PLoS ONE. 2007;2:e930. doi: 10.1371/journal.pone.0000930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wiśniewski J.R., Zougman A., Nagaraj N., Mann M. Universal sample preparation method for proteome analysis. Nat. Methods. 2009;6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 22.Kel A., Voss N., Jauregui R., Kel-Margoulis O., Wingender E. Beyond microarrays: Find key transcription factors controlling signal transduction pathways. BMC Bioinformatics. 2006;7(Suppl 2):S13. doi: 10.1186/1471-2105-7-S2-S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Day R., Lazure C., Basak A., Boudreault A., Limperis P., Dong W., Lindberg I. Prodynorphin processing by proprotein convertase 2. Cleavage at single basic residues and enhanced processing in the presence of carboxypeptidase activity. J. Biol. Chem. 1998;273:829–836. doi: 10.1074/jbc.273.2.829. [DOI] [PubMed] [Google Scholar]
- 24.Silberring J., Castello M.E., Nyberg F. Characterization of dynorphin A-converting enzyme in human spinal cord. An endoprotease related to a distinct conversion pathway for the opioid heptadecapeptide? J. Biol. Chem. 1992;267:21324–21328. [PubMed] [Google Scholar]
- 25.Hauser K.F., Foldes J.K., Turbek C.S. Dynorphin A (1-13) neurotoxicity in vitro: Opioid and non-opioid mechanisms in mouse spinal cord neurons. Exp. Neurol. 1999;160:361–375. doi: 10.1006/exnr.1999.7235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tan-No K., Cebers G., Yakovleva T., Hoon Goh B., Gileva I., Reznikov K., Aguilar-Santelises M., Hauser K.F., Terenius L., Bakalkin G. Cytotoxic effects of dynorphins through nonopioid intracellular mechanisms. Exp. Cell Res. 2001;269:54–63. doi: 10.1006/excr.2001.5309. [DOI] [PubMed] [Google Scholar]
- 27.Goody R.J., Martin K.M., Goebel S.M., Hauser K.F. Dynorphin A toxicity in striatal neurons via an alpha-amino-3-hydroxy-5-methylisoxazole-4-propionate/kainate receptor mechanism. Neuroscience. 2003;116:807–816. doi: 10.1016/s0306-4522(02)00563-8. [DOI] [PubMed] [Google Scholar]
- 28.Mansour A., Fox C.A., Akil H., Watson S.J. Opioid-receptor mRNA expression in the rat CNS: Anatomical and functional implications. Trends Neurosci. 1995;18:22–29. doi: 10.1016/0166-2236(95)93946-u. [DOI] [PubMed] [Google Scholar]
- 29.Schadrack J., Willoch F., Platzer S., Bartenstein P., Mahal B., Dworzak D., Wester H.J., Zieglgänsberger W., Tölle T.R. Opioid receptors in the human cerebellum: Evidence from [11C]diprenorphine PET, mRNA expression and autoradiography. Neuroreport. 1999;10:619–624. doi: 10.1097/00001756-199902250-00032. [DOI] [PubMed] [Google Scholar]
- 30.Sherwood T.W., Askwith C.C. Dynorphin opioid peptides enhance acid-sensing ion channel 1a activity and acidosis-induced neuronal death. J. Neurosci. 2009;29:14371–14380. doi: 10.1523/JNEUROSCI.2186-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin X., Antalffy B., Kang D., Orr H.T., Zoghbi H.Y. Polyglutamine expansion down-regulates specific neuronal genes before pathologic changes in SCA1. Nat. Neurosci. 2000;3:157–163. doi: 10.1038/72101. [DOI] [PubMed] [Google Scholar]
- 32.Ikeda Y., Dick K.A., Weatherspoon M.R., Gincel D., Armbrust K.R., Dalton J.C., Stevanin G., Dürr A., Zühlke C., Bürk K. Spectrin mutations cause spinocerebellar ataxia type 5. Nat. Genet. 2006;38:184–190. doi: 10.1038/ng1728. [DOI] [PubMed] [Google Scholar]
- 33.Merg F., Filliol D., Usynin I., Bazov I., Bark N., Hurd Y.L., Yakovleva T., Kieffer B.L., Bakalkin G. Big dynorphin as a putative endogenous ligand for the kappa-opioid receptor. J. Neurochem. 2006;97:292–301. doi: 10.1111/j.1471-4159.2006.03732.x. [DOI] [PubMed] [Google Scholar]
- 34.Herman B.H., Leslie F., Goldstein A. Behavioral effects and in vivo degradation of intraventricularly administered dynorphin-(1-13) and D-Ala2-dynorphin-(1-11) in rats. Life Sci. 1980;27:883–892. doi: 10.1016/0024-3205(80)90084-3. [DOI] [PubMed] [Google Scholar]
- 35.Walker J.M., Moises H.C., Coy D.H., Baldrighi G., Akil H. Nonopiate effects of dynorphin and des-Tyr-dynorphin. Science. 1982;218:1136–1138. doi: 10.1126/science.6128791. [DOI] [PubMed] [Google Scholar]
- 36.Nakazawa T., Ikeda M., Kaneko T., Yamatsu K., Kitagawa K., Kiso Y. Bestatin potentiates the antinociception but not the motor dysfunction induced by intracerebrally administered dynorphin-B in mice. Neuropeptides. 1989;13:277–283. doi: 10.1016/0143-4179(89)90082-6. [DOI] [PubMed] [Google Scholar]
- 37.Caudle R.M., Mannes A.J. Dynorphin: Friend or foe? Pain. 2000;87:235–239. doi: 10.1016/S0304-3959(00)00360-2. [DOI] [PubMed] [Google Scholar]
- 38.Lai J., Ossipov M.H., Vanderah T.W., Malan T.P., Jr., Porreca F. Neuropathic pain: The paradox of dynorphin. Mol. Interv. 2001;1:160–167. [PubMed] [Google Scholar]
- 39.Tan-No K., Takahashi H., Nakagawasai O., Niijima F., Sato T., Satoh S., Sakurada S., Marinova Z., Yakovleva T., Bakalkin G. Pronociceptive role of dynorphins in uninjured animals: N-ethylmaleimide-induced nociceptive behavior mediated through inhibition of dynorphin degradation. Pain. 2005;113:301–309. doi: 10.1016/j.pain.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 40.Nguyen X.V., Masse J., Kumar A., Vijitruth R., Kulik C., Liu M., Choi D.Y., Foster T.C., Usynin I., Bakalkin G., Bing G. Prodynorphin knockout mice demonstrate diminished age-associated impairment in spatial water maze performance. Behav. Brain Res. 2005;161:254–262. doi: 10.1016/j.bbr.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 41.Yakovleva T., Marinova Z., Kuzmin A., Seidah N.G., Haroutunian V., Terenius L., Bakalkin G. Dysregulation of dynorphins in Alzheimer disease. Neurobiol. Aging. 2007;28:1700–1708. doi: 10.1016/j.neurobiolaging.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 42.Krude H., Biebermann H., Luck W., Horn R., Brabant G., Grüters A. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat. Genet. 1998;19:155–157. doi: 10.1038/509. [DOI] [PubMed] [Google Scholar]
- 43.Marinova Z., Vukojevic V., Surcheva S., Yakovleva T., Cebers G., Pasikova N., Usynin I., Hugonin L., Fang W., Hallberg M. Translocation of dynorphin neuropeptides across the plasma membrane. A putative mechanism of signal transmission. J. Biol. Chem. 2005;280:26360–26370. doi: 10.1074/jbc.M412494200. [DOI] [PubMed] [Google Scholar]
- 44.Hugonin L., Vukojević V., Bakalkin G., Gräslund A. Membrane leakage induced by dynorphins. FEBS Lett. 2006;580:3201–3205. doi: 10.1016/j.febslet.2006.04.078. [DOI] [PubMed] [Google Scholar]
- 45.Hugonin L., Vukojević V., Bakalkin G., Gräslund A. Calcium influx into phospholipid vesicles caused by dynorphin neuropeptides. Biochim. Biophys. Acta. 2008;1778:1267–1273. doi: 10.1016/j.bbamem.2008.02.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.