

Abstract
The congenital dyserythropoietic anemias (CDAs) are inherited red blood cell disorders whose hallmarks are ineffective erythropoiesis, hemolysis, and morphological abnormalities of erythroblasts in bone marrow. We have identified a missense mutation in KLF1 of patients with a hitherto unclassified CDA. KLF1 is an erythroid transcription factor, and extensive studies in mouse models have shown that it plays a critical role in the expression of globin genes, but also in the expression of a wide spectrum of genes potentially essential for erythropoiesis. The unique features of this CDA confirm the key role of KLF1 during human erythroid differentiation. Furthermore, we show that the mutation has a dominant-negative effect on KLF1 transcriptional activity and unexpectedly abolishes the expression of the water channel AQP1 and the adhesion molecule CD44. Thus, the study of this disease-causing mutation in KLF1 provides further insights into the roles of this transcription factor during erythropoiesis in humans.
Main Text
The CDAs represent a heterogeneous group of rare congenital anemias predominantly caused by dyserythropoiesis in the bone marrow.1 Three major (types I to III) and several minor subgroups have been differentiated, mainly according to the morphological abnormalities of erythroblast nuclei observed in bone marrow smears (e.g., chromatin bridges or double nuclei).2,3 The gene responsible for CDA I (MIM 224120) was identified by positional cloning in 20024 and coined CDAN1 (MIM 607465), but its function remains to be elucidated. The gene responsible for CDA II (MIM 224100) has recently been shown to encode SEC23B (MIM 610512), which was known to be involved in the vesicular transport between the endoplasmic reticulum and Golgi apparatus but whose erythroid-specific role was unsuspected.5,6 Although CDA I and CDA II represent most cases, the identification of causative genetic defects in other CDA subgroups or in patients with unclassified CDA may offer further insights into the different pathways underlying erythropoiesis.
The first CDA patient investigated in this study (male patient ME) was born at 28 weeks of gestation to nonconsanguineous healthy parents in a context of acute fetal distress. Hydrops fetalis-associated anemia had been detected at 23 weeks of gestation and treated with two intrauterine transfusions; the karyotype of the fetus was normal. The neonatal examination revealed severe hyperbilirubinemia, hepatomegaly, hypertrophic cardiomyopathy, and several dysmorphic features (micropenis, hypospadia, large anterior fontanel, and hypertelorism). Anemia did not improve after birth and required transfusions. At 4 months of age, the analysis of bone marrow smears showed marked hyperplasia of the erythroid lineage, leading to a diagnosis of CDA, but the dysplastic changes in the erythroblasts did not clearly fit any classification of CDA (Figure S1). Despite treatments with erythropoietin or interferon-alpha, the hemolytic anemia persisted and required recurrent transfusions (at 2–3 week intervals) until a splenectomy was performed at 4 years of age (the enlarged spleen showed no pathologic features). Shortly thereafter, transfusion independence was achieved, and hemoglobin levels were stabilized at around 8.0 g/dl (Table S1). At 13 years of age, patient ME showed short stature (height −3 SD, weight −2 SD) despite growth-hormone therapy and treatment for hypothyroidism and thalassemic facies.
A striking feature of patient ME's CDA was the very large number of nucleated red blood cells in his peripheral blood (there were 210% the number of white blood cells before splenectomy and up to 1,000% thereafter; Figure 1A and Table S1). Most of these circulating nucleated red blood cells were orthochromatic erythroblasts, but only a few of them were enucleating, which suggested a failure of terminal erythroid differentiation. Analysis of these cells by electron microscopy revealed various ultrastructural abnormalities, especially atypical cytoplasmic inclusions and enlarged nuclear pores (Figure S2). The in vitro study of erythroid differentiation of CD34+ cells7 isolated from patient ME's peripheral blood showed normal proliferation and differentiation but impaired enucleation capacity (Figure 1B). Furthermore, when we analyzed a panel of markers on the surface of his erythrocytes by flow cytometry (Figure S3), we noticed the absence of CD44, which was confirmed by immunoblot analysis (Figure 1C), as well as reduced expression of two other adhesion molecules, BCAM and ICAM4. CD44 was similarly absent from his mature erythrocytes and circulating erythroblasts, but it was present on his granulocytes and all his other leukocyte populations (Figure 1D and Figure S4), suggesting that only the erythroid lineage was affected, consistent with a CDA trait. We also found that patient ME's erythrocytes were deficient in the water channel AQP1 (Figure 1C) and, consequently, had a reduced water permeability similar to that of erythrocytes in the very rare AQP1−/− individuals8 (Figure S5). Patient ME's CDA was unique and certainly different from CDA I and CDA II, as suggested by bone marrow analysis and later confirmed by the absence of mutations in CDAN1 and SEC23B (data not shown).
Figure 1.
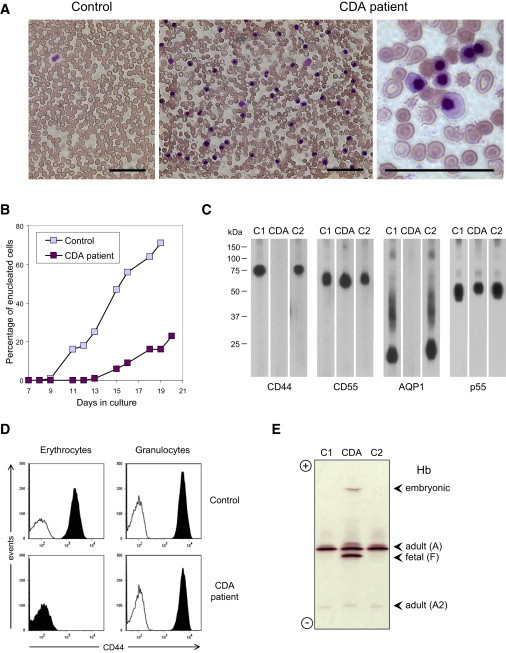
Analysis of the Peripheral Blood of CDA Patient ME Shows Unique Abnormalities
(A) Peripheral blood smears from the patient (right panels; sample taken on 10/28/2008) and a control (left panel; sample taken at the same time) stained with May-Grünwald Giemsa. Note the large number of circulating erythroblasts (purple nuclei) as well as poikilocytosis, anisocytosis and fragmented erythrocytes in the patient. The scale bars represent 40 μm.
(B) Study of the number of enucleated cells during in vitro erythroid differentiation of CD34+ cells from the patient (purple; sample taken on 10/25/2005) or a control donor (blue). Note the markedly reduced enucleation capacity of the patient's cell culture.
(C) Immunoblot analysis of CD44, CD55, AQP1, and p55 in erythrocyte membrane lysates from the patient (CDA; sample taken on 6/7/2004), his healthy mother (C2), or a random control (C1). Note the combined deficiency of CD44 and AQP1 in the patient's erythrocytes.
(D) Flow-cytometry analysis of CD44 on mature erythrocytes (left panels) and granulocytes (right panels) from the patient (bottom panels; sample taken on 1/5/2010) and a control (top panels; sample taken at the same time). Note the erythroid-specific deficiency of CD44 in the patient. For the analysis of erythrocytes, whole blood samples were costained with fluorochrome-conjugated anti-CD44 (black histogram) or isotype control antibody (white histogram) and anti-CD71, and the mature erythrocytes were gated on FSS, SCC, and CD71− so that the reticulocytes and erythroblasts would be eliminated; for the analysis of granulocytes, nucleated blood cells were first isolated by hypotonic erythrocyte lysis, then costained as above and directly gated on FSC and SSC.
(E) Isoelectric focusing analysis of the different hemoglobin (Hb) variants in the patient (CDA; sample taken on 10/28/2008) and two controls (C1 and C2). Note the atypical globin expression in the patient; he exhibited very high levels of fetal Hb (α2γ2 tetramer, 37.3%; normal range, less that 1%) as well as embryonic Hb Portland (ζ2γ2 tetramer, 2.9%; normal range, absent) as ascertained by reverse-phase liquid chromatography; adult HbA and HbA2 were at 55.5% (α2β2 tetramer, normal range: 90%–100%) and 1.2% (α2δ2 tetramer, normal range: 2%–3%), respectively. Extensive sequencing of patient ME's globin loci detected no gross abnormalities but a heterozygous mutation in the α1-globin gene (c.62_63insT, p.His21fsX36), which could not be responsible for the profound β-globin locus dysregulation and was indeed present in his healthy paternal aunt. Of note, patient SF was a carrier of a 4 bp deletion in the promoter of Aγ-globin gene, as was her healthy father.14
All analyses presented herein were performed on blood samples taken from patient ME after splenectomy and at least 6 months after transfusion.
In order to identify the causative genetic defect, we took fresh blood samples from patient ME and his relatives after obtaining a signed informed consent under an institutional-review-board-approved protocol, and we extracted genomic DNA. After unsuccessfully exploring the possibility of an inherited recessive mutation by performing a SNP-based genome-wide screen in patient ME's family (Affymetrix GeneChip Human Mapping 250K Nsp Array, data not shown), we thought that this unique CDA might be caused by a de novo mutation in a transcription factor essential for expression of CD44 (MIM 107269) and AQP1 (MIM 107776), among others. Because CD44 deficiency was apparently restricted to the erythroid lineage, we first focused our analysis on erythroid transcription factors such as GATA1 and KLF1 (also known as EKLF).9,10 GATA1 represented an attractive candidate,11,12 but no mutations in GATA1 (MIM 305371) were detected (data not shown). Sequencing of KLF1 (MIM 600599) (Table S2) in patient ME revealed the presence of two heterozygous mutations: a T-to-C transition in exon 2 (NM_006563.3:c.304T>C, NP_006554.1:p.Ser102Pro) and a G-to-A transition in exon 3 (NM_006563.3:c.973G>A, NP_006554.1:p.Glu325Lys) (Figure S6). A comparison with the NCBI dbSNP database (build 131) showed that c.304T>C mutation corresponded to a previously reported SNP (rs2072597) and was unlikely to be pathogenic; this was consistent with rs2072597 heterozygosity of his healthy mother (data not shown). In contrast, c.973G>A mutation had never been reported and was not present in 96 regular blood donors or in patient ME's relatives (Figure 2A and data not shown), suggesting it was the disease-causing mutation.
Figure 2.
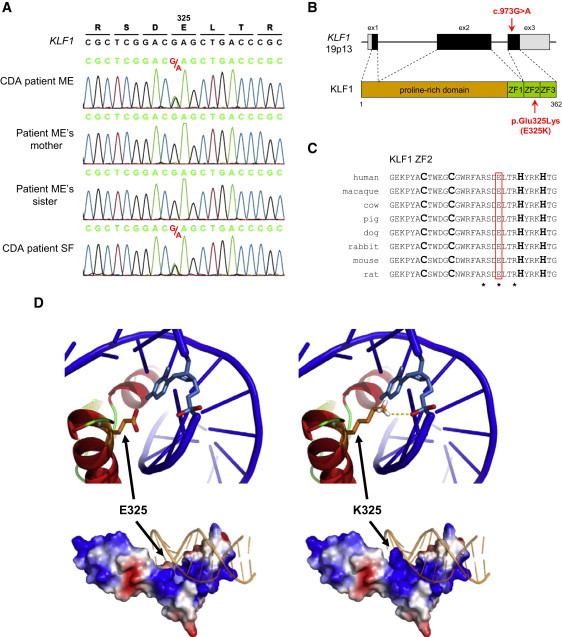
Identification of the KLF1 Mutation Associated with this CDA and Structural Analysis of the Variant Transcription Factor
(A) A detail of KLF1 sequencing in patient ME, his unaffected mother and sister, and unrelated patient SF show the same heterozygous KLF1 mutation in both CDA patients. The experimental sequences were aligned with the NCBI reference sequence of KLF1 (NG_013087.1). Genomic DNA samples from patient ME's father and patient SF's parents were not available.
(B) A diagram shows the structure of KLF1 (based on NG_013087.1) and its products and highlights the localization of the mutation c.973G>A, p.Glu325Lys (E325K) was found in patients ME and SF. KLF1 consists of three exons (boxes; black represents coding regions, and gray represents untranslated regions) and encodes a 362 amino acid peptide with a proline-rich domain at the amino-terminus (the transactivation domain is in brown) and three C2H2-type zinc fingers (ZF) at the carboxy-terminus (the DNA-binding domain is in green). The pathogenic mutation is located in the third exon and encodes a single amino acid change in the second zinc finger of the transcription factor.
(C) A sequence alignment of the second zinc finger of KLF1 from various mammalian species shows the conservation of a glutamate at amino acid position 325 (red box). The two cysteines and the two histidines contacting Zn2+ are indicated in bold, and the three conserved residues contacting the DNA are indicated by stars.
(D) A modeling structure of wild-type (left) and variant E325K (right) zinc-finger domain of KLF1 shows the enhanced electrostatic interaction between the E325K variant and the DNA backbone. The change of glutamate to lysine at position 325 exerts a double effect by reversing the side-chain charge from negative to positive and extending the side-chain length toward the negatively charged DNA backbone; the putative hydrogen bond created by the E325K variant is shown as a yellow dashed line. The bottom panels show the overall structure of the protein-DNA complexes, as well as the electrostatic potential surfaces (negative is in red, positive is in blue) of wild-type and variant proteins. The top panels show a close-up view of a cartoon representation of the region around residue 325, highlighting as sticks the side chain of residue 325 (orange) and the closest nucleotide (light blue); of note, residue 325 is not oriented toward the nucleotide base but toward the phosphate.
To verify that the KLF1 mutation c.973G>A was responsible for this unique CDA, we searched for it in other patients. Female patient SF was extensively studied in the 90s, and her atypical CDA showed striking similarities with that of patient ME: combined deficiency of CD44 and AQP1 on erythrocytes,13 circulating erythroblasts,14 unique ultrastructural abnormalities in erythroblasts,15 and increased electrophoretic mobility of the erythrocyte membrane protein Band 313 (Figure S7). When we sequenced KLF1 in patient SF, we found the same heterozygous KLF1 mutation, c.973G>A, as in patient ME (Figure 2A), verifying that this mutation was responsible for this type of CDA. Of note, and consistent with a de novo mutation, analysis of KLF1 haplotypes of patients ME and SF does not support the hypothesis of a founder effect for the pathogenic KLF1 mutation c.973G>A. In addition to the previously mentioned unique features of this CDA, patient SF was shown to express high levels of fetal hemoglobin, along with embryonic ζ-globin chain, in the majority of her erythrocytes.14 When we analyzed patient ME's hemoglobin by isoelectric focusing (Figure 1E), we similarly found large amounts of fetal hemoglobin as well as an unusual hemoglobin, migrating like embryonic hemoglobin Portland (ζ2γ2 tetramer), which was confirmed by reverse-phase liquid chromatography (Figure S8). Thus, the KLF1 mutation c.973G>A was associated with a profound dysregulation of globin gene expression and provided in vivo evidence of the critical role of KLF1 in this complex transcriptional regulation in humans, as predicted by thalassemia-associated mutations in the proximal KLF1 binding sites of β-globin gene16 and extensive studies in mouse models.17–19 Interestingly, while this manuscript was being finalized, Borg et al.20 described a large Maltese family in which a defective KLF1 allele segregates with the persistence of high levels of fetal hemoglobin in adults—a benign and usually asymptomatic condition known to alleviate the severity of β-thalassemia and sickle cell disease—and they further demonstrated that KLF1 indirectly downregulates fetal globin gene expression by activating BCL11A expression.
The pathogenic KLF1 mutation c.973G>A results in the substitution of the evolutionarily conserved glutamate 325 by a lysine (E325K) in the second zinc finger (ZF2) of KLF1 (Figure 2B). As with arginine 322 and arginine 328, glutamate 325 is predicted to contact DNA16 (Figure 2C). In order to investigate the possible structural effect of the E325K variant, we built a new structural model for the zinc-finger domain of KLF1 on the basis of the X-ray structures of Wilms' tumor protein in complex with DNA.21 The currently used models of KLF1 are based on the structure of Zif268 bound to DNA,22 but alignment of their respective zinc-finger domains requires a 2 amino acid gap in KLF1 ZF2, which significantly changes the local topology. According to the new structural model, glutamate 325 is located on a helix and is directed toward the DNA backbone (Figure 2D, upper left panel). The single amino acid variant E325K does not alter the overall topology of the KLF1 zinc-finger domain but mainly affects the side chain of residue 325 (Figure 2D, lower panels). Actually, this charge-reversal variant enhances the electrostatic interaction between KLF1 and the DNA and potentially creates a novel hydrogen bond (Figure 2D, upper right panel). Thus, the E325K variant is predicted to stabilize, rather than disrupt, the binding of KLF1 to its DNA target sequences.
Before testing the potential effect of the E325K variant on KLF1 transcriptional activity, we first wanted to check whether it affected the stability or cellular localization of the variant protein. For this purpose, we constructed vectors encoding either wild-type or E325K variant KLF1 tagged with a Flag epitope at the N terminus to allow its detection and then transfected them into human erythroid K-562 cells. Flag-tagged KLF1 E325K had the same expression level as the wild-type protein by immunoblot analysis (Figure 3A) and showed the same nuclear localization by immunofluorescence analysis (Figure 3B). The combined CD44 and AQP1 deficiencies observed in this unique CDA suggested that AQP1 and CD44 were direct targets of KLF1, which was consistent with the presence of several potential KLF1 binding sites (CCNCNCCCN)16 upstream of their respective initiating codons. Therefore, we studied the effect of the E235K variant on KLF1 transcriptional activity with CD44 and AQP1 promoter-reporter assays in K-562 cells. Flag-tagged KLF1 wild-type was able to activate CD44 and AQP1 promoters, whereas KLF1 E235K showed markedly reduced transcriptional activity (Figures 3C and 3D). Furthermore, when we coexpressed KLF1 E325K with KLF1 wild-type in order to mimic heterozygosity of patients ME and SF, we observed that KLF1 E325K was able to inhibit the activation of CD44 promoter induced by KLF1 wild-type (Figure 3D). On the basis of these data, we conclude that the E325K variant has a dominant-negative effect on the transcriptional activity of KLF1, in total agreement with the phenotype of heterozygous patients.
Figure 3.
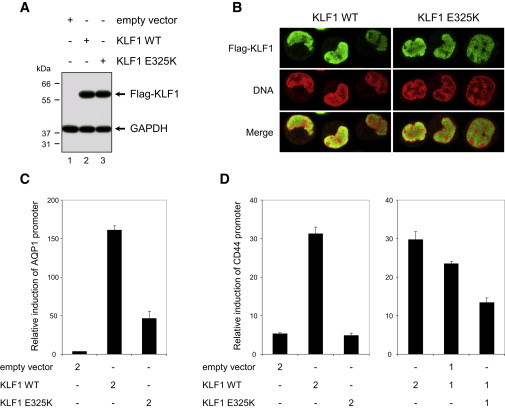
Characterization of the Effect of the E325K Variant on KLF1 Function
(A) Immunoblot analysis of transfected K-562 cells shows that the E325K variant does not affect the stability of KLF1. Constructs encoding Flag-tagged KLF1 wild-type (WT), variant E325K, or empty vector were transfected in K-562 cells, and protein expression was analyzed after 24 hr by immunoblot with anti-Flag and anti-GAPDH (loading control).
(B) Immunofluorescence analysis of transfected K-562 cells shows that the E325K variant does not affect the nuclear localization of KLF1. K-562 cells transfected as in (a) were analyzed after 24 hr by immunofluorescence with anti-Flag (green) along with propidium iodide for DNA staining (red). No anti-Flag staining was detected in K-562 cells transfected with the empty vector (data not shown).
(C) An AQP1 promoter-reporter assay in K-562 cells shows that the E325K variant affects the transcriptional activity of KLF1. K-562 cells were cotransfected with an AQP1 promoter-Photinus luciferase (Pluc) construct and a HSV-TK promoter-Renilla luciferase (Rluc) construct for normalization, along with 2 μg of the indicated KLF1 constructs, and the luciferase activities were analyzed after 24 hr; the results are shown as means ± SD (n = 3) of Pluc activity normalized by Rluc activity.
(D) A CD44 promoter-reporter assay in K-562 cells shows that the E325K variant has a dominant-negative effect on the transcription activity of KLF1. Not only does KLF1 E325K have a markedly reduced transcriptional activity, but it is also able to impede the transcriptional activity of coexpressed KLF1 wild-type. K-562 cells were cotransfected with a CD44 promoter-Pluc construct and a HSV-TK promoter-Rluc construct for normalization, along with 2 or 1 μg of the KLF1 constructs as indicated, and the luciferase activities were analyzed after 24 hr; the results are shown as means ± SD (n = 3) of Pluc activity normalized by Rluc activity.
This study describes a missense KLF1 mutation responsible for a human pathology, a hitherto unclassified CDA. Interestingly, while this manuscript was being finalized, a new CDA patient with KLF1 mutation c.973G>A was identified (female patient SE; A.I., unpublished data), suggesting that this type of CDA might be less rare than expected. The previously reported KLF1 mutations, found in the heterozygous state too, are responsible for the blood-group phenotype In(Lu)23 (MIM 111150), which is mainly characterized by a reduced expression of Lu (also known as BCAM) on erythrocytes but which is not associated with any pathology. Of note, patient SF does not show a reduced expression of Lu (Figure S9), indicating that the KLF1 mutation c.973G>A is not directly responsible for the reduced Lu expression observed in patient ME (Figure S3). We assume that distinct KLF1 mutations might differently affect the gene repertoire of this transcription factor and thus lead to different phenotypes, as observed with GATA1 mutations.11,12 Fortuitously, while this manuscript was being finalized, Siatecka et al.24 reported that the mouse mutation Nan, a dominant ethylnitrosourea-induced mutation causing hemolytic anemia, corresponds to a missense Klf1 mutation encoding the E339D variant and affecting the expression of only a subset of Klf1 target genes, and they further showed that the E339D variant indeed alters Klf1 DNA binding to only a subset of its target sequences. Of note, glutamate 339 in mouse Klf1 is the equivalent of glutamate 325 in human KLF1; however, Klf1 variant E339D is intrinsically different from KLF1 variant E325K because of the opposite charge of the variant residue (see structure prediction above), which is consistent with the distinct resulting pathologies in mice and humans. Extensive studies with mouse models suggested that KLF1 played a global role during erythropoiesis by regulating a wide spectrum of genes19,25,26 but also organizing nuclear hubs for efficient and coordinated transcription of genes coregulated with the β-globin gene.27,28 This study provides in vivo evidence that human KLF1 plays a critical role in the regulation of fetal globin genes, as recently demonstrated by Borg et al.20 (see above), but also of other unexpected genes such as AQP1 and CD44. Although AQP1 deficiency in humans doesn't cause dyserythropoiesis,8 we cannot exclude that CD44 deficiency contributes, even though CD44 is dispensable for mouse erythropoiesis.29 Future studies will need to identify the KLF1 target genes whose expression is essential for human erythropoiesis but that are deficient in this form of CDA. Finally, we propose that KLF1 should be systematically sequenced as a novel candidate gene in all CDA cases with unknown genetic cause; such sequencing might eventually lead to the discovery of other pathogenic KLF1 mutations.
Acknowledgments
First, we would like to thank the patients and patient ME's relatives for providing blood samples for this study. We would like also to thank D. Sommelet, J. Buisine, L. Douay, M. Goossens, C. Etchebest, Y. Colin, T. Zelinski, P. Gane, T. Cynober, M. Feneant-Thibault, C. Schmitt, R. Russo, M.R. Esposito, C. Menanteau, S. Kappler-Gratias, B.-N. Pham, P.-Y. Le Pennec, E. Vera, C. Verheyde, G. Nicolas, B.A. Ballif, and M. Le Gall for their contributions to this study. L.A. and J.-P.C. were supported by the National Institute of Blood Transfusion (INTS), the National Institute for Health and Medical Research (INSERM), and Paris Diderot University (Paris 7). G.C. was supported by an operating grant from the Winnipeg Rh Institute Foundation to T. Zelinski. H.T. was supported by an Israel Science Foundation grant (No. 699/_03-18.4), by an Israeli Ministry of Science, Culture, and Sport grant in the framework of the Israel-France Program, and by an Israeli Ministry of Science-Eshkol Fellowship. A.I. was supported by the Italian Ministry of University and Research (grant MUR-PS 35-126/Ind), the Italian Telethon Foundation (project GGP09044), and Regione Campania (DGRC 1901/2009).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Entrez Gene browser, http://www.ncbi.nlm.nih.gov/gene/
Entrez Single Nucleotide Polymorphism (SNP) browser, http://www.ncbi.nlm.nih.gov/snp/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/omim/
Structure modeling of KLF1, http://www.dsimb.inserm.fr/∼debrevern/KLF1
References
- 1.Renella R., Wood W.G. The congenital dyserythropoietic anemias. Hematol. Oncol. Clin. North Am. 2009;23:283–306. doi: 10.1016/j.hoc.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 2.Heimpel H., Wendt F., Klemm D., Schubothe H., Heilmeyer L. Congenital dyserythropoietic anemia. Arch. Klin. Med. 1968;215:174–194. [PubMed] [Google Scholar]
- 3.Heimpel H., Kellermann K., Neuschwander N., Högel J., Schwarz K. The morphological diagnosis of congenital dyserythropoietic anemia: Results of a quantitative analysis of peripheral blood and bone marrow cells. Haematologica. 2010;95:1034–1036. doi: 10.3324/haematol.2009.014563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dgany O., Avidan N., Delaunay J., Krasnov T., Shalmon L., Shalev H., Eidelitz-Markus T., Kapelushnik J., Cattan D., Pariente A. Congenital dyserythropoietic anemia type I is caused by mutations in codanin-1. Am. J. Hum. Genet. 2002;71:1467–1474. doi: 10.1086/344781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwarz K., Iolascon A., Verissimo F., Trede N.S., Horsley W., Chen W., Paw B.H., Hopfner K.P., Holzmann K., Russo R. Mutations affecting the secretory COPII coat component SEC23B cause congenital dyserythropoietic anemia type II. Nat. Genet. 2009;41:936–940. doi: 10.1038/ng.405. [DOI] [PubMed] [Google Scholar]
- 6.Bianchi P., Fermo E., Vercellati C., Boschetti C., Barcellini W., Iurlo A., Marcello A.P., Righetti P.G., Zanella A. Congenital dyserythropoietic anemia type II (CDAII) is caused by mutations in the SEC23B gene. Hum. Mutat. 2009;30:1292–1298. doi: 10.1002/humu.21077. [DOI] [PubMed] [Google Scholar]
- 7.Giarratana M.C., Kobari L., Lapillonne H., Chalmers D., Kiger L., Cynober T., Marden M.C., Wajcman H., Douay L. Ex vivo generation of fully mature human red blood cells from hematopoietic stem cells. Nat. Biotechnol. 2005;23:69–74. doi: 10.1038/nbt1047. [DOI] [PubMed] [Google Scholar]
- 8.Preston G.M., Smith B.L., Zeidel M.L., Moulds J.J., Agre P. Mutations in aquaporin-1 in phenotypically normal humans without functional CHIP water channels. Science. 1994;265:1585–1587. doi: 10.1126/science.7521540. [DOI] [PubMed] [Google Scholar]
- 9.Kim S.I., Bresnick E.H. Transcriptional control of erythropoiesis: Emerging mechanisms and principles. Oncogene. 2007;26:6777–6794. doi: 10.1038/sj.onc.1210761. [DOI] [PubMed] [Google Scholar]
- 10.Sankaran V.G., Xu J., Orkin S.H. Advances in the understanding of haemoglobin switching. Br. J. Haematol. 2010;149:181–194. doi: 10.1111/j.1365-2141.2010.08105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hollanda L.M., Lima C.S., Cunha A.F., Albuquerque D.M., Vassallo J., Ozelo M.C., Joazeiro P.P., Saad S.T., Costa F.F. An inherited mutation leading to production of only the short isoform of GATA-1 is associated with impaired erythropoiesis. Nat. Genet. 2006;38:807–812. doi: 10.1038/ng1825. [DOI] [PubMed] [Google Scholar]
- 12.Nichols K.E., Crispino J.D., Poncz M., White J.G., Orkin S.H., Maris J.M., Weiss M.J. Familial dyserythropoietic anaemia and thrombocytopenia due to an inherited mutation in GATA1. Nat. Genet. 2000;24:266–270. doi: 10.1038/73480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parsons S.F., Jones J., Anstee D.J., Judson P.A., Gardner B., Wiener E., Poole J., Illum N., Wickramasinghe S.N. A novel form of congenital dyserythropoietic anemia associated with deficiency of erythroid CD44 and a unique blood group phenotype [In(a-b-), Co(a-b-)] Blood. 1994;83:860–868. [PubMed] [Google Scholar]
- 14.Tang W., Cai S.P., Eng B., Poon M.C., Waye J.S., Illum N., Chui D.H. Expression of embryonic zeta-globin and epsilon-globin chains in a 10-year-old girl with congenital anemia. Blood. 1993;81:1636–1640. [PubMed] [Google Scholar]
- 15.Wickramasinghe S.N., Illum N., Wimberley P.D. Congenital dyserythropoietic anaemia with novel intra-erythroblastic and intra-erythrocytic inclusions. Br. J. Haematol. 1991;79:322–330. doi: 10.1111/j.1365-2141.1991.tb04541.x. [DOI] [PubMed] [Google Scholar]
- 16.Feng W.C., Southwood C.M., Bieker J.J. Analyses of beta-thalassemia mutant DNA interactions with erythroid Krüppel-like factor (EKLF), an erythroid cell-specific transcription factor. J. Biol. Chem. 1994;269:1493–1500. [PubMed] [Google Scholar]
- 17.Nuez B., Michalovich D., Bygrave A., Ploemacher R., Grosveld F. Defective haematopoiesis in fetal liver resulting from inactivation of the EKLF gene. Nature. 1995;375:316–318. doi: 10.1038/375316a0. [DOI] [PubMed] [Google Scholar]
- 18.Perkins A.C., Sharpe A.H., Orkin S.H. Lethal beta-thalassaemia in mice lacking the erythroid CACCC-transcription factor EKLF. Nature. 1995;375:318–322. doi: 10.1038/375318a0. [DOI] [PubMed] [Google Scholar]
- 19.Drissen R., von Lindern M., Kolbus A., Driegen S., Steinlein P., Beug H., Grosveld F., Philipsen S. The erythroid phenotype of EKLF-null mice: defects in hemoglobin metabolism and membrane stability. Mol. Cell. Biol. 2005;25:5205–5214. doi: 10.1128/MCB.25.12.5205-5214.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Borg J., Papadopoulos P., Georgitsi M., Gutiérrez L., Grech G., Fanis P., Phylactides M., Verkerk A.J., van der Spek P.J., Scerri C.A. Haploinsufficiency for the erythroid transcription factor KLF1 causes hereditary persistence of fetal hemoglobin. Nat. Genet. 2010;42:801–805. doi: 10.1038/ng.630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stoll R., Lee B.M., Debler E.W., Laity J.H., Wilson I.A., Dyson H.J., Wright P.E. Structure of the Wilms tumor suppressor protein zinc finger domain bound to DNA. J. Mol. Biol. 2007;372:1227–1245. doi: 10.1016/j.jmb.2007.07.017. [DOI] [PubMed] [Google Scholar]
- 22.Elrod-Erickson M., Benson T.E., Pabo C.O. High-resolution structures of variant Zif268-DNA complexes: Implications for understanding zinc finger-DNA recognition. Structure. 1998;6:451–464. doi: 10.1016/s0969-2126(98)00047-1. [DOI] [PubMed] [Google Scholar]
- 23.Singleton B.K., Burton N.M., Green C., Brady R.L., Anstee D.J. Mutations in EKLF/KLF1 form the molecular basis of the rare blood group In(Lu) phenotype. Blood. 2008;112:2081–2088. doi: 10.1182/blood-2008-03-145672. [DOI] [PubMed] [Google Scholar]
- 24.Siatecka M., Sahr K.E., Andersen S.G., Mezei M., Bieker J.J., Peters L.L. Severe anemia in the Nan mutant mouse caused by sequence-selective disruption of erythroid Kruppel-like factor. Proc. Natl. Acad. Sci. USA. 2010;107:15151–15156. doi: 10.1073/pnas.1004996107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hodge D., Coghill E., Keys J., Maguire T., Hartmann B., McDowall A., Weiss M., Grimmond S., Perkins A. A global role for EKLF in definitive and primitive erythropoiesis. Blood. 2006;107:3359–3370. doi: 10.1182/blood-2005-07-2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tallack M.R., Whitington T., Yuen W.S., Wainwright E.N., Keys J.R., Gardiner B.B., Nourbakhsh E., Cloonan N., Grimmond S.M., Bailey T.L., Perkins A.C. A global role for KLF1 in erythropoiesis revealed by ChIP-seq in primary erythroid cells. Genome Res. 2010;20:1052–1063. doi: 10.1101/gr.106575.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patrinos G.P., de Krom M., de Boer E., Langeveld A., Imam A.M., Strouboulis J., de Laat W., Grosveld F.G. Multiple interactions between regulatory regions are required to stabilize an active chromatin hub. Genes Dev. 2004;18:1495–1509. doi: 10.1101/gad.289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schoenfelder S., Sexton T., Chakalova L., Cope N.F., Horton A., Andrews S., Kurukuti S., Mitchell J.A., Umlauf D., Dimitrova D.S. Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nat. Genet. 2010;42:53–61. doi: 10.1038/ng.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schmits R., Filmus J., Gerwin N., Senaldi G., Kiefer F., Kundig T., Wakeham A., Shahinian A., Catzavelos C., Rak J. CD44 regulates hematopoietic progenitor distribution, granuloma formation, and tumorigenicity. Blood. 1997;90:2217–2233. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.