

Abstract
Next Generation Sequencing Technology has revolutionized our ability to study the contribution of rare genetic variation to heritable traits. However, existing single-marker association tests are underpowered for detecting rare risk variants. A more powerful approach involves pooling methods that combine multiple rare variants from the same gene into a single test statistic. Proposed pooling methods can be limited because they generally assume high-quality genotypes derived from deep-coverage sequencing, which may not be available. In this paper, we consider an intuitive and computationally efficient pooling statistic, the cumulative minor-allele test (CMAT). We assess the performance of the CMAT and other pooling methods on datasets simulated with population genetic models to contain realistic levels of neutral variation. We consider study designs ranging from exon-only to whole-gene analyses that contain noncoding variants. For all study designs, the CMAT achieves power comparable to that of previously proposed methods. We then extend the CMAT to probabilistic genotypes and describe application to low-coverage sequencing and imputation data. We show that augmenting sequence data with imputed samples is a practical method for increasing the power of rare-variant studies. We also provide a method of controlling for confounding variables such as population stratification. Finally, we demonstrate that our method makes it possible to use external imputation templates to analyze rare variants imputed into existing GWAS datasets. As proof of principle, we performed a CMAT analysis of more than 8 million SNPs that we imputed into the GAIN psoriasis dataset by using haplotypes from the 1000 Genomes Project.
Introduction
The Genome-Wide Association Study (GWAS) is a powerful tool for analyzing common variation across the human genome.1 In recent years, GWASs have identified risk alleles for a wide range of complex human diseases.2 However, most of these alleles provide only small to moderate increases in risk and contribute little to the overall heritability of the disease.3 Because it is unlikely that the remaining heritability can be completely explained by undetected common variants with even lower effects,4 heritable factors besides common variation must contribute to complex diseases. The Common Disease-Rare Variant Hypothesis proposes that some of the missing heritability can be explained by low frequency variants with larger effect sizes.5,6 Under this model, the contribution of individual variants to population prevalence is small, but the combined effect of numerous rare variants can account for an appreciable fraction of the prevalence. This model is feasible if risk variants are subject to weak purifying selection and is supported by the fact that allele frequencies for protein-altering mutations are more heavily skewed toward rare variants than those for neutral variants.7
Previously, technological limitations hampered the ability to affordably assay and test rare variants in large population-based samples. However, recent advances in next-generation sequencing technology now provide the potential to detect all polymorphisms in a genomic region.8 Thus, it is possible to test rare variants directly rather than rely on indirect linkage disequilibrium (LD)-based methods. Already, candidate-region resequencing has led to the discovery of numerous rare variants contributing to phenotypic variation and complex disease in humans. Resequencing of coding regions and consensus splice sites in NPC1L1 and PCSK9 has led to the identification of multiple rare nonsynonymous mutations collectively associated with variation in sterol absorption and plasma levels of LDL-C.9,10
Individually testing each variant identified by resequencing is not a powerful strategy because it requires stringent multiple testing correction and power diminishes with decreasing allele frequencies.11 To avoid these issues, several groups have proposed various statistical methods that instead pool together multiple rare variants from the same gene and jointly test them for association.9,12–14 The recent literature has addressed two related questions in rare-variant testing: first, the question of how to effectively combine multiple rare variants in a gene into a single test and, second, how to weight variants on the basis of some prior assumption about the likelihood of functionality. Cohen et al. performed a pooled analysis of rare variants in NPC1L1 and identified nonsynonymous variants observed only in cases or only in controls and used Fisher's exact test to compare the distributions of cases and controls carrying these variants.9 Li and Leal proposed the Combined Multivariate and Collapsing method that pools variants below a specified minor-allele frequency (maf) and then dichotomizes individuals on the basis of whether they carry a variant allele at one of the pooled sites.12 A multivariate statistic is used for jointly analyzing the set of pooled variants together with more common variants in the region.
Madsen and Browning introduced two features in the weighted sum statistic (WSS).13 First, the WSS accumulates rare-variant counts within the same gene for each individual rather than collapsing on them. Second, it introduces a weighting term to emphasize alleles with a low maf in controls. The result is that each individual receives a quantitative genetic score that is more informative than a qualitative score, especially for individuals harboring more than one rare allele in the region. The scores for all samples are ordered, and the WSS is computed as the sum of ranks for cases. One determines significance by permuting affection status and re-ranking. The ranking protects against outliers but becomes computationally expensive for large sample sizes.
Price et al.14 showed that the power gain of weights based on minor allele frequency is dependent on the relationship between risk-allele frequency and likely effect size; this relationship is in turn is dependent on selection strength. The weights used by Madsen and Browning, for example, correspond to strong purifying selection. If this model is correct, the WSS provides a significant power gain over the previous methods. To generate a test that is powerful under multiple evolutionary models, Price et al. proposed a variable maf-threshold approach. For a given frequency threshold, one computes a likelihood ratio statistic to compare summed minor-allele counts for variants below the maf threshold for cases and controls. The likelihood ratio statistic is maximized across a range of frequency thresholds so that the statistic is adapted to the underlying model of selection.
All pooling statistics are subject to variant misspecification—that is, potential inclusion of neutral variants or exclusion of risk variants. Study designs to date have opted to minimize inclusion of neutral variants by limiting analysis to nonsynonymous coding variants of candidate genes.11 The power of this strategy depends on the cumulative effect of rare risk variants that are exonic. Although coding variants are most likely to be functional, they account for only a tiny fraction of variation in the genome. Numerous pieces of evidence indicate that noncoding variants play an extensive role in disease etiology. Eighty-eight percent of trait-associated variants identified by GWAS have occurred outside of known coding regions.2 Large portions of noncoding regions in the human genome are subject to negative selection, indicating a functional purpose to the sequence.15 In addition, noncoding risk variants have already been verified for numerous diseases.16–18 Resequencing noncoding intronic and regulatory regions could enable detection of these more elusive risk variants but also presents new technical and analytical challenges to rare-variant analysis. In particular, noncoding sequence contains substantially more neutral variation than coding regions.
Existing pooling methods have not been carefully assessed under a paradigm where many risk variants reside outside exons. Instead, these methods have only been considered for fairly optimal testing conditions in which each gene is assumed to have few variants, most of which are causative.12,13 Moreover, previously published pooling methods assume high-quality rare-variant genotypes that are only available through deep-coverage sequencing. Exon-only studies can attain high-quality genotype calls because sequencing is limited to relatively small regions. Generating high-quality sequence data of larger genomic regions (including whole-genome sequencing) is still expensive, which limits the number of samples that can be sequenced at deep coverage for a given study. Instead, cost-effective strategies such as low-coverage sequencing19 and genotype imputation20 will be used to produce sample sizes large enough to powerfully analyze rare variants. Genotype calls from these methods are less precise than deep sequencing, generating probabilistic rather than exact genotypes. Thus, tests applied to whole-gene sequence data containing both coding and noncoding regions must accept probabilistic genotypes and be robust to potentially high inclusion rates for neutral variants.
In this article, we consider a simple pooling statistic, the cumulative minor-allele test (CMAT) and show that it is easily extended to accommodate practical analysis considerations such as qualitative covariates and probabilistic genotypes. The CMAT is closely related to the tests described in Madsen and Browning13 and in Price et al.14 in that it aggregates allele counts rather than collapsing on them. Like these methods, the CMAT jointly analyzes sets of variants that occur in the same gene and that would otherwise be missed by a standard single-marker analysis. Because the power of single-marker tests is dependent on study sample size and risk-allele frequency, the CMAT is computed on variants with a maf below a preset threshold. In this paper we especially focus on markers with a maf <5% and hereafter refer to these as rare variants.
One computes the CMAT statistic by summing rare-allele counts for sites predicted to be functionally relevant separately for cases and controls. Our test statistic is analogous to the single marker allelic χ2 statistic typically used to test for allele frequency difference between cases and controls. Significance is determined by permutation to account for correlation between pooled variants.
We compare the power of several pooling methods on case-control sequencing datasets simulated with population genetic models designed to mimic the overall level of diversity seen in European HapMap samples. We create a disease model of allelic heterogeneity by placing multiple rare risk variants in the population. The effect size for each risk variant is determined by allele frequency to ensure low power for a single marker test. Because our datasets contain realistic levels of neutral variation, we can consider the effect of variant misspecification, both inclusion of neutral variants and exclusion of causal variants, in study designs ranging from exon-only to whole-gene analysis. We show that, depending on the proportion of noncoding risk variants, whole-gene designs can be more powerful than exon-only designs even if they include a large number of neutral variants.
The form of the CMAT statistic conveniently allows for categorical covariates and probabilistic genotypes. These extensions allow rare-variant analysis for datasets containing imputed genotypes or low-coverage sequence data as well as common confounding variables such as population stratification. We demonstrate the importance of these extensions by analyzing two previously unconsidered rare-variant study designs. First, we simulate rare-variant datasets containing spurious associations created by population stratification. Ignoring the stratification leads to an elevated Type I Error rate, and controlling for it with the covariate form of the CMAT maintains the desired α-level.
Second, we present a study design consisting of both sequenced and imputed samples. We assume that the sequenced samples are used for identification of novel rare variants in a region of interest and that they serve as templates for imputation of genotypes for these variants into the remaining (non-sequenced) samples. While carefully accounting for the uncertainty involved in imputing rare variants, we simulate datasets for this study design and analyze them with the CMAT. We show that using imputation to increase the sample size of a sequencing dataset can substantially improve power. Hence we predict that imputation will provide a powerful cost-saving strategy for future resequencing studies. Moreover, our results suggest that one could use existing resources such as the 1000 Genomes Project to reanalyze existing GWAS datasets by imputing rare variants and performing tests such as the CMAT.
Finally, we illustrate the possibility of reanalyzing GWAS datasets without resequencing samples. As a proof of principle, we imputed more than 8 million SNPs into the GAIN psoriasis GWAS dataset by using CEU haplotypes from the 1000 Genomes Project. This dataset had previously been augmented with genotypes imputed from HapMap haplotypes and analyzed with a single-marker association test.21 That analysis identified numerous common risk loci that were subsequently replicated; these included several variants in the HLA region on chromosome 6. We reanalyzed 3000 genes with at least two rare variants (maf ≤ 5%) by using the CMAT. One gene, SKIV2L, located on chromosome 6 near the HLA region, maintained a significant test statistic after we corrected for multiple testing.
Methods
Below, we develop notation for exact and probabilistic genotype calls, then introduce the CMAT along with three alternative rare-variant tests. Subsequently, we describe our algorithm for simulating case-control sequencing data on the basis of population genetic models. Finally, we provide details for our application of the CMAT to the GAIN Psoriasis dataset.
Data Structure
We assume a dataset of NA cases and NU controls. Let be the true number of minor alleles at the jth variant site in the ith case. Let yij be the same value for the ith control. We consider two possible types of genotype calls in the data: exact calls, discrete values from giving the observed minor-allele count, and probabilistic calls, consisting of a posterior probability mass function giving the likelihood for each possible minor-allele count. Exact genotypes reflect the high-confidence calls possible in deep-coverage sequencing data, whereas the probabilistic calls represent the uncertainty in low-coverage sequencing and imputation. In the dataset, we define the observed value for the jth variant site in the ith case to be
That is, we assume the true minor-allele count is observed if an exact call is made; otherwise, we observe the minor-allele count that is expected on the basis of the posterior probability distribution. Similarly, we define Yij for the jth variant site in the ith control and replace xij with yij.
Cumulative Minor-Allele Test
We assume the genetic data are partitioned into a collection of discrete testing units, genomic regions to be individually tested for association with disease susceptibility. The most natural choice for a testing unit is a single gene, but highly conserved nongenic regions or pathways containing multiple genes are also suitable. Assume variants in the testing unit, each with a weighting factor , . It is possible to filter a variant out of the analysis by setting the respective weight to zero or emphasize its presence by assigning a large weight. For this paper, wj is a simple indicator function that identifies variants included in the analysis (it is described in more detail later). Note that a testing unit containing only a single variant with positive weight is equivalent to a single-marker test on that variant.
We first describe application of the CMAT to a dataset containing exact genotype calls for all NA cases and all NU controls. Let and be the weighted minor-allele counts across all sites in the testing unit for cases and controls, respectively. Then and are therefore the weighted major-allele counts across all sites for cases and controls, respectively. We define the CMAT statistic to be
| (1) |
The statistic is derived from the standard Pearson χ2 statistic for testing independence between allele frequency and disease status in a single-marker association test. However, does not have an asymptotic χ2 distribution because independent counts are required for the asymptotic properties to be valid. Because we sum over multiple sites in a testing unit, and because some of these sites might be in LD with each other, the counts are not independent. Instead, we determine the statistical significance of by permuting affection status while holding the genetic data fixed. For each permuted realization, is recomputed, and the p value is defined as the proportion of permutations with a test statistic greater than or equal to the observed statistic.
In the presence of qualitative covariate data on potential confounders, the weighted allele counts are computed separately within each covariate level, and the form of is changed to a Cochran-Mantel-Haenszel-like statistic. Assume a qualitative covariate . Using similar notation, we define the observed value for the jth variant site in the ith case of the cth covariate class to be
Similarly, we define Yijc for the jth variant site in the ith control of the cth covariate class and replace xijc with yijc. Assume cases and controls within the cth covariate class and . Weighted allele counts are then computed within each covariate class separately. Let and be the weighted minor-allele counts across all sites in the testing unit for cases and controls, respectively, in the cth covariate class. Then and are the weighted major-allele counts across sites for cases and controls, respectively, of the cth covariate class. We define the covCMAT statistic to be
| (2) |
Statistical significance is determined by permuting case-control status while keeping the genetic and covariate data fixed. Equation (2) resembles the Cochran-Mantel-Haenszel χ2 statistic and simplifies to Equation (1) when .
We now consider a dataset containing cases and controls with exact genotype calls and cases and controls with probabilistic calls. Computation of (Equation 1) remains the same except expected minor allele counts replace exact counts for imputed samples. One again determines significance by permuting affection status. However, to account for the difference in quality between the two data types, one must shuffle affection status separately for exact and probabilistic calls. That is, for all permutations, the number of cases and controls with exact genotype counts must remain constant. Failure to modify the permutation method in this manner can affect type I error, especially for unbalanced designs ().
Alternative Rare-Variant Methods
We compared the performance of the CMAT to three alternative rare-variant methods. First, we implemented Li and Leal's collapsing method,12 which compares number of rare-variant carriers in cases to the number in controls. Let the indicator variable Xi denote whether the ith case carries at least one rare variant at a site of interest, as follows
Yi is analogously defined to indicate controls carrying at least one rare variant. Then and are, respectively, the number of cases and controls carrying at least one rare variant. The Pearson χ2 statistic,
tests the null hypothesis that cases and controls are equally likely to be carriers of a rare variant. has an asymptotic χ2 distribution with one degree of freedom.
Next, we considered a private-allele test similar to the method used by Cohen et al,9 to compare the number of rare variants unique to either cases or controls. For this test we required an equal number of cases and controls (). A site is defined to be private if it is polymorphic in either cases or controls but monomorphic in the other group. The minor allele at a private site is called a private allele. For example, the minor allele at the jth site is private to cases if but . Under the null hypothesis, rare variants are not associated with disease risk, and private alleles are therefore equally likely to occur in cases and controls. This is tested formally with a χ2 test in the following manner: Let npriv be the total number of private alleles in the dataset and nA and nU the number of private alleles unique to cases and controls, respectively (). Define
Under the null distribution of no association, is asymptotically χ2 distributed with one degree of freedom. As with the CMAT and collapsing test, the private-allele test considers only variants with positive weighting terms.
Finally, we implemented the WSS as described by Madsen and Browning.13 For the ith individual in the dataset, one computes a genetic score defined as . The genetic scores for all samples in the dataset (cases and controls combined) are sorted, and the sum of ranks of genetic scores for cases, , is computed. Statistical significance of x is determined by permutation. Madsen and Browning recommend increasing the weight of rare variants by defining weighting terms according to maf in controls. We do not directly consider the question of how to weight rare variants in this paper. Therefore, we applied a simple uniform weighting scheme to all tests. However, for comparative purposes, we include application of the WSS and CMAT in which the weights defined in Madsen and Browning are used (Figure S2). The three alternative methods have been formally defined only for exact genotypes; thus, we limit power comparisons to datasets containing only exact genotype calls.
Simulations
Deep-Sequence Datasets
We simulated deep-sequence datasets containing exact genotype calls for an equal number N of cases and controls. We first created a population of ten thousand 100 kb haplotypes by using the coalescent simulator cosi with parameters chosen to reflect characteristics seen in the European HapMap samples.22 Let ntot be the total number of polymorphic sites among the ten thousand population haplotypes. Denote the allele at the jth site on the ith haplotype as Aij, where if the major allele is present and if the minor allele is present and . We fixed a maximum allele frequency pmax for risk alleles and randomly chose k sites with maf to be causative. Let if the jth variant site is selected to be causative and if it is neutral.
For each risk variant, we assigned an effect size that ensured that a single-marker association test would have a low probability of being statistically significant. Specifically, we computed the relative risk γp necessary for a risk variant with maf to have power in a 1 degree of freedom χ2 test of 1000 cases and 1000 controls performed at (Figure 1). As a result, rarer variants are assigned larger relative risks, although we capped relative risks for variants with maf at six. Assuming the maf for the jth variant is p, we set the relative risk at that site to be .
Figure 1.
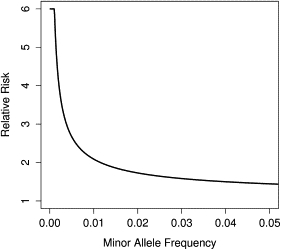
Relationship between Minor-Allele Frequency and Relative Risk in Our Disease Model
The relative risk is chosen such that a single marker test of 1000 cases and 1000 controls performed at on a risk variant with the specified maf has a maximum power of 10%. Relative risks for variants with a maf were truncated to 6.
Assuming a multiplicative effect between causative variants, the penetrance for haplotype i is
where b is the risk for an individual with wild-type (non-risk) alleles at all causative sites and is set to ensure that the population prevalence remains fixed at a desired level.
We then sampled diploid cases and controls by using Bayes’ Theorem to randomly drawing two haplotypes conditional on disease status. For example, if we assume unconditionally that each of the ten thousand population haplotypes is equally likely to be selected, the probability that the ith and jth haplotypes will be chosen for a case is
We treat the unconditional probability of being a case (population prevalence) as a fixed parameter in our simulations.
After the construction of a dataset, we mimicked a bioinformatic annotation process to determine the set of variants predicted to be functional and therefore included in the analysis. Each observed variant was randomly labeled as either “included” or “excluded” from the analysis conditional on whether it was causative or neutral. Define pc to be the probability that a causative variant is predicted to be functional and therefore included in the analysis. Likewise, pn is the same probability for neutral variants. Then, if we let Ij be an indicator for inclusion in the analysis, the jth variant is included with probability
We treated the values pc and pn as parameters to simulate study designs with alternative inclusion thresholds. Using the functional annotations, we defined the weighting terms used in our simulations
| (3) |
This weighting scheme therefore acted as a filter to retain variants that had a maf and were predicted to be functional in the annotation step.
Imputation Datasets
Next, we created datasets containing exact genotypes for Nseq cases and controls that were assumed to have been sequenced at deep coverage. We also created datasets containing probabilistic genotypes for an additional imputed cases and controls. Thus, in contrast to our deep-sequence simulations, where we assumed deep-sequence data for all samples, here we assumed deep-sequence data for only a fraction of the total sample size. It was computationally infeasible to phase and impute genotypes for each simulated dataset; therefore, we drew haplotypes for N cases and controls by using the previously described method and replaced the true minor-allele counts with expected minor-allele counts for the imputed portion of the sample. Expected minor-allele counts were drawn from empirical sampling distributions created via independent imputation runs (see Appendix A). Individual draws were made conditional on the true minor-allele count at the locus to be imputed and the number of times the minor allele at that site was observed in the sequenced samples. We created separate empirical distributions for Nseq = 100, 200, and 400. Only sites polymorphic among the sequenced samples were eligible for inclusion in the analysis. Singletons in the sequenced samples cannot be accurately phased and were therefore not imputed. Hence, the minor allele must be observed at least twice in the sequenced samples to be imputed.
Stratified Datasets
To demonstrate the covariate form of the CMAT statistic, we simulated datasets containing population stratification. To do so, we used cosi to simulate sets of haplotypes that reflect variation observed in European and African populations.22 We drew datasets containing N cases and N controls under the null hypothesis of no risk variants (); however, we preferentially chose haplotypes from the African population to be cases. For each sample in a dataset, we first chose a population of origin for the sample. We let p be the probability that a control is derived from the African population and , , to be the probability that a case is derived from the African population. Controls and cases are therefore drawn from the European population with probability and , respectively. Once population of origin was determined, we randomly selected two haplotypes from the appropriate population to create a diploid sample. We analyzed each simulated dataset with both the CMAT and the covCMAT and controlled for population of origin in the latter. When applying the covCMAT, we assumed that the true population of origin was known for each sample.
Simulation Settings
We fixed the population disease prevalence at 1% throughout the simulations. Under our disease model, increasing the number of causative sites k while holding prevalence constant increases the proportion of disease prevalence explained by variation at the locus. We focused our analysis on risk alleles with a maf by setting parameters pmax and β to 0.05. However we repeated our analysis while restricting risk variants to a maf and report those results as well. Because causative sites were chosen at random and the allele frequency spectrum was heavily shifted toward extremely low frequencies, approximately of risk alleles in our simulations have frequency even for simulations with . We estimated power for each test at different parameter settings as the proportion of simulated datasets with statistically significant p values (based on a minimum of at least 1000 simulated datasets). We report power at a critical level of , for which we assume that the sequenced region contains several genes to be tested.
GWAS Application
We imputed 8.2 million autosomal SNPs into the GAIN Psoriasis dataset by using 112 CEU haplotypes from the August 2009 release of the 1000 Genomes Project as a reference. We filtered the imputed SNPs by removing all variants with very low estimated imputation accuracy ( < 0.3). We annotated SNPs discovered in the 1000 Genomes Project Pilot by using a custom Perl script. The tool reports for each SNP the gene locus (if available) and the predicted protein effect, based on a set of curated transcripts from Refseq and GenBank. We included in our analysis SNPs annotated as missense, nonsense, or splice-site mutation or an untranslated region (UTR). We filtered out variants with a maf and pooled the remaining variants together by genes. That is, we used the following weighting strategy:
Results
Deep-Coverage Sequencing Datasets
To evaluate the performance of the CMAT, we used coalescent simulations to generate realistic case-control sequence data for a 100 kb region of interest, representing the exons, introns, and surrounding regulatory regions for a large gene. A dataset of cases and controls drawn from a population with rare (maf ) causative sites contained, on average, segregating variable sites with a mean pairwise sequence difference . Of the observed sites, 1272 had a maf , and we observed 12.4 of the 15 risk alleles. A larger dataset with cases and controls contained an average of 1556 polymorphic sites with frequency and 14.1 of the 15 risk alleles. Larger sample sizes therefore increase both the number of risk alleles observed in the sample and the number of neutral variants.
We mimicked functional filtering by analyzing only a subset of the variants observed in a dataset. If they were observed, causative variants were “predicted” to be functional and therefore were included in the analysis with probability pc ; neutral variants were included with probability pn. Because few of the observed variants are actually causative, pn is approximately the overall proportion of rare variants included in the analysis, and pc can be thought of as the success rate for including causal variants.
We determined practical values for pc and pn by investigating the distribution of functional annotations for genic SNPs in the dbSNP database.23 Of genic SNPs with at least one annotation, approximately 1.6% were denoted as nonsynonymous coding or splicing variants (nonsense, missense, frameshift, or altered splice-site mutations), 1% were synonymous coding variants, 2.7% occurred in the UTR, and 5.3% occurred outside the transcribed region of the gene. Intronic SNPs accounted for the remaining class of variants. Thus, an overall inclusion rate of 1%–2% roughly corresponds to analyzing only nonsynonymous variants, whereas extending the analysis to include variants in the UTR and outside the transcribed region has an inclusion rate of approximately 10%.
We computed power for the rare-variant methods on a misspecification grid with values of pn between 0 and 0.1 and pc between 0.2 and 1.0. First we computed the type I error for each test by setting . The CMAT, collapsing method, and WSS each maintained the desired false-positive rate for all values of pn. Type I error for the private allele test was initially conservative for smaller values of pn, and then increased with the number of variants included until it became anti-conservative for larger values of pn (Figure S1). Increased false positives for the private-allele test were probably due to the inclusion of variants in high pairwise LD in the calculation, in violation of the independence assumption required for the asymptotic distribution.
In the presence of causative variants (), the power to identify a gene depended on the inclusion parameters pc and pn (Figure 2). We discuss results generated with a sample size of and causative variants; results for and were similar (not shown). When all variants were correctly specified (), the CMAT, WSS, and collapsing test attained power near 100%, and the private test attained a power of 72%, indicating that each test is quite powerful under perfect filtering. However, power for each test dropped when we allowed for misspecification. Increasing the probability of including neutral variants () reduced power. Decreasing the probability of including causative variants () also lowered power.
Figure 2.
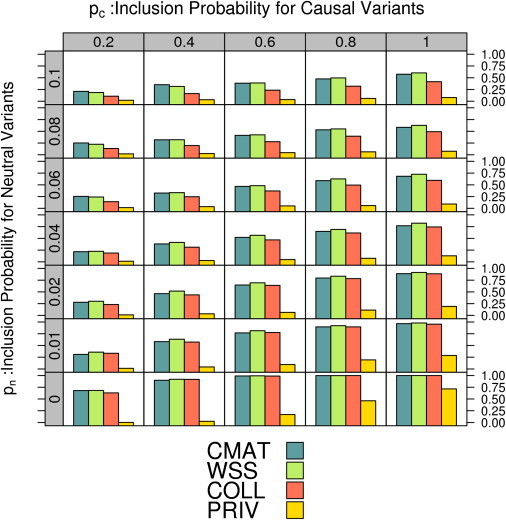
Power to Analyze Deep-Sequencing Datasets for a Range of Inclusion Probabilities
Each dataset contains exact genotypes for cases and controls based on causative variants in the population. Along the vertical axis, we vary the probability of (incorrectly) including a neutral variant (pn) in the analysis, and along the horizontal axis we vary the probability of (correctly) including a causative variant (pc). The height of the bars in each cell indicates the power for the four tests at .
A comparison of power between tests illustrates that the CMAT and WSS had nearly identical performance and were the most powerful tests at all levels of misspecification considered. The private-allele test had power <20% for most parameter settings. Power for the CMAT, WSS, and collapsing test was nearly identical when only a small number of neutral variants were included in the test statistic (). Here, the absolute power for the three tests was heavily dependent on the inclusion rate for causal variants; it increased from 30% up to 95% as the number of included causal variants increased.
The CMAT and WSS showed a clear power gain over the collapsing method for larger neutral variant-inclusion probabilities. In fact, the power gain was greatest when filtering accuracy was poorest. The CMAT had a power of 24%, as opposed to 11% for the collapsing test when pn = 0.1 and pc = 0.2. This trend continued for values of (data not shown). This difference is caused by the way the different tests account for individuals with more than one rare variant of interest. For larger values of pn, individual samples are increasingly likely to contain multiple rare variants. By directly testing the number of rare variants rather than the number of rare-variant carriers, the CMAT and WSS have additional power over the collapsing test.
Appropriately weighting variants in the test statistic might further improve power. However, it is presently unclear which weighting strategy is the most powerful, and it is likely that it will differ from case to case. Although we do not directly address the issue of most powerful weighting scheme in this paper, we computed power for both the CMAT and WSS by using the weighting scheme described by Madsen and Browning.13 Under this scheme, allele counts for the jth variant are weighted by the inverse of the standard deviation of allele count in controls. To facilitate comparison, we included only variants with a maf below our predetermined threshold () in the analysis. The maf-based weights correspond more closely to our disease model (Figure 1) than do the simple uniform weights and therefore provided a more powerful analysis for both methods except when misspecification rates were highest (Figure S2). Conditional on weighting scheme, the CMAT and WSS had similar power across the grid.
To assess the influence of variants with a maf of 1%–5% on the presented results, we repeated all simulations while restricting attention to variants with a maf (ie ). The misspecification grid for these settings (Figure S3) showed that overall power for each test was slightly lower than in the presented results. The noticeable change was that for the largest values of pn and pc, the WSS showed a power advantage, whereas the CMAT and the collapsing test had similar power.
For the remainder, simulation results are based on inclusion parameters of pn = 0.1 and pc = 0.8 so that they reflect a whole-gene analysis strategy that includes nonsynonymous coding and splice-site mutations plus variants in the UTR and potential regulatory regions flanking the gene.
Covariate Correction
Next, we created datasets in which samples were drawn from two distinct populations meant to resemble European and African haplotypes. We simulated the datasets under the null hypothesis of no association () but preferentially drew cases from the African population. Because the African haplotypes contain more rare variation than do the European haplotypes, the datasets contain a spurious association between disease status and an excess of rare variants. Datasets contained cases and controls drawn from the African population with probability and p, respectively. We analyzed each dataset at with the CMAT and the covCMAT and controlled for population of origin in the latter.
We present results for and (Figure 3). Ignoring the population stratification resulted in an elevated CMAT type I error, which increased sharply for . The magnitude of this increase is affected by the inclusion probability for the summary statistics. For strategies that attempt to capture all variants near a gene (shown here), the false-positive rate is substantially larger than for strategies focusing on exonic variation. Controlling for ancestry by including it as a covariate into the covCMAT maintained the desired type I error across all values of δ we considered.
Figure 3.
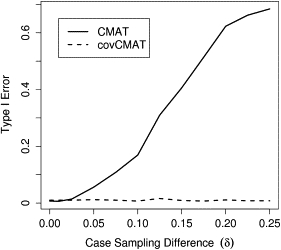
Application of the covCMAT to Control for Population Stratification
Cases were preferentially sampled from a population containing a larger number of rare variants. Failure to account for population stratification leads to inflated false-positive rates for the CMAT. When applied with the covariate correction, the covCMAT maintained the appropriate type I error.
Imputation Datasets
The CMAT is easily applied to imputation datasets containing probabilistic genotype calls. To consider the potential of a study design combining sequenced and imputed samples, we simulated exact genotype calls for the sequenced samples and probabilistic genotypes for the remaining samples. We considered a design with an equal number of cases and controls sequenced in a 100 kb region of interest and genotyped for tagSNPs in a 1Mb encompassing region. Imputed samples were assumed to be genotyped for the same set of tagSNPs. In this design, variants observed at least twice in the sequenced samples were imputed in the nonsequenced samples.
We found that the addition of imputed samples to a fixed number of sequenced samples can provide a considerable power gain over analyzing only the sequenced samples (Figure 4). A whole-gene CMAT analysis of datasets drawn from a population containing causative variants and constrained to sequenced cases and controls has a power of 14%. Augmenting these sequenced samples with an additional 1800 imputed cases and controls (total sample size ) increases power to 48%. This compares favorably with the optimal design that sequences all samples and has a power of 66%. Thus, the additional information from imputed samples recovered much but not all of the power of a fully sequenced dataset.
Figure 4.
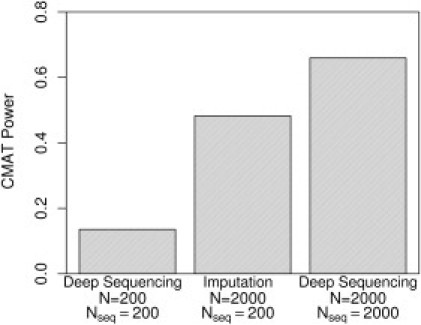
Comparison of CMAT Power for Deep Sequencing and Imputation Study Designs
From left to right, the bars show power at for a deep-sequencing dataset with , an imputation dataset with Nseq = 200 and N = 2000, and a deep-sequencing dataset with . For each, we used the whole-gene inclusion threshold ().
We extended our analysis to a wide range of sample sizes with N from 200 to 5000 and considered the effect of sequencing Nseq = 100, 200, or 400 samples for each N. The CMAT had a well controlled type I error when it was applied to datasets simulated with causative variants (data not shown). We present results for an analysis involving whole-genome inclusion parameters and k = 15 causative variants in the population (Figure 5). Power curves for inclusion thresholds that reflect an exon-only analysis () were slightly lower across all considered values of N (data not shown). For comparison, we also computed CMAT power for a dataset containing exact genotypes for all samples (i.e., ). We found that for a given total sample size N, CMAT power increased with the number of sequenced samples. At , datasets containing 100, 200, and 400 sequenced samples had powers of 48%, 56%, and 65%, respectively. Attaining similar power in a set of fully sequenced samples requires N = Nseq = 1000, 1500, and 2000 samples, respectively.
Figure 5.
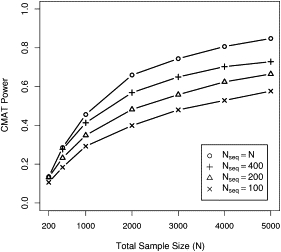
CMAT Power for Imputation Datasets
Datasets contain exact genotypes for Nseq sequenced cases and controls and probabilistic genotypes based on imputation for the remaining samples. The top line shows CMAT power when all samples are sequenced and serves as an upper bound for power at a fixed total sample size N. We report power at by using the whole-gene inclusion threshold ().
The dependence of power on the number of sequenced individuals is driven by three factors. First, replacing an exact genotype with a probabilistic genotype results in a loss of information. Thus, for a fixed sample size, datasets containing fewer sequenced samples suffer a larger information loss. Second, increasing the number of sequenced samples increases the chance that a risk allele is observed at least twice and can therefore be imputed. Of risk alleles in the simulations with maf <5%, an average of 3.2 were observed at least twice among 100 sequenced cases and controls. This number increased to 5.0 and 7.5 for datasets with 200 and 400 sequenced cases and controls, respectively. Third, imputation accuracy for an individual allele improves as that allele is observed more often in the sequenced samples. Sequencing a larger number of samples increases the number of times a risk allele is observed, and thus improves imputation accuracy for that allele.
We repeated the imputation simulations by using 1% maf parameter settings (Figure S4). We observed only a small reduction in power compared to the analysis with maf . Only datasets with 100 sequenced cases and 100 sequenced controls showed a notable reduction in power. For Nseq = 100, 200, and 400 sequenced cases and controls, a study with total sample size of had powers of , and , respectively. Hence, provided there is a sufficiently large set of sequenced templates, imputation of rare variants is a useful strategy, even if variants with a maf are of particular interest.
Application to GAIN Psoriasis Data
Our simulation study assumed that imputation templates were sequenced individuals from the study sample. It is feasible to instead use haplotypes from a public dataset as the imputation templates. This has the advantage that it allows rare-variant analysis in any existing GWAS dataset without requiring additional sequencing by the investigator.
As proof of principle for this approach, we applied the CMAT to the GAIN Psoriasis (MIM 177900) dataset consisting of 1,359 psoriasis cases and 1,400 unaffected controls of white European ancestry. We imputed 8.2 million autosomal SNPs into the dataset by using 112 CEU haplotypes from the August 2009 release of the 1000 Genomes Project as our reference panel. Previously, others had imputed this dataset for 2.5 million SNPs by using the CEU HapMap samples and analyzed it with a standard single-marker test for association.21 The strongest signal for association (, single marker p = 4 × 10−53) was located 13 kb upstream of the HLA-C (MIM 142840) gene, a previously known psoriasis locus on chromosome 6. Ten of the top 18 loci identified in the initial analysis were subsequently replicated in a larger, independent sample.
To apply the CMAT, we assigned the imputed SNPs to genes and retrieved functional annotations for genic variants (see Methods). We retained only SNPs with maf and annotated these as nonsynonymous, splice-site, or UTR. In total, 2889 genes containing two or more SNPs after filtering were analyzed with the CMAT. Of the genes tested, 55% contained two SNPs, 23% contained three SNPs, 11% contained four SNPs, and the remaining 11% contained five or more SNPs. None of the ten replicated SNPs from the original analysis remained after filtering, and only three genes (IL12B [MIM 161561], TSC1 [MIM 605284] and TNFAIP3 [MIM 191163]) near a replicated signal were included in the CMAT analysis.
After Bonferroni correction for the number of genes tested, one gene, SKIV2L, achieved statistical significance after Bonferroni correction) (MIM 600478). SKIV2L is located on 6p21.33, 700 kb away from HLA-C, the previously implicated psoriasis-susceptibility locus. The SKIV2L testing unit contained four imputed variants with a maf < 0.05 (Table 1). Although each variant trended toward significance in the single-marker test, no individual p value is sufficient to explain the level of significance observed in the CMAT. Genotypes for these variants were uncorrelated, indicating they are probably on different haplotype backgrounds and therefore independently contribute to the CMAT statistic. Thus, the significance of the SKIV2L CMAT statistic is driven by the cumulative effect of the four variants. Because imputation accuracy, indicated by , is high for each variant, it is unlikely that the observed signal is the result of low imputation quality.
Table 1.
Summary of the Top Result from CMAT Analysis of the GAIN Psoriasis Dataset, into which 8.2 Million SNPs were imputed on the basis of 112 CEU Haplotypes from the 1000 Genomes Project as a Reference
| SKIV2L Variant | maf | Function |
Imputation |
Single-Marker p Value | Correlation between Imputed Genotypes | ||||
|---|---|---|---|---|---|---|---|---|---|
| rs17201466 | 0.0496 | UTR | 0.98 | 0.0018 | 1.000 | ||||
| rs36038685 | 0.0109 | R324W | 0.99 | 0.1210 | −0.016 | 1.000 | |||
| rs3911893 | 0.0427 | D887N | 0.94 | 0.0029 | −0.055 | −0.006 | 1.000 | ||
| rs106287 | 0.0359 | V917M | 0.91 | 0.0888 | −0.059 | −0.027 | −0.034 | 1.000 | |
SKIV2L was statistically significant after Bonferroni correction (CMAT p < 10−6). SKIV2L is located on 6p21.33, 700 kb away from HLA-C, a known psoriasis susceptibility locus. The table lists the maf, functional annotation, imputation accuracy , and single-marker p value of individual variants included in the pooled statistic. The last columns contain the pairwise correlations between imputed minor-allele counts.
Analysis of common variation in the HLA region indicated the potential for additional functional variants in the same or different genes after conditioning on . Our result for SKIV2L might indicate such an additional psoriasis locus in this region and makes SKIV2L an interesting candidate for further investigation.
Discussion
We described the CMAT, a simple method for jointly testing multiple rare variants in case-control sequence data; the CMAT can be easily extended to deal with typical challenges of modern genomic studies. Notably, our statistic accepts expected minor-allele counts from probabilistic genotypes, making it applicable to both low-coverage sequencing and imputed data. The statistic can incorporate qualitative covariates and thus allow correction for confounders such as population stratification. Moreover, the CMAT is both computationally fast and straightforward to implement.
We assessed the CMAT by applying it to simulated case-control sequencing datasets specifically designed to contain realistic levels of neutral variation. We also considered three alternative testing strategies, a private-allele test similar to the one used by Cohen et al.,9 the collapsing test described by Li and Leal,12 and the weighted sum statistic (WSS) of Madsen and Browning.13 We considered levels of variant misspecification that are representative of exon-only sequencing to entire genic regions. Our results indicated that the strategy of focusing on exonic variants is appropriate if most rare risk variants are located in exons. However, if the majority of rare risk variants are located in regulatory regions, then analyzing all rare variants together, both exonic and nonexonic, can be more powerful than analyzing only the exonic variants. That is, the increase in signal from including noncoding risk variants can outweigh the additional noise of noncoding neutral variants. Comparing the different tests, we noticed that the CMAT, WSS, and collapsing test were equally powerful for the exon-only model. However, the CMAT and the WSS were more robust to variant misspecification and were therefore significantly more powerful when we analyzed data representative of whole-gene analysis. The CMAT provides similar power to that of the WSS and is computationally more efficient. Because the WSS is based on ranking individuals, its computation time is bounded by the theoretical maximum of O(nlog(n)); the computation time of the CMAT is linear with sample size. This difference can be substantial for analysis of large sample sizes in genome-wide studies.
A pooling statistic that accepts probabilistic genotypes dramatically increases the range of possible rare-variant study designs. Our simulations demonstrated the potential of including genotypes from both direct sequencing and imputation in the test statistic. Because genotypes for rare variants are generally imputed with higher error rates than common variants, it is important to propagate this uncertainty into the analysis by using expected minor-allele counts, as opposed to most likely genotype, in the CMAT. Our simulation results show that one can gain substantial power by augmenting sequencing datasets with imputed samples. In particular, sequencing only a fraction of available individuals and imputing the remainder can recoup much of the power of a study that sequences all samples and provide a major cost reduction. Other methods for testing rare variants can most likely be adapted so that there is a comparable gain of efficiency from imputed data. Note that we modeled the sequencing of an equal number of cases and controls, but more powerful sequencing strategies for observing risk alleles might exist, for example, one such strategy might involve sequencing mainly cases.24
We also provided an example of a rare-variant analysis that does not require sequencing. Instead, rare variants can be imputed into existing GWAS datasets from publicly available reference panels. Single-marker tests have limited power to detect an association at these imputed variants because of both low maf and high uncertainty in imputing rare variants.25 Pooling these variants and testing their cumulative effect is more powerful and could uncover additional signals in the data. We used the haplotypes from the CEU samples in the 1000 Genomes Project to impute rare variants into the existing GAIN Psoriasis GWAS dataset. Our analysis shows that the CMAT can identify interesting genes that cannot be found by single-marker tests. The identified gene (SKIV2L) contains multiple rare variants, none of which achieved genome-wide significance in a single-marker test. SKIV2L resides in the HLA region of chromosome 6, which is thought to harbor multiple psoriasis susceptibility genes. However, the biological interpretation is not clear. SKIV2L is not an obvious candidate for psoriasis. Although SKIV2L might be a psoriasis locus, it is also conceivable that multiple rare variants in SKIV2L tag the same functional common variant in another gene, and the observed signal might be the result of reverse synthetic association.26 Further analysis is necessary to validate this finding. The analysis was limited by the size of our reference panel, which contained only 112 haplotypes. Only 2889 genes contained two or more coding variants with a maf <0.05 in this panel and were thus eligible for the pooled analysis. Future releases from the 1000 Genomes Project should provide low-coverage sequencing of 2500 individuals and deep-exome resequencing of the same 2500 individuals.19 This will increase the number of imputable rare variants and make this analysis method more powerful.
Accurate prediction of functionally relevant sites and appropriate weighting will reduce variant misspecification and could further improve the power of pooling methods. The weighting scheme proposed by Madsen and Browning13 is based on allele frequency and is most powerful for risk variants under relatively high purifying selection.14 Alternatively, variants can be weighted according to predictions of molecular function. In practice, bioinformatic tools such as PolyPhen27 and SIFT28 are useful in predicting deleterious potential but are typically limited to coding variants. Determining functionality of noncoding variants is more difficult, and although databases containing known phenotype-altering noncoding variants exist (PupaSuite,29 for example), these are not applicable to novel variants. Instead, identifying conserved regulatory regions within noncoding portions of a gene will be crucial in determining which noncoding variants have phenotype-altering potential and should be included in an analysis.30 For this paper, rather than attempting to optimize weights for our specific disease model, we assumed very simple uniform weights and focused on the overall performance of our test with respect to variant misspecification and imputation. However, we have included a general weighing term into the statistic to allow any desired scheme to be incorporated.
Our simulation results are based on several underlying assumptions. Like other methods, our method assumes that all rare variants pooled together have the same type of effect. That is, either all are causative, the likely model if risk variants are under purifying selection,5 or they are all protective. If this assumption is violated and causal and protective alleles are combined into a single statistic, pooling methods will lose power. Our results also depend on our disease model, specifically the range of allele frequencies and effect sizes for risk variants. The true frequency spectrum for risk alleles will depend on the strength of purifying selection at the locus and can range from extremely rare family-specific mutations to so-called ‘goldilocks’ alleles that segregate at low frequency in the population.14 We evaluated a combination of both models; this combination allowed frequencies between and for risk variants. However, we showed that our results also apply to analyses restricted to rarer variants between and . Because we are interested in variants that would not be detected by existing association methods, we assigned larger relative risks to rarer alleles. Our results therefore apply to this class of risk variants and do not generalize to extremely rare variants with Mendelian inheritance patterns. In particular, we note that our choice of disease model explains the poor performance of the private allele test, which is best suited for testing highly penetrant Mendelian-like risk alleles segregating within families. We have included it in our analysis because it is currently one of the few statistical tests that has successfully provided evidence for rare-variant associations.
In summary, the CMAT is a powerful and versatile tool for analyzing the contribution of rare variants to the heritability of common complex diseases. The test accounts for the uncertainty that imputation methods can confer to genotypes and can be used for reanalyzing existing GWAS datasets.
Acknowledgments
We thank Tanya Teslovich, Paul Scheet, Mike Boehnke, and Gonçalo Abecasis for helpful discussions, Derek Chiang for the 1000 genomes annotation and Matthew Flickinger for assistance with graphical layouts. This work was supported by National Institutes of Health grants R01 HL90564 and R01 HG005855.
Appendix
Empirical Distributions for Expected Minor-Allele Counts
We assume a set of cases and controls genotyped for a set of tagSNPs across a 1 Mb segment that contains a 100 kb region of interest. We assume that Nseq cases and controls are randomly selected and sequenced at deep coverage in the 100 kb region. Variants observed among the sequenced samples in the region of interest are imputed into the remaining samples.
We created empirical distributions of expected minor-allele counts for imputed genotypes by assuming sequence data for Nseq = 100, 200, and 400 cases and controls and tagSNPs for the remainder of the sample. For each, we first simulated ten independent populations of ten thousand 1 Mb haplotypes by using cosi and for each region selected a set of tagSNPsthat mimicked real-world tagging properties.22 For each 1 Mb region, the 100 selected tagSNPs resulted in of the common variants having an with one of the selected tagSNPs, similar to the tagging properties of the Illumina HumanHap300 BeadChip SNP genotyping platform. From each population, we drew a random subset of 4000 haplotypes and treated the first 2 × Nseq as sequenced in the middle 100 kb region of interest (these sample sizes correspond to datasets with and Nseq sequenced cases and controls).
We statistically phased the 2 × Nseq haplotypes across the entire 1 Mb region. These phased haplotypes then served as a reference panel for imputation of the variants observed in the middle 100 kb into the remaining haplotypes. Phasing and imputation were performed with the software program MaCH.20 MaCH includes a “states” option that speeds computation by limiting the number of haplotypes considered at each iteration of phasing or imputation. Because our analysis focused on rare variants that might only appear on a few haplotypes, we did not use the states shortcut. This probably prolonged computation time but improved imputation accuracy.
We observed that imputation accuracy for rare variants was dependent on the allele frequency, the total number of haplotypes in the reference panel (2 × Nseq), and the number of times a variant was observed in the reference panel (M.Z. and S.Z., unpublished data). Therefore, we created empirical sampling distributions by binning the observed expected minor-allele counts (dosage) by true underlying genotype and the number of times the minor allele was observed in the reference panel. We pooled analogous distributions across all ten realizations to average over varying degrees of LD. The distributions for true heterozygotes were bimodal and had peaks at 1.0, the true dosage for a heterozygote, and 0.0, the true dosage for a major-allele homozygote. Because the minor allele is observed more often in the reference panel, imputation was more accurate, as indicated by fact that the density of the peak at 0.0 shifted to larger dosage values. Table 2 summarizes these empirical distributions for . The and columns capture the density in the two peaks. The distributions for true major-allele homozygotes consist of a point mass at 0.0 and a small amount of density just above 0.0. As the number of minor alleles observed in the reference panel increases, the density shifts slightly away from the point mass.
Table 2.
Summary of Empirical Distributions of Minor-Allele Dosage for True Heterozygotes
| Minor-Allele Count in Reference Haplotypes |
Fraction of Heterozygote Minor-Allele Dosages |
|||
|---|---|---|---|---|
| <0.1 | [0.1, 0.5) | [0.5, 0.9) | ≥ 0.9 | |
| 1 | 0.729 | 0.120 | 0.063 | 0.088 |
| 2 | 0.331 | 0.188 | 0.133 | 0.349 |
| 3 | 0.291 | 0.169 | 0.128 | 0.413 |
| 4 | 0.199 | 0.170 | 0.162 | 0.469 |
| 5 | 0.327 | 0.176 | 0.162 | 0.335 |
| 6 | 0.255 | 0.180 | 0.136 | 0.428 |
| 7 | 0.166 | 0.149 | 0.132 | 0.553 |
| 8 | 0.203 | 0.195 | 0.179 | 0.422 |
| 9 | 0.091 | 0.114 | 0.128 | 0.667 |
| 10 | 0.100 | 0.159 | 0.195 | 0.546 |
| 11–20 | 0.061 | 0.094 | 0.118 | 0.727 |
| 21–30 | 0.043 | 0.054 | 0.092 | 0.811 |
| 31–40 | 0.016 | 0.039 | 0.081 | 0.865 |
| 41–50 | 0.016 | 0.051 | 0.100 | 0.834 |
| 51–60 | 0.006 | 0.023 | 0.050 | 0.921 |
| 61–70 | 0.011 | 0.039 | 0.087 | 0.863 |
| 71–80 | 0.009 | 0.026 | 0.072 | 0.893 |
| 81–90 | 0.007 | 0.017 | 0.054 | 0.923 |
| 91–100 | 0.005 | 0.019 | 0.065 | 0.911 |
Each distribution is conditional on the indicated minor-allele count in the reference haplotypes. Here we report results for sequenced cases and controls.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
References
- 1.Risch N., Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- 2.Hindorff L.A., Sethupathy P., Junkins H.A., Ramos E.M., Mehta J.P., Collins F.S., Manolio T.A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. USA. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manolio T.A., Collins F.S., Cox N.J., Goldstein D.B., Hindorff L.A., Hunter D.J., McCarthy M.I., Ramos E.M., Cardon L.R., Chakravarti A. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goldstein D.B. Common genetic variation and human traits. N. Engl. J. Med. 2009;360:1696–1698. doi: 10.1056/NEJMp0806284. [DOI] [PubMed] [Google Scholar]
- 5.Pritchard J.K. Are rare variants responsible for susceptibility to complex diseases? Am. J. Hum. Genet. 2001;69:124–137. doi: 10.1086/321272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pritchard J.K., Cox N.J. The allelic architecture of human disease genes: Common disease-common variant…or not? Hum. Mol. Genet. 2002;11:2417–2423. doi: 10.1093/hmg/11.20.2417. [DOI] [PubMed] [Google Scholar]
- 7.Gorlov I.P., Gorlova O.Y., Sunyaev S.R., Spitz M.R., Amos C.I. Shifting paradigm of association studies: Value of rare single-nucleotide polymorphisms. Am. J. Hum. Genet. 2008;82:100–112. doi: 10.1016/j.ajhg.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Metzker M.L. Sequencing technologies—The next generation. Nat. Rev. Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 9.Cohen J.C., Pertsemlidis A., Fahmi S., Esmail S., Vega G.L., Grundy S.M., Hobbs H.H. Multiple rare variants in NPC1L1 associated with reduced sterol absorption and plasma low-density lipoprotein levels. Proc. Natl. Acad. Sci. USA. 2006;103:1810–1815. doi: 10.1073/pnas.0508483103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cohen J., Pertsemlidis A., Kotowski I.K., Graham R., Garcia C.K., Hobbs H.H. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat. Genet. 2005;37:161–165. doi: 10.1038/ng1509. [DOI] [PubMed] [Google Scholar]
- 11.Kryukov G.V., Shpunt A., Stamatoyannopoulos J.A., Sunyaev S.R. Power of deep, all-exon resequencing for discovery of human trait genes. Proc. Natl. Acad. Sci. USA. 2009;106:3871–3876. doi: 10.1073/pnas.0812824106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li B., Leal S.M. Methods for detecting associations with rare variants for common diseases: Application to analysis of sequence data. Am. J. Hum. Genet. 2008;83:311–321. doi: 10.1016/j.ajhg.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Madsen B.E., Browning S.R. A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet. 2009;5:e1000384. doi: 10.1371/journal.pgen.1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Price A.L., Kryukov G.V., de Bakker P.I.W., Purcell S.M., Staples J., Wei L.-J., Sunyaev S.R. Pooled association tests for rare variants in exon-resequencing studies. Am. J. Hum. Genet. 2010;86:832–838. doi: 10.1016/j.ajhg.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Asthana S., Noble W.S., Kryukov G., Grant C.E., Sunyaev S., Stamatoyannopoulos J.A. Widely distributed noncoding purifying selection in the human genome. Proc. Natl. Acad. Sci. USA. 2007;104:12410–12415. doi: 10.1073/pnas.0705140104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Emison E.S., McCallion A.S., Kashuk C.S., Bush R.T., Grice E., Lin S., Portnoy M.E., Cutler D.J., Green E.D., Chakravarti A. A common sex-dependent mutation in a RET enhancer underlies Hirschsprung disease risk. Nature. 2005;434:857–863. doi: 10.1038/nature03467. [DOI] [PubMed] [Google Scholar]
- 17.Haller G., Torgerson D.G., Ober C., Thompson E.E. Sequencing the IL4 locus in African Americans implicates rare noncoding variants in asthma susceptibility. J. Allergy Clin. Immunol. 2009;124:1204–1209. doi: 10.1016/j.jaci.2009.09.013. e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pauws E., Moore G.E., Stanier P. A functional haplotype variant in the TBX22 promoter is associated with cleft palate and ankyloglossia. J. Med. Genet. 2009;46:555–561. doi: 10.1136/jmg.2009.066902. [DOI] [PubMed] [Google Scholar]
- 19.The 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y., Willer C., Sanna S., Abecasis G. Genotype imputation. Annu. Rev. Genomics Hum. Genet. 2009;10:387–406. doi: 10.1146/annurev.genom.9.081307.164242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nair R.P., Duffin K.C., Helms C., Ding J., Stuart P.E., Goldgar D., Gudjonsson J.E., Li Y., Tejasvi T., Feng B.-J., Collaborative Association Study of Psoriasis Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat. Genet. 2009;41:199–204. doi: 10.1038/ng.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schaffner S.F., Foo C., Gabriel S., Reich D., Daly M.J., Altshuler D. Calibrating a coalescent simulation of human genome sequence variation. Genome Res. 2005;15:1576–1583. doi: 10.1101/gr.3709305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sherry S.T., Ward M.H., Kholodov M., Baker J., Phan L., Smigielski E.M., Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li B., Leal S.M. Discovery of rare variants via sequencing: implications for the design of complex trait association studies. PLoS Genet. 2009;5:e1000481. doi: 10.1371/journal.pgen.1000481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang L., Wang C., Rosenberg N.A. The relationship between imputation error and statistical power in genetic association studies in diverse populations. Am. J. Hum. Genet. 2009;85:692–698. doi: 10.1016/j.ajhg.2009.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dickson S.P., Wang K., Krantz I., Hakonarson H., Goldstein D.B. Rare variants create synthetic genome-wide associations. PLoS Biol. 2010;8:e1000294. doi: 10.1371/journal.pbio.1000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramensky V., Bork P., Sunyaev S. Human non-synonymous SNPs: Server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ng P.C., Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reumers J., Conde L., Medina I., Maurer-Stroh S., Van Durme J., Dopazo J., Rousseau F., Schymkowitz J. Joint annotation of coding and non-coding single nucleotide polymorphisms and mutations in the SNPeffect and PupaSuite databases. Nucleic Acids Res. 2008;36(Database issue):D825–D829. doi: 10.1093/nar/gkm979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lomelin D., Jorgenson E., Risch N. Human genetic variation recognizes functional elements in noncoding sequence. Genome Res. 2010;20:311–319. doi: 10.1101/gr.094151.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.