

Abstract
Distinctive facial features consisting of hypertelorism, telecanthus, blepharophimosis, blepharoptosis, epicanthus inversus, periumbilical defects, and skeletal anomalies are seen in autosomal-recessive Carnevale, Malpuech, Michels, and oculo-skeletal-abdominal (OSA) syndromes. The gene or genes responsible for these syndromes were heretofore unknown. We report on three individuals from two consanguineous Turkish families with findings characteristic of these syndromes, including facial dysmorphism, periumbilical depression, mixed hearing loss, radioulnar synostosis, and coccygeal appendage. Homozygosity mapping yielded an autozygous region on chromosome 3q27 in both families. In one family, whole exome sequencing revealed a missense mutation, MASP1 c.2059G>A (p.G687R), that cosegregated with the phenotype. In the second family, Sanger sequencing of MASP1 revealed a nonsense mutation, MASP1 c.870G>A (p.W290X), that also cosegregated with the phenotype. Neither mutation was found in 192 Turkish controls or 1200 controls of various other ancestries. MASP1 encodes mannan-binding lectin serine protease 1. The two mutations occur in a MASP1 isoform that has been reported to process IGFBP-5, thereby playing a critical role in insulin growth factor availability during craniofacial and muscle development. These results implicate mutations of MASP1 as the cause of a human malformation syndrome and demonstrate the involvement of MASP1 in facial, umbilical, and ear development during the embryonic period.
Main Text
A phenotype consisting of ptosis of the eyelids, downslanting palpebral fissures, hypertelorism, developmental delay, radioulnar synostosis, and periumbilical depression was reported by Carnevale et al. in 1989 in two siblings from a consanguineous Italian family (MIM 265050).1 In 1996, Mingarelli et al. reported two sisters with similar ocular, facial, and abdominal defects and additional skeletal anomalies, but with normal intelligence, and suggested that this phenotype be called oculo-skeletal-abdominal (OSA) syndrome (also MIM 265050).2 Because of clinical overlap with Michels syndrome3 (MIM 257920) and Malpuech syndrome4 (MIM 248340), it was subsequently suggested that these four syndromes might belong to the phenotypic spectrum of the same disorder, which could be referred to as the 3MC (Malpuech-Michels-Mingarelli-Carnevale) syndrome.5 Distinctive features have been noted. The presence of anterior chamber eye anomalies was suggested to be limited to Michels syndrome, and growth and mental retardation, caudal appendage, and cleft lip or palate were more frequently associated with Malpuech syndrome.6 However, the combination of characteristic facial and umbilical findings with anterior chamber anomalies and caudal appendage in a further family again suggested that these syndromes could be causally related.7 The gene or genes responsible for these phenotypes remained unknown.
We ascertained two families from Turkey with phenotypes very similar to those described by Carnevale et al. (Figure 1A).1 The two patients in family 1 are sisters aged 15 years (individual 1-101; IV-1 in Figure 1B) and 10 years (individual 1-102; IV-2 in Figure 1B). Their parents are first cousins. Sisters 1-101 and 1-102 were born after uncomplicated pregnancies with apparently small birth weights (no measurement was available). Initial gross and fine motor skills, as well as speech development, were delayed. Height and weight of 1-101 and 1-102 were 160 cm (between the 25th and 50th centiles) and 51 kg (25th centile) and 148 cm (75th centile) and 46 kg (75th centile), respectively. Both siblings had mild mental retardation with full-scale IQ scores of 60–65. Pure tone audiograms indicated that 1-101 had moderate (right) and profound (left) mixed hearing loss and that 1-102 had mild mixed (right) and profound sensorineural (left) hearing loss. High-resolution CT scans of the temporal bone showed large vestibules, large superior semicircular canals, and absent lateral semicircular canals in both siblings. The facial phenotype included highly arched eyebrows, hypertelorism, blepharoptosis, broad and high nasal bridge with epicantus inversus, downslanting palpebral fissures, and limited upward gaze in both siblings and bifid nasal tip in 1-102 (Figure 1A and Table 1). Hypertelorism was considered when interpupillar distance was greater than +2 standard deviation above the mean, and telecanthus was diagnosed when the ratio of inner canthal distance to interpupillary distance was bigger than 0.6.8 In 1-101, interpupillar, outer, and inner canthal distances and palpebral fissure lengths were 68 mm (>97th centile) and 10 cm (97th centile) and 37 mm (95th centile) and 31 mm (between the 50th and 75th centiles), respectively. In 1-102, interpupillar, outer, and inner canthal distances and palpebral fissure lengths measured 67 mm (>97th centile) and 10.2 cm (>97th centile) and 40 mm (>95th centile) and 30 mm (75th centile), respectively. Severe hypermetropy without any anterior chamber anomalies, excessive skin over the coccygeal area, and supraumbilical depression were present in both siblings (Figure 1A). The older sister (1-101) was found to have right radioulnar synostosis and solitary ectopic kidney. Echocardiography was normal in both siblings. The parents and three siblings appeared to be healthy, with no distinctive features.
Figure 1.
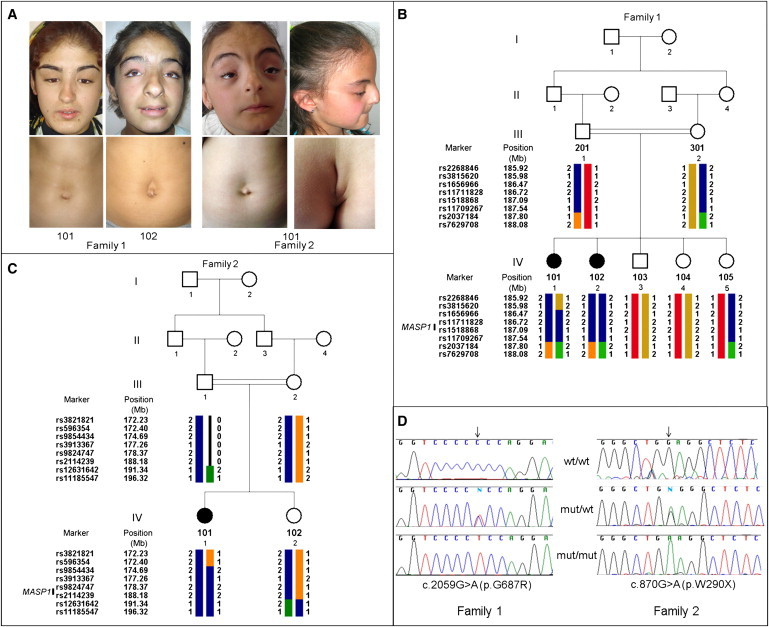
Clinical Findings, Pedigrees, and MASP1 Mutations
(A) Phenotypic findings in affected individuals. Note hypertelorism, blepharoptosis, telecanthus, downslanting palpebral fissures, arched eyebrows, and supraumbilical depression in all three patients. Bifid nasal tip and limitation of upward gaze are seen in individual 1-102 of family 1 and individual 2-101 of family 2. A coccygeal skin appendage with a groove, characteristics of Malpuech syndrome, and high nasal bridge with posteriorly rotated ear are seen in individual 2-101 of family 2.
(B) Family 1 pedigree and longest autozygous run at chromosome 3q27 that cosegregates with the phenotype.
(C) Family 2 pedigree and haplotypes flanking the MASP1 gene.
(D) Confirmation of mutations in the MASP1 gene in families 1 and 2.
Table 1.
Phenotypic Findings of Carnevale, Michels, OSA, and Malpuech Syndromes and of Patients Reported in This Study
| Phenotypic Findinga | Michels3,5,25–28 | Malpuech4,29–33 | Carnevale1,6 | OSA2 | Family Reported by Leal et al. as 3MC7 |
Family 1 |
Family 2, 2-101 | |
|---|---|---|---|---|---|---|---|---|
| 1-101 | 1-102 | |||||||
| Short stature | 6/10 | 10/13 | 0/4 | 0/2 | 0/2 | − | − | − |
| Mental retardation | 4/9 | 11/13 | 1/4 | 0/2 | 0/2 | + | + | + |
| Craniofacial Findings | ||||||||
| Arched eyebrows | 10/10 | 13/13 | 4/4 | 2/2 | 2/2 | + | + | + |
| Telecanthus | 9/10 | 12/14 | 4/4 | 2/2 | 0/2 | − | + | + |
| Hypertelorism | 9/10 | 14/14 | 4/4 | 2/2 | 0/2 | + | + | − |
| Blepharophimosis | 9/10 | 2/13 | 0/4 | 0/2 | 1/2 | − | − | − |
| Blepharoptosis | 10/10 | 7/13 | 4/4 | 2/2 | 2/2 | + | + | + |
| Downslanting palpebral fissures | 7/10 | 8/13 | 4/4 | 2/2 | 2/2 | + | + | + |
| Anterior chamber anomalies | 5/10 | 1/14 | 0/4 | 0/2 | 1/2 | − | − | − |
| Hearing loss | 8/10 | 6/8 | 0/4 | 0/2 | 2/2 | + | + | − |
| Cleft lip/cleft palate | 7/10 | 14/14 | 1/4 | 0/2 | 0/2 | − | − | − |
| Abdominal Findings | ||||||||
| Periumbilical depression | 8/9 | 0/4 | 3/4 | 2/2 | 1/2 | + | + | + |
| Omphalocele/umbilical hernia | 2/9 | 4/6 | 0/4 | 0/2 | 2/2 | − | − | − |
| Skeletal Findings | ||||||||
| Hip anomaly | NA | 0/5 | 3/4 | 0/2 | 0/2 | − | − | − |
| Deviated or short fingers | 6/9 | 3/4 | 1/4 | 2/2 | 0/2 | + | − | + |
| Limitation of elbow movements | 4/10 | NA | 3/4 | 1/2 | 1/2 | + | − | − |
| Vertebral anomalies | 2/5 | 2/6 | 0/2 | 2/2 | 1/2 | − | − | − |
| Genitourinary Anomalies | ||||||||
| Genital | 3/9 | 10/14 | 2/4 | 0/2 | 1/2 | − | − | − |
| Vesicorenal | 5/7 | 8/13 | 0/2 | 0/2 | 0/2 | + | − | + |
| Caudal appendage | 0/5 | 5/9 | NA | NA | 2/2 | + | + | + |
NA denotes that information is not available.
Numbers indicate patients with a specific finding among patients with published information on that finding.
The patient in family 2 (individual 2-101; IV-1 in Figure 1C) is a female aged 9 years, the only affected child of first-cousin parents. She was born at term with a birth weight of 2800 g. Her initial developmental skills were within the normal limits, with walking and starting to talk at around 15 months of age. Her height, weight, and head circumference at 9 years of age were 124.5 cm (10th centile), 25 kg (25th centile), and 51 cm (25th centile), respectively. Her interpupillar, outer, and inner canthal distances and palpebral fissure lengths were 55 mm (75th centile) and 10 cm (>97th centile) and 41 mm (>95th centile) and 29 mm (between the 50th and 75th centiles), respectively. The clinical phenotype included highly arched eyebrows, telecanthus, high nasal bridge, blepharoptosis, epicanthus inversus, limited upward gaze, downslanting palpebral fissures, ectropion of lateral third of lower eyelids, synophyris, downturned corners of the mouth, slightly bifid nasal tip, supraumbilical depression, short and tapering fingers with fifth finger clinodactyly, coccygeal skin appendage with a deep groove (Figure 1A), and syndactyly between the second and third toes in the right side. She was found to have mild mental retardation (full-scale IQ was 54). Pure tone audiogram was normal. Abdominal ultrasound showed fused cross ectopia of the left kidney. Echocardiography showed small patent ductus arteriosus at 9 years of age. The parents and the patient's sister had no phenotypic features of the syndrome.
Phenotypic features of these three patients were similar to those reported in Carnevale, OSA, Malpuech, and Michels syndromes (Table 1). Absence of anterior chamber anomalies was not characteristic of Michels syndrome, and normal height was not characteristic of Malpuech syndrome, but both Michels and Malpeuch syndromes are phenotypically heterogeneous. Overall, our patients appeared to represent the combination of clinical findings reported in Carnevale, OSA, Malpuech, and Michels syndromes, similar to those reported by Leal et al. as 3MC syndrome (Table 1).7 A research protocol to evaluate the families genetically was approved by the Ethics Committee at the University of Ankara, Turkey and the Institutional Review Boards at the University of Miami and the University of Washington. All participants provided informed consent or assent prior to enrollment.
Seven members of family 1 (individuals III-1, III-2, and IV-1–5 in Figure 1B) and three members of family 2 (individuals III-2, IV-1, and IV-2) were enrolled (Figures 1B and 1C). Chromosomal karyotype was normal in all three affected individuals. DNA was extracted from peripheral leukocytes, and homozygosity mapping was carried out as previously described9 with Affymetrix 6.0 arrays. In family 1, only one homozygous segment longer than 1.5 Mb was shared by both affected children but none of their unaffected siblings (Figure 1B). This region was 1,812,702 bp between SNP markers rs3815620 and rs2037184. In family 2, this 1.81 MB autozygous region was embedded in a homozygous region of approximately 24 Mb that segregated with the phenotype (Figure 1C).
In order to identify the critical gene, genomic DNA of the proband of family 1 was evaluated by whole exome sequencing, as previously described.10 A total of 3.28 Gb of sequence (215,986,000 pairs of reads) aligned to the exome target. The proportion of the entire targeted exome covered by >10 reads was 91.2%. DNA variants were filtered against dbSNP131 and phase 3 of the 1000 Genomes project (March 2010 data release), then classified by predicted function to include all missense, nonsense, frameshift, or splice-site alleles. Because dbSNP includes both disease-associated and benign alleles, the analysis was designed to include known SNPs identified as clinically associated.
The 1.81 Mb autozygous region on chromosome 3q27 includes 20 genes, all of which were fully targeted by the SureSelect All Exon kit. Whole exome sequencing yielded >50× median coverage for this region. Four rare changes were identified in the region (Table 2). One missense was chr3:186,953,600C>T (hg19), supported by 78 sequence reads, all of the mutant genotype, corresponding to MASP1 c.2059G>A (NM_139125.3), leading to p.G687R (NP_624302.1; Table 3). The other missense was chr3:187,447,696T>G (hg19), corresponding to BCL6 c.497A>C (NM_001706.4), leading to p.N166T (NP_001124317.1) at a nonconserved site, with SIFT score 0.69 and PolyPhen2 classification benign (score 0.000). The other two rare changes in the region were chr3:186,791,888G>C, at a nonconserved intronic site in ST6GAL1 (NM_173216.2), and chr3:186,410,219C>T, at a nonconserved intergenic site.
Table 2.
Rare Variants in the 1.813 Mb Homozygous Region on Chromosome 3q27 Revealed by Whole Exome Sequencing in Proband 1-101
| SBP | Indel | Total | |
|---|---|---|---|
| Number of variants with ≥10 reads and variant ≥15% of total reads | 4 | 0 | 4 |
| Number of nonsense, missense, frameshift, or splice variants | 2 | 0 | 2 |
| Nonsense, frameshift, splice | 0 | 0 | 0 |
| Missense at conserved site | 1 | 0 | 1 |
SBP denotes single base pair substitutions. Indel denotes insertions or deletions up to 20 bp in length.
Table 3.
Amino Acid Substitutions in the MASP-3-Specific Exon Harboring p.G687R in Family 1
| Locale (hg19) | Substitution | dbSNP131 | 1000 Genomes Pilot 3a | PolyPhen2 HumVar scoreb |
|---|---|---|---|---|
| chr3:186954107C>T | p.V518I | rs73068950 | LWK, YRI | 0.028 |
| chr3:186953975C>A | p.P562A | rs72549155 | − | 0.038 |
| chr3:186953932C>A | p.R576M | rs72549154 | LWK, CEU, YRI, CHB, CHD, TSI | 0.081 |
| chr3:186953812A>G | p.V616A | rs2461280 | − | 0.044 |
| chr3:186953749C>A | p.R637L | − | YRI | 0.496 |
| chr3:186953600C>T | p.G687R | − | − | 0.902 |
| chr3:186953491T>C | p.E723G | − | JPT, CHD | 0.079 |
Populations are defined on the HapMap website.
Range is from 0.000 (most probably benign) to 0.999 (most probably damaging).
MASP1 encodes mannan-binding lectin serine protease 1 (MIM 600521).11 MASP1 p.G687R occurs at a highly conserved site in an exon specific to MASP1 isoform 2, also known in the literature as MASP-3.12 MASP-3 p.G687R is predicted to be a damaging change, with SIFT score 0.00 and PolyPhen2 classification “probably damaging” (HumDiv score 0.983; HumVar score 0.902). Data from the 980 individuals of the 1000 Genomes Project Pilot 3 and data from dbSNP131 can be evaluated to reveal the spectrum and predicted consequences of mutations in control populations in this exon. Six amino acid substitutions are reported (Table 3). All that appear on dbSNP are rare, and those specific to 1000 Genomes are also likely to be rare, given that they do not appear on dbSNP. None are in conserved sites or are predicted to be damaging to the protein. Therefore, in at least 1200 controls from multiple populations, this exon does not sustain mutations with consequences predicted to be as deleterious as p.G687R.
Sanger sequencing of the entire MASP1 gene in all relatives of family 1 confirmed that both affected children were homozygous for p.G687R. Both parents and unaffected sibling 1-105 were heterozygous, and the two other healthy siblings (1-103 and 1-104) were homozygous for wild-type alleles (Figure 1D). Primer sequences used for sequencing are available in Table S1 available online.
Sanger sequencing of the complete MASP1 gene in affected child 2-101 from family 2 revealed chr3:186,970,978C>T, corresponding to MASP1 c.870G>A, leading to a stop at p.W290X (Figure 1D; Table 4). This mutation was homozygous in the affected child and heterozygous in both her mother and an unaffected sister. Neither mutation in MASP1 was detected in 192 healthy Turkish controls or 1200 controls of various other ancestries.
Table 4.
Mutations in MASP1
| Family | Chr. | Coordinate | Variant | Wild-Type Reads | Variant Reads | Effect | Genotype of Affected Individuals |
|---|---|---|---|---|---|---|---|
| 1 | 3 | 186,953,600 | c.2059G>A | 0 | 78 | p.G687R | Homozygous |
| 2 | 3 | 186,970,978 | c.870G>A | Sanger sequence | p.W290X | Homozygous |
The MASP-1 protein was first identified as a serine protease activating the classical complement pathway by binding to lectin.11 The MASP1 genetic locus is complex, encoding three isoforms that share amino acid residues in their amino terminal regions but differ in their carboxy terminals containing serine protease domains. The first and second isoforms (MASP-1 and MASP-3) contain different serine protease domains, and the third isoform (also referred to as MAp44) lacks a serine protease domain (Figure 2A). The nonsense mutation p.W290X in family 2 lies in coding exon 6, which is shared by all MASP-1 isoforms. The missense mutation p.G687R in family 1 is in an exon unique to MASP-3. The mutant MASP-3 site p.G687 is completely conserved from fish to humans (Figure 2B) and lies at a predicted substrate binding site that is highly conserved across a wide variety of serine protease domains (Figure 2C). MASP-3 is 728 amino acids in length.12 The locale of the missense mutation suggests that the phenotype associated with the two MASP1 mutations may be due specifically to impairment of the MASP-3 isoform.
Figure 2.
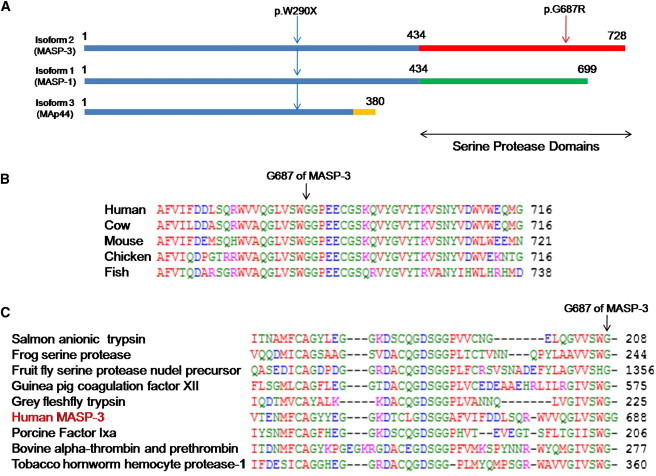
Three Isoforms of the MASP1 Gene and Mutation Sites
(A) MASP1-encoded isoforms MASP-1, MASP-3, and MAp44, indicating that the N-terminal amino acids are identical but the trypsin-like serine protease domains differ between MASP-1 and MASP-3 and that MAp44 does not contain a serine protease domain. The p.W290X alteration is present in all three isoforms; p.G687R is present only in MASP-3.
(B) Conservation of residue G687 (arrow) in MASP-3 among vertebrates.
(C) Conservation of residue G687 (arrow) in the predicted substrate-binding site of various trypsin-like serine proteases. Alignment was obtained through the NCBI protein database.
To further understand the physiological significance of the p.G687R alteration, we generated 3D atomic models of the serine protease domain of human MASP-3 on the basis of amino acid sequence homology with the known crystal structure of human MASP-1 (PDB# 3GOV; Figure 3).13 Homology modeling was carried out with MODELER software.14,15 The serine protease domains of MASP-1 and MASP-3 share 35% amino acid sequence identity in the core catalytic regions. Thus, the atomic models of the wild-type and the G687R-variant serine protease domains of MASP-3 would be expected to adopt 3D folds fairly similar to the template structure. For each case, 100 atomic models were calculated, and the structures with the lowest energies, as judged by the MODELER Objective Function, were selected for further analysis. The atomic models were rendered with RIBBONS.16 The modeled structures display the canonical serine protease fold comprised of two six-stranded β-barrels tightly packed against each other. The enzyme active site contains the catalytic triad H497/D553/S664, characteristic of all serine proteases and located at the junction of the two β-barrels within a deep cleft. Residue G687 is located within the active site close to the junction of the two β-barrels and in close proximity to D658, an acidic residue key to proper organization of the active site and to stabilization of incoming protein substrates.13,17 Given the close proximity of D658 to G687, the p.G687R variant would likely result in the formation of an intermolecular salt bridge through ion pairing of the carboxylic side chain group of D658 and the guanidino side chain moiety of p.G687R. A D658-G687R salt bridge could lead to disorganization and distortion of the active site, with misalignment and disorientation of the catalytic triad H497/D553/S664. In consequence, both the protein recognition function and catalytic function of the serine protease domain of MASP-3 could be compromised. Similar internal salt bridges within the active sites of MASP-1 and complement factor D are known to render them catalytically inactive.12,18–20 Although it remains to be tested experimentally, the atomic model suggests that the p.G687R variant in MASP-3 may be catalytically lethal.
Figure 3.
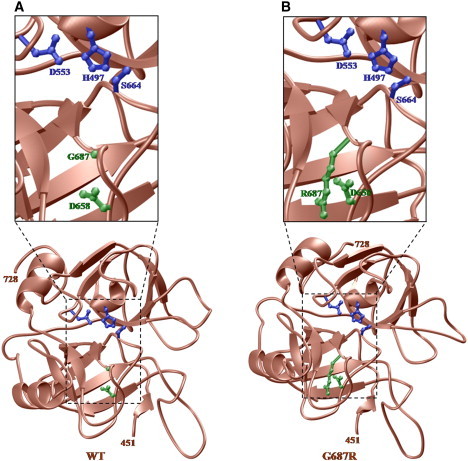
Ribbon Representation of 3D Atomic Models of the Serine Protease Domain of Human MASP-3
(A and B) Wild-type (A) and mutant (B) G687R. The serine protease canonical fold is comprised of two six-stranded β-barrels packed against each other (brown). Side chains of the catalytic triad H497/D553/S664 within the enzyme active site are shown in blue; side chains of D658 and G687/R687 are in green. Amino acid residues 451–728 are the boundaries of the serine protease domain of human MASP-3.
MASP-1 and MASP-3 share a domain structure with C1r and C1s, two serine proteases of the classical complement pathway. C1s selectively cleaves insulin-like growth factor binding protein (IGFBP-5), which is thought to regulate physiologic processes in kidney, bone, and elsewhere. IGFBP-5 also binds to IGFs, such as IGF-I, in extracellular space and, in this way, regulates the interaction of IGFs with their receptors on cell surfaces.21 Although binding of IGFBPs to IGFs generally leads to downmodulation of IGF signaling, it can also stabilize IGF, resulting in a net increase in signaling.22 It has been suggested that MASP-3 processes IGFBP-5.23 Interestingly, IGFBP-5 is expressed in the craniofacial region during mouse embryonic development, and IGFBP-5 mRNA was found in mouse epithelium at early stages, corresponding to completion of neural crest migration. Also, IGFBP5-expressing cells were present around developing cartilage and bone during differentiation of craniofacial mesenchyme into skeletal tissue. Overexpression of IGFBP-5 in mice causes decreased ossification or deletion of head bones.22 Finally, IGFBP-5 is also thought to promote muscle cell differentiation by switching on the IGF-II autoregulation loop.24 In total, these results suggest that processing of IGFBP-5 by MASP-3 may play a critical role in IGF availability during craniofacial and muscle development.
Involvement of MASP-3 in the IGF signaling pathway could explain the dysmorphic facial features, skeletal abnormalities, and defects in muscular development that are present in our patients. It will be informative to learn the consequences to embryonic development of different mutations in MASP1. Sequencing MASP1 in more patients with these phenotypes will also illuminate both locus and allelic heterogeneity—in particular, whether Michels, Malpuech, OSA, and Carnevale syndromes are different genetic entities, represent different phenotypic manifestations of a spectrum of mutations in the same gene, or both.
Acknowledgments
The authors are grateful to Fatos Yalcinkaya and Suat Fitoz at Ankara University School of Medicine for clinical and radiological evaluation and to Ming K. Lee and Anne M. Thornton at the University of Washington for advice and assistance. This work was supported in part by National Institutes of Health grants R01DC009645, R01DC005641, and R01GM083897, with a supplement from the American Recovery and Reinvestment Act.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
International HapMap Project, http://www.hapmap.org/
NCBI Protein Database, http://www.ncbi.nlm.nih.gov/protein/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Carnevale F., Krajewska G., Fischetto R., Greco M.G., Bonvino A. Ptosis of eyelids, strabismus, diastasis recti, hip defect, cryptorchidism, and developmental delay in two sibs. Am. J. Med. Genet. 1989;33:186–189. doi: 10.1002/ajmg.1320330210. [DOI] [PubMed] [Google Scholar]
- 2.Mingarelli R., Castriota Scanderbeg A., Dallapiccola B. Two sisters with a syndrome of ocular, skeletal, and abdominal abnormalities (OSA syndrome) J. Med. Genet. 1996;33:884–886. doi: 10.1136/jmg.33.10.884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Michels V.V., Hittner H.M., Beaudet A.L. A clefting syndrome with ocular anterior chamber defect and lid anomalies. J. Pediatr. 1978;93:444–446. doi: 10.1016/s0022-3476(78)81154-8. [DOI] [PubMed] [Google Scholar]
- 4.Malpuech G., Demeocq F., Palcoux J.B., Vanlieferinghen P. A previously undescribed autosomal recessive multiple congenital anomalies/mental retardation (MCA/MR) syndrome with growth failure, lip/palate cleft(s), and urogenital anomalies. Am. J. Med. Genet. 1983;16:475–480. doi: 10.1002/ajmg.1320160405. [DOI] [PubMed] [Google Scholar]
- 5.Titomanlio L., Bennaceur S., Bremond-Gignac D., Baumann C., Dupuy O., Verloes A. Michels syndrome, Carnevale syndrome, OSA syndrome, and Malpuech syndrome: Variable expression of a single disorder (3MC syndrome)? Am. J. Med. Genet. A. 2005;137A:332–335. doi: 10.1002/ajmg.a.30878. [DOI] [PubMed] [Google Scholar]
- 6.Al Kaissi A., Klaushofer K., Safi H., Chehida F.B., Ghachem M.B., Chaabounni M., Hennekam R.C. Asymmetrical skull, ptosis, hypertelorism, high nasal bridge, clefting, umbilical anomalies, and skeletal anomalies in sibs: Is Carnevale syndrome a separate entity? Am. J. Med. Genet. A. 2007;143:349–354. doi: 10.1002/ajmg.a.31610. [DOI] [PubMed] [Google Scholar]
- 7.Leal G.F., Silva E.O., Duarte A.R., Campos J.F. Blepharophimosis, blepharoptosis, defects of the anterior chamber of the eye, caudal appendage, radioulnar synostosis, hearing loss and umbilical anomalies in sibs: 3MC syndrome? Am. J. Med. Genet. A. 2008;146A:1059–1062. doi: 10.1002/ajmg.a.32252. [DOI] [PubMed] [Google Scholar]
- 8.Hall J.G., Allanson J.E., Gripp K., Slavotinek A.M. Second Edition. Oxford University Press; New York: 2007. Handbook of Physical Measurements. pp. 127. [Google Scholar]
- 9.Sirmaci A., Erbek S., Price J., Huang M., Duman D., Cengiz F.B., Bademci G., Tokgöz-Yilmaz S., Hişmi B., Ozdağ H. A truncating mutation in SERPINB6 is associated with autosomal-recessive nonsyndromic sensorineural hearing loss. Am. J. Hum. Genet. 2010;86:797–804. doi: 10.1016/j.ajhg.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walsh T., Shahin H., Elkan-Miller T., Lee M.K., Thornton A.M., Roeb W., Abu Rayyan A., Loulus S., Avraham K.B., King M.C., Kanaan M. Whole exome sequencing and homozygosity mapping identify mutation in the cell polarity protein GPSM2 as the cause of nonsyndromic hearing loss DFNB82. Am. J. Hum. Genet. 2010;87:90–94. doi: 10.1016/j.ajhg.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matsushita M., Fujita T. Activation of the classical complement pathway by mannose-binding protein in association with a novel C1s-like serine protease. J. Exp. Med. 1992;176:1497–1502. doi: 10.1084/jem.176.6.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dahl M.R., Thiel S., Matsushita M., Fujita T., Willis A.C., Christensen T., Vorup-Jensen T., Jensenius J.C. MASP-3 and its association with distinct complexes of the mannan-binding lectin complement activation pathway. Immunity. 2001;15:127–135. doi: 10.1016/s1074-7613(01)00161-3. [DOI] [PubMed] [Google Scholar]
- 13.Dobó J., Harmat V., Beinrohr L., Sebestyén E., Závodszky P., Gál P. MASP-1, a promiscuous complement protease: Structure of its catalytic region reveals the basis of its broad specificity. J. Immunol. 2009;183:1207–1214. doi: 10.4049/jimmunol.0901141. [DOI] [PubMed] [Google Scholar]
- 14.Martí-Renom M.A., Stuart A.C., Fiser A., Sánchez R., Melo F., Sali A. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- 15.John B., Sali A. Comparative protein structure modeling by iterative alignment, model building and model assessment. Nucleic Acids Res. 2003;31:3982–3992. doi: 10.1093/nar/gkg460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carson M. Ribbons 2.0. J. Appl. Cryst. 1991;24:958–961. [Google Scholar]
- 17.Harmat V., Gál P., Kardos J., Szilágyi K., Ambrus G., Végh B., Náray-Szabó G., Závodszky P. The structure of MBL-associated serine protease-2 reveals that identical substrate specificities of C1s and MASP-2 are realized through different sets of enzyme-substrate interactions. J. Mol. Biol. 2004;342:1533–1546. doi: 10.1016/j.jmb.2004.07.014. [DOI] [PubMed] [Google Scholar]
- 18.Narayana S.V., Carson M., el-Kabbani O., Kilpatrick J.M., Moore D., Chen X., Bugg C.E., Volanakis J.E., DeLucas L.J. Structure of human factor D. A complement system protein at 2.0 A resolution. J. Mol. Biol. 1994;235:695–708. doi: 10.1006/jmbi.1994.1021. [DOI] [PubMed] [Google Scholar]
- 19.Ambrus G., Gál P., Kojima M., Szilágyi K., Balczer J., Antal J., Gráf L., Laich A., Moffatt B.E., Schwaeble W. Natural substrates and inhibitors of mannan-binding lectin-associated serine protease-1 and -2: A study on recombinant catalytic fragments. J. Immunol. 2003;170:1374–1382. doi: 10.4049/jimmunol.170.3.1374. [DOI] [PubMed] [Google Scholar]
- 20.Rossi V., Cseh S., Bally I., Thielens N.M., Jensenius J.C., Arlaud G.J. Substrate specificities of recombinant mannan-binding lectin-associated serine proteases-1 and -2. J. Biol. Chem. 2001;276:40880–40887. doi: 10.1074/jbc.M105934200. [DOI] [PubMed] [Google Scholar]
- 21.Salih D.A., Tripathi G., Holding C., Szestak T.A., Gonzalez M.I., Carter E.J., Cobb L.J., Eisemann J.E., Pell J.M. Insulin-like growth factor-binding protein 5 (Igfbp5) compromises survival, growth, muscle development, and fertility in mice. Proc. Natl. Acad. Sci. USA. 2004;101:4314–4319. doi: 10.1073/pnas.0400230101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bobola N., Engist B. IGFBP5 is a potential regulator of craniofacial skeletogenesis. Genesis. 2008;46:52–59. doi: 10.1002/dvg.20360. [DOI] [PubMed] [Google Scholar]
- 23.Cortesio C.L., Jiang W. Mannan-binding lectin-associated serine protease 3 cleaves synthetic peptides and insulin-like growth factor-binding protein 5. Arch. Biochem. Biophys. 2006;449:164–170. doi: 10.1016/j.abb.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 24.Ren H., Yin P., Duan C. IGFBP-5 regulates muscle cell differentiation by binding to IGF-II and switching on the IGF-II auto-regulation loop. J. Cell Biol. 2008;182:979–991. doi: 10.1083/jcb.200712110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cunniff C., Jones K.L. Craniosynostosis and lid anomalies: Report of a girl with Michels syndrome. Am. J. Med. Genet. 1990;37:28–30. doi: 10.1002/ajmg.1320370108. [DOI] [PubMed] [Google Scholar]
- 26.De La Paz M.A., Lewis R.A., Patrinely J.R., Merin L., Greenberg F. A sibship with unusual anomalies of the eye and skeleton (Michels' syndrome) Am. J. Ophthalmol. 1991;112:572–580. doi: 10.1016/s0002-9394(14)76861-8. [DOI] [PubMed] [Google Scholar]
- 27.Guion-Almeida M.L., Rodini E.S. Michels syndrome in a Brazilian girl born to consanguineous parents. Am. J. Med. Genet. 1995;57:377–379. doi: 10.1002/ajmg.1320570302. [DOI] [PubMed] [Google Scholar]
- 28.Leal G.F., Baptista E.V. Three additional cases of the Michels syndrome. Am. J. Med. Genet. A. 2007;143A:2747–2750. doi: 10.1002/ajmg.a.32029. [DOI] [PubMed] [Google Scholar]
- 29.Chinen Y., Naritomi K. Malpuech facial clefting syndrome in a Japanese boy with cardiac defects. Jpn. J. Hum. Genet. 1995;40:335–338. doi: 10.1007/BF01900601. [DOI] [PubMed] [Google Scholar]
- 30.Guion-Almeida M.L. Apparent Malpuech syndrome: Report on three Brazilian patients with additional signs. Am. J. Med. Genet. 1995;58:13–17. doi: 10.1002/ajmg.1320580104. [DOI] [PubMed] [Google Scholar]
- 31.Crisponi G., Marras A.R., Corrias A. Two sibs with Malpuech syndrome. Am. J. Med. Genet. 1999;86:294–299. doi: 10.1002/(sici)1096-8628(19990917)86:3<294::aid-ajmg20>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 32.Kerstjens-Frederikse W.S., Brunner H.G., van Dael C.M., van Essen A.J. Malpuech syndrome: Three patients and a review. Am. J. Med. Genet. A. 2005;134:450–453. doi: 10.1002/ajmg.a.30662. [DOI] [PubMed] [Google Scholar]
- 33.Priolo M., Ciccone R., Bova I., Campolo G., Laganà C., Zuffardi O. Malpuech syndrome: Broadening the clinical spectrum and molecular analysis by array-CGH. Eur. J. Med. Genet. 2007;50:139–143. doi: 10.1016/j.ejmg.2006.10.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.