

Abstract
Autism spectrum disorders (ASD) and schizophrenia are neurodevelopmental disorders for which recent evidence indicates an important etiologic role for rare copy number variants (CNVs) and suggests common genetic mechanisms. We performed cytogenomic array analysis in a discovery sample of patients with neurodevelopmental disorders referred for clinical testing. We detected a recurrent 1.4 Mb deletion at 17q12, which harbors HNF1B, the gene responsible for renal cysts and diabetes syndrome (RCAD), in 18/15,749 patients, including several with ASD, but 0/4,519 controls. We identified additional shared phenotypic features among nine patients available for clinical assessment, including macrocephaly, characteristic facial features, renal anomalies, and neurocognitive impairments. In a large follow-up sample, the same deletion was identified in 2/1,182 ASD/neurocognitive impairment and in 4/6,340 schizophrenia patients, but in 0/47,929 controls (corrected p = 7.37 × 10−5). These data demonstrate that deletion 17q12 is a recurrent, pathogenic CNV that confers a very high risk for ASD and schizophrenia and show that one or more of the 15 genes in the deleted interval is dosage sensitive and essential for normal brain development and function. In addition, the phenotypic features of patients with this CNV are consistent with a contiguous gene syndrome that extends beyond RCAD, which is caused by HNF1B mutations only.
Introduction
Autism is the most severe end of a spectrum of neurodevelopmental conditions collectively known as autism spectrum disorders (ASD [MIM 209850]), which also include Asperger syndrome and pervasive developmental disorder not otherwise specified (PDD-NOS). Autism is characterized by impairments in social interaction, deficits in communication, and repetitive and stereotypical patterns of interests and behavior that manifest before 3 years of age. Affecting males four times as often as females, the prevalence of autism is considered to be 1 in 500 individuals, rising to 1 in 110 when all ASD are included.1
Schizophrenia (MIM 181500) is also a neurodevelopmental disorder involving a broad range of social, cognitive, and perceptual functions. With a prevalence of 1 in 100 to 1 in 200, the disorder typically comes to clinical attention when frank “positive” psychotic symptoms (hallucinations, delusions, disordered thinking, and bizarre behavior) or “negative” symptoms (profound social withdrawal, anhedonia, or apathy) emerge during late adolescence or early adulthood. Males are affected 1.4 times as often as females, and the age of onset is typically somewhat earlier, and course of illness more severe, in males.2
The genetic overlap between ASD and schizophrenia has been the focus of several recent studies. Both conditions are strongly genetic, as evidenced by a heritability higher than 80% for each of these disorders.3 Furthermore, despite the etiological and phenotypic variability of autism and schizophrenia, the same structural variants are being identified in both diagnoses.3,4 However, none of the individual mutations and copy number variants (CNVs) identified to date in both disorders account for more than 1% to 2% of the cases.5,6 This substantial etiologic heterogeneity resembles that observed in intellectual disability, where many different rare mutations account for the majority of cases with a known genetic cause.7
One approach to addressing this heterogeneity is to analyze subgroups of individuals with specific recurrent or overlapping CNVs to identify genotype-phenotype correlations. Although individually rare, these genomic imbalances are invaluable tools for uncovering new candidate genes and shared molecular pathways that could provide a better understanding of the pathophysiology of neurodevelopmental disorders, such as ASD and schizophrenia.8,9
Recurrent CNVs, which occur through nonallelic homologous recombination mediated by segmental duplications, are frequently identified via clinical genetic testing.10 The expansion of cytogenomic array technologies has led to greater recognition of this type of imbalance over the past few years, and in turn the discovery of several new genomic disorders caused by CNVs that were previously undetected, with ASD or schizophrenia as part of the phenotype. Recurrent CNVs associated with both diagnoses include deletions in 1q21 (MIM 612474),11,12 3q29 (MIM 609425),13,14 15q13.3 (MIM 612001),12,15,16 and 22q11.2 (MIM 188400),12,17 as well as duplications in 16p11.2 (MIM 611913)18,19 and 16p13.1.20,21 As more arrays are performed and collaboration among genetic laboratories is fostered, more CNVs that cause or predispose to neurodevelopmental or psychiatric disorders will be identified.
Here we describe the association between a recurrent deletion at 17q12, which harbors HNF1B (hepatocyte nuclear factor 1 homeobox B [MIM 189907]), the gene responsible for renal cysts and diabetes syndrome (RCAD [MIM 137920]), and both ASD and schizophrenia, giving further support to a shared genetic etiology in a subset of cases and pointing to one or more of the 15 genes in the deleted interval as strong candidates for neurodevelopmental and psychiatric disorders.
Subjects and Methods
International Standards for Cytogenomic Arrays Consortium
Subjects
We used whole-genome cytogenomic array analysis to search for pathogenic CNVs in a discovery sample from the International Standards for Cytogenomic Arrays (ISCA) Consortium, an international collaboration among clinical cytogenomic laboratories whose purpose is to standardize cytogenomic array testing and facilitate public data sharing.22 We assessed 15,749 patients referred for clinical genetic testing because of ASD, developmental delay, and/or intellectual disability. There was a referring diagnosis of ASD in approximately 15%–20% of the cases, in line with reports on similar populations.22 We analyzed data from seven ISCA laboratories: Emory University, GeneDx, University of Nebraska Medical Center, ARUP Laboratories, Mayo Clinic, Michigan Medical Genetics Laboratories, and Wessex Regional Genetics Laboratory. Research-based informed consent was not required because results were anonymous. We carried out additional clinical assessments on nine patients with 17q12 deletions identified by Emory University and GeneDx, from whom we obtained written informed consent. All procedures followed are in accordance with the ethical standards of the Institutional Review Board at Emory University.
Cytogenomic Array and Confirmation of Findings
We performed cytogenomic array testing according to the manufacturer's protocol, via custom-designed oligonucleotide arrays on 4×44K, 2×105K, and 4×180K platforms (Agilent Technologies, Santa Clara, CA). These designs combine both backbone coverage across the whole genome (probes spaced every 25–75 kb, depending on the design) and targeted coverage of clinically relevant regions (a minimum of 10–20 probes per gene or region, depending on the design), as described elsewhere.23
We used fluorescence in situ hybridization (FISH) and quantitative PCR (qPCR) to confirm all 17q12 deletions and to test for parental inheritance. For FISH analyses, we used homebrew FISH probes (BACs; bacterial artificial chromosome clones) as previously described.23 For confirmation by qPCR, we assessed gene dosage for HNF1B by genomic real-time PCR by using TaqMan probe chemistry and the Universal ProbeLibrary system (Roche Applied Science, Indianapolis, IN) with SOD1 (MIM 147450) as a control.
Clinical Assessments
All nine patients for whom we obtained written informed consent had a comprehensive medical and behavioral evaluation, performed by a clinical geneticist and either a developmental pediatrician, a psychiatrist, or a clinical psychologist. Previous medical records from all nine patients were reviewed. We used DSM-IV criteria to assess for ASD. When possible, additional supporting tests were performed, including the Autism Diagnostic Interview-Revised (ADI-R), the Autism Diagnostic Observation Schedule (ADOS), the Childhood Autism Rating Scale (CARS), and the Quantitative Checklist for Autism in Toddlers (Q-CHAT).
SGENE Consortium, deCODE Genetics, and GeneSTAR
Subjects
We included 4,173 patients with schizophrenia, 376 patients with ASD and/or neurocognitive impairment, and 43,899 ethnically matched control individuals. All subjects gave written informed consent. The populations of patients and controls we analyzed and their sources are summarized below (Table 2).
Table 2.
Statistical Analyses of 17q12 Deletions in ASD/Neurodevelopmental Disorders and Schizophrenia
|
Uncorrected |
Cochran-Mantel-Haenszel |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample | Phenotype | Case Sample Source | Control Sample Source | Cases | Controls | OR | 95% CI Lower | 95% CI Upper | p | OR | 95% CI Lower | 95% CI Upper | p |
| Discovery | ASD/neurodevelopmental disorders | ISCA Consortium | literature (43,44) | 18/15,749 | 0/4,519 | ∞ | 1.26 | ∞ | 1.96 × 10−2 | ||||
| Follow-up | ASD/neurodevelopmental disorders | SSC Chinese | SSC Chinese | 0/31 | 0/51 | ||||||||
| SSC European American | SSC and GeneSTAR European American | 1/728 | 0/3,465 | ||||||||||
| SSC Japanese | SSC Japanese | 0/6 | 0/31 | ||||||||||
| SSC African American | SSC and GeneSTAR African American | 0/41 | 0/1,306 | ||||||||||
| Iceland | Iceland | 1/376 | 0/33,645 | ||||||||||
| Overall | 2/1,182 | 0/38,498 | ∞ | 6.12 | ∞ | 8.87 × 10−4 | ∞ | 4.73 | ∞ | 1.92 × 10−3 | |||
| schizophrenia | SGENE Denmark | SGENE Denmark | 0/565 | 0/493 | |||||||||
| SGENE Scotland | SGENE Scotland | 0/648 | 0/663 | ||||||||||
| SGENE Germany | SGENE Germany | 1/1,086 | 0/1435 | ||||||||||
| SGENE UK | SGENE UK | 1/91 | 0/83 | ||||||||||
| SGENE Italy | SGENE Italy | 0/84 | 0/86 | ||||||||||
| SGENE Finland A | SGENE Finland A | 0/126 | 0/49 | ||||||||||
| SGENE Finland B | SGENE Finland B | 0/61 | 0/146 | ||||||||||
| SGENE Holland | SGENE Holland | 1/613 | 0/3,687 | ||||||||||
| SGENE Norway | SGENE Norway | 0/274 | 0/371 | ||||||||||
| SGENE Iceland | SGENE Iceland | 0/625 | 0/33,645 | ||||||||||
| GAIN African American | GAIN African American | 0/952 | 0/978 | ||||||||||
| GAIN European American | GAIN European American | 1/1,215 | 0/1,440 | ||||||||||
| Overall | 4/6,340 | 0/43,076 | ∞ | 4.49 | ∞ | 2.71 × 10−4 | ∞ | 1.25 | ∞ | 1.47 × 10−2 | |||
| ASD/neurodevelopmental disorders and schizophrenia | Combined follow-up samples | 6/7,522 | 0/47,929 | ∞ | 7.51 | ∞ | 6.22 × 10−6 | ∞ | 5.99 | ∞ | 7.37 × 10−5 | ||
| Total discovery and follow up | 24/23,271 | 0/52,448 | ∞ | 13.58 | ∞ | 5 × 10−13 | |||||||
Iceland
The Icelandic sample consisted of 625 cases with schizophrenia, 376 cases with developmental delay (autistic spectrum disorders, intellectual disability, or slow expressive language development [SELD]), and 33,645 controls. Patients and controls were all Icelandic and were recruited from all over Iceland. Diagnoses were assigned according to Research Diagnostic Criteria (RDC24) through the use of the Schedule for Affective Disorders and Schizophrenia Lifetime Version (SADS-L25). The 33,645 controls were recruited as a part of various genetic programs at deCODE and were not screened for psychiatric disorders. Subjects with known attention deficit and hyperactive disorder, dyslexia, and bipolar were omitted from the control group. Furthermore, parents of subjects with schizophrenia and developmental delay were also omitted from the control group. Apart from this, the control group is in line with what has been reported previously.26 All subjects gave a written informed consent for their participation.
Finland
The Finnish genome-wide typed sample consisted of 187 schizophrenia patients and 195 controls that had no medical history of schizophrenia. The controls were selected from among participants of the Health 2000 survey27,28 and were matched with the cases with regard to age and sex. The schizophrenia patients were drawn from a nationwide collection of families with schizophrenia spectrum disorders. Two independent psychiatrists blind to family structures made a consensus diagnosis to give best-estimate lifetime diagnoses according to the criteria of Diagnostic and Statistical Manual of Mental Disorders, 4th edition (DSM-IV).29–31 Approximately half of the sample originated from an internal isolate of Finland (Kuusamo-Finland A) having a 3.0% age-corrected lifetime risk for schizophrenia compared to 1.1% in the general population.30
Scotland
The Scottish sample was comprised of 648 schizophrenia cases and 663 controls. All participants self-identified as born in the British Isles (95% in Scotland). All cases met DSM-IV and International Classification of Diseases, 10th revision (ICD-10) criteria for schizophrenia. Diagnosis was made by the Operational Criteria Checklist (OPCRIT32). Controls were volunteers recruited through general practices in Scotland. Practice lists were screened for potentially suitable volunteers by age and sex and by exclusion of subjects with major mental illness or use of antipsychotic medication.
England
Samples from the English subjects (n = 91) were drawn from the Maudsley Family Study of psychosis,33 the psychosis twin study,34 and the genetics and psychosis (GAP) study for which cases were derived from the Camberwell case register.35 All controls were unrelated European whites (n = 83). Patients were interviewed with the SADS-L24 or the Item Group Checklist (IGC) of the Schedule for Clinical Assessment in Neuropsychiatry (SCAN36) and only patients with an ICD-10 research diagnosis of schizophrenia were finally included as cases. The study received approval from the Ethics Committee of South London and the Maudsley Trust, and after complete description of the study to the participants, written informed consent was obtained.
Italy
Diagnosis of the 84 Italian subjects was identical to that for the GAP sample (see England), via the IGC. Patients with a diagnosis of psychotic disorders (ICD-10, F20-F25) attending the South Verona CMHS were identified from the South Verona Psychiatric Case Register, and cases with ICD-10 research diagnosis of schizophrenia were finally included. The controls (n = 86) were unrelated volunteers randomly selected from the general population of South Verona. The study received ethical approval and after complete description of the study to the participants, written informed consent was obtained.
Germany
The German sample consisted of 1086 schizophrenia cases and 1435 controls. All cases and controls were whites of German descent. All cases diagnosed according to DSM-IV criteria based on SCID,37 SADS-L,24 and OPCRIT. The controls were unrelated volunteers from the general population. Ethical approval was obtained from the local Ethics Committees, and all participants gave written informed consent.
The Netherlands
The Dutch sample consisted of 614 patients and 469 controls obtained from the University Medical Center Utrecht and an additional 3218 control individuals collected by the Radboud University Nijmegen Medical Centre (RUNMC) in the Netherlands. In-patients and out-patients were recruited from different psychiatric hospitals and institutions throughout the Netherlands, coordinated via academic hospitals in Amsterdam, Groningen, Maastricht, and Utrecht. Detailed medical and psychiatric histories were collected, including the Comprehensive Assessment of Symptoms and History (CASH), an instrument for assessing diagnosis and psychopathology. Only patients with a DSM-IV diagnosis of schizophrenia were included as cases. The University Medical Center Utrecht controls were volunteers and were free of any psychiatric history. The RUNMC controls were collected as cancer and control samples. As for SGENE-plus, STRUCTURE runs were carried out on the genome-wide typed samples, and individuals with less than 90% estimated European white ancestry were removed. Ethical approval was obtained from the local Ethics Committees (including the Institutional Review Board of Radboud University) and all participants gave written informed consent.
Denmark
The Danish sample included 565 patients who had been recruited to the Danish Psychiatric Biobank from the psychiatric departments at the six hospitals in the Copenhagen region. All patients had been clinically diagnosed with schizophrenia according to ICD-10 (F20) without ever having received a diagnosis of mania or bipolar illness (F30-31). An experienced research and consultant psychiatrist verified high reliability of the clinical diagnoses via OPCRIT. Healthy control subjects (n = 888) were recruited through the Danish Blood Donor Corps in the Copenhagen area. Apparent behavioral abnormality was an exclusion criterion and all individuals stated that they felt completely healthy and were able to discuss health-related issues with a physician. An additional 493 population control samples from the Copenhagen area were recruited by the Danish Headache Center. The Danish Scientific Committees and the Danish Data Protection Agency approved the study and all the patients had given written informed consent prior to inclusion into the project.
Norway
The Norwegian sample included 274 schizophrenia patients who had been recruited to the TOP (Tematisk område psykoser) study at Oslo University Hospital-Ulleval from all the psychiatric hospitals in the Oslo area. The patients were diagnosed according to the SCID as having schizophrenia. The healthy control subjects (n = 371) were randomly selected from statistical records of persons from the same catchment area as the patient groups. Only subjects born in Norway, all of European white origin, were contacted by letter and invited to participate. All subjects had given written informed consent prior to inclusion into the project and the Norwegian Scientific-Ethical Committee and the Norwegian Data Protection Agency approved the study.
GeneSTAR
We analyzed copy number data from 3241 control individuals from USA participating in genetic studies carried out by deCODE genetics (2021 whites and 1220 African Americans).
Genetic Analyses
All samples, except the Norwegian, were genotyped with Illumina (Illumina Inc., San Diego, CA) SNP microarrays (317K, 370K, 550K, 1M) as previously described.38 PennCNV39 was used for CNV detection, and only the subset of SNPs present on all the Illumina microarrays used in this study were taken into account for CNV detection (a total of 305,381 SNPs). In this subset of SNPs, 137 are within the 17q12 deletion. The Norwegian sample was genotyped via the Affymetrix 6.0 SNP microarrays (Affymetrix, Santa Clara, CA). PennCNV39 was used to analyze copy number variants in the full Affymetrix array and then the 17q12 region was manually inspected for large deletions.
Simons Foundation Autism Research Initiative Simplex Collection
Subjects
Genetic dosage information was available for 806 individuals with ASD (698 males and 108 females). All the patients came from USA, from families with a single individual affected with ASD. Additionally, the 1612 unaffected parents of these individuals were included as control samples, as well as matched control individuals from GeneSTAR. All subjects gave written informed consent.
Genetic Analyses
Samples were run on Illumina Human 1M and Human 1M Duo microarrays (Illumina Inc.) and analyzed by the SSC (Simons Foundation Autism Research Initiative Simplex Collection) Genetics Consortium (SSCGC). The microarray data were reclustered in Beadstudio (Illumina Inc.) against 200 unaffected parents from the SSC; genotypes and intensity data were exported for analysis. Reported family structure was checked by assessing gender, mendelian errors, and identity-by-descent via PLINK v1.06.40
CNV detection and quality control was performed with CNVision v1.0, a CNV analysis package that runs and merges the results from three detection algorithms: PennCNV,39 QuantiSNP v1.1,41 and GNOSIS (a CNV detection algorithm within CNVision). The inheritance of CNVs was determined by comparing CNVs in offspring to the CNVs in their parents and re-examining parental genotypes and intensity data within each detected CNV for evidence of an inherited CNV.
Genetic Association Information Network Collection
Subjects and Genetic Analyses
We analyzed raw data for CNV analysis, based on the Affymetrix 6.0 array platform, from 2167 schizophrenia cases and 2418 controls. Normalization and log ratio data calculation was obtained with the Affymetrix power tools (APT) software (Affymetrix). Log2 ratio data for chromosome 17 was extracted and analyzed with the BEAST algorithm (A.S. Allen, M.I. Ikeda, I. Morna, J.G.M., G.A. Satten, and S.T.W., unpublished data). All subjects gave written informed consent.
Statistical Analyses
All p values and odds ratios were calculated via Fisher's exact test in R. We used the Cochran-Mantel-Haenszel model42 to correct for ethnic origin when possible.
Results
Discovery Sample
17q12 Deletion Identified in a Clinical Testing Setting
We identified 18 patients with the same 17q12 deletion (13 males and 5 females) out of the 15,749 we assessed within the ISCA consortium (dbVar; accession number NSTD37). We confirmed all 18 deletions with FISH or qPCR. This CNV was absent from 4519 previously published control individuals43,44 and from the Database of Genomic Variants,45 which serves as a repository for CNVs in several control populations. We found the reciprocal 17q12 duplication, which has been associated with intellectual disability and epilepsy,46,47 in 21 cases and 2 controls within the same population. Additionally, this duplication was inherited in 7 out of the 11 cases for whom we had parental copy number information. For this study, we chose to focus on the 17q12 deletion given that among this discovery sample, we observed an empirical association between this recurrent CNV and autism as a referral diagnosis (4 out of the 16 patients with referring diagnosis information were sent for ASD). To confirm this observation and to comprehensively assess the clinical spectrum associated with this CNV, we obtained additional clinical information on eight individuals and the affected mother of one of them, who also had the 17q12 deletion. We performed high-resolution cytogenomic array on these nine patients (eight probands and the affected mother) by using a custom 180K array design to standardize the array results of these patients and to exclude the presence of smaller genomic anomalies elsewhere. We confirmed a 1.4 Mb region deleted in all patients (Figure 1) (chr17:31,893,783-33,277,865; hg18 genome assembly) containing 15 genes (AATF [MIM 608463], ACACA [MIM 200350], C17orf78, DDX52 [MIM 612500], DHRS11, DUSP14 [MIM 606618], GGNBP2 [MIM 612275], HNF1B, LHX1 [MIM 601999], MRM1, MYO19, PIGW [MIM 610275], SYNRG [MIM 607291], TADA2A [MIM 602276], ZNHIT3 [MIM 604500]) consistent with previous reports.46 The breakpoints of each of the deletions lie within a cluster of highly homologous segmental duplications that range from 300 kilobases (kb) to 400 kb in size and vary in copy number, which hampered the precise identification of the boundaries of the deletion. At the 75 kb backbone resolution of our genome-wide 180K array, we detected no other pathogenic genomic rearrangements22 in any of these individuals. The deletion was de novo in seven out of the eight cases we studied in detail. In one case, the deletion was inherited from an affected mother who had polycystic kidneys (MIM 173900) and diabetes mellitus (MIM 125853), features previously observed in patients with this CNV.46 Remarkably, she also had a long history of bipolar disorder (MIM 125480).
Figure 1.
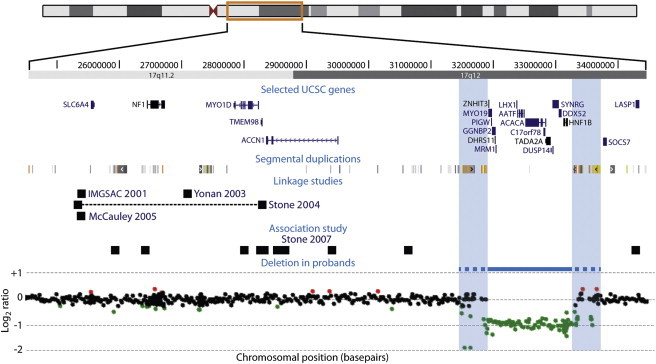
17q12 Region
Chromosome 17 schematic showing a close-up of 17q11.2-17q12, including selected genes and segmental duplications in the region, along with the results of four different autism linkage studies51–54 and one association study on male-only autism families55 (black squares [in Stone et al.,54 black squares correspond to markers flanking the linkage peak and are connected by a dotted horizontal black line]). The recurrent 17q12 deletion found in our patients is depicted as a horizontal blue line, flanked by segmental duplications (blue shaded rectangles). The 180K cytogenomic array results of one of the patients are shown below.
These nine patients shared a number of consistent phenotypic features (Table 1 and Figure 2); a more extensive clinical comparative description of patients is available online (Table S1). Macrocephaly, defined for this study as head circumference over the 90th percentile, or relative macrocephaly and dolichocephaly were observed in most of the patients. Similar mild facial dysmorphic features were present in all cases (Figure 2). Genitourinary tract anomalies were present in most of the probands and included prenatal echogenic renal calyces, prenatal hydronephrosis, unilateral and bilateral cystic kidneys, urethral stenosis, and uterus didelphis. Only the eldest of the patients, a 37-year-old woman, had diabetes. In addition, recurrent infections of the ear, upper respiratory system, and urinary tract, scoliosis, and hypermetropia were each observed in more than two cases. One of the patients (Case 6) had short stature.
Table 1.
Neurocognitive and Behavioral Phenotype of Nine Patients with 17q12 Deletions
| Case 1 | Case 2 | Case 3 | Case 4 | Case 5 | Case 6 | Case 7 | Case 8 | Case 9 | Total | |
|---|---|---|---|---|---|---|---|---|---|---|
| Inheritance | de novo | de novo | de novo | de novo | U | maternal (son of Case 5) | de novo | de novo | de novo | |
| Sex | female | male | male | male | female | male | female | male | male | |
| Current age | 19 y | 4 y | 7 y | 12 y | 37 y | 1 y, 9 months | 1 y, 10 months | 10 y | 22 y | |
| Autism or autistic features | - | + | + | + | - | + | - | + | + | 6/9 in total (66%); 6/6 males (100%); 0/3 females (0%) |
| Deficits in coordination and motor skills | U | + | + | + | U | + | + | + | + | 7/7 (100%) |
| Intellectual impairment | - | + | + | + | + | + | + | + | + | 8/9 (89%) |
| Language impairment | - | + | + | + | - | + | + | + | - | 6/9 (67%) |
| Anxiety | - | - | + | + | + | - | - | + | + | 5/9 (56%) |
| Aggression | - | + | - | + | - | - | - | - | - | 2/9 (22%) |
| Other behaviors and neuropsychiatric comorbidities | depression, migraines | unusual phobias, hyperactivity, short attention span, extremely selective diet | occasional unmotivated giggling, extremely selective diet | mood changes, obsessive-compulsive behaviors | bipolar disorder | irritability, minimal response to pain | - | self-injurious behaviors, pica, short attention span | mood changes |
Abbreviations: +, present; -, absent; U, unknown; y, years.
Figure 2.

Facial Phenotype of Patients with 17q12 Deletions
Macrocephaly, epicanthal folds, downslanting palpebral fissures, arched and high eyebrows, slightly depressed nasal bridge, and malar flattening are the features shared most consistently among these patients. Profile pictures and photographs at different ages are provided when available, showing the progression of the phenotype over time.
Mild to moderate neurocognitive impairment was present in most patients, primarily affecting speech. We observed different degrees of difficulties in sensory integration and motor skills, specifically regarding coordination and motor tasks. Remarkably, autism or autistic features were present in all six male patients. Four of them met DSM-IV and/or ADI-ADOS diagnostic criteria for autism, and the other two had autistic behaviors but did not meet the threshold for a formal diagnosis of autism. Case 6 was too young at the time of assessment for a conclusive diagnosis, though he had been previously referred for evaluation because of delayed development and autistic features, including stereotypic behaviors and relative deficits in social responsivity and joint attention. Case 9 did not meet formal criteria for autism, but he presented with autistic features, comprising excessive insistence on performing tasks to perfection and difficulties in disengaging from those tasks, emotional irritability, and unusual affect. Difficulties in social skills were present but had a late onset, on the background of nonverbal learning disability. Additionally, he had marked anxiety, which was seen in most of the patients as well. Obsessive-compulsive behaviors and prominent anxiety were seen along with the autism diagnosis in most of these patients. Neither of the two females nor the mother with the deletion had autistic features at the time of assessment. However, mild neurocognitive impairment was observed in all three, and one of them had a history of severe bipolar disorder, and depression (MIM 608516) was present in another one.
Follow-up Sample
Deletions of 17q12 in ASD/Neurodevelopmental Disorders
We were intrigued to find that six out of the nine patients we studied in detail had autism or autistic features. We therefore expanded our assessment of 17q12 deletions to two independent existing ASD collections. Funded by the Simons Foundation Autism Research Initiative (SFARI), the SFARI Simplex Collection (SSC) is a sample of well-phenotyped patients with ASD from families with a single affected individual. We obtained CNV information, based on the Illumina 1M SNP array platform, from 806 patients for this analysis. In this sample, we identified one female with the 17q12 deletion out of 806 individuals with ASD.
Additionally, we assessed the deCODE neurodevelopmental collection, composed of 376 patients with ASD and/or neurocognitive impairment for copy number imbalances by using Illumina SNP microarrays (317K, 370K, 550K, 1M) as previously described.38 Among this sample, we identified one female with the 17q12 CNV. To estimate the risk for the combined ASD/neurocognitive impairment sample, we used the Cochran-Mantel-Haenszel model because of the different ethnic compositions of the samples, and we demonstrated a high risk of ASD associated with this CNV (Table 2; OR ∞, 4.73-∞, p = 1.92 × 10−3).
Deletions of 17q12 in Schizophrenia
Having shown the 17q12 deletion to be a strong risk factor for ASD and intellectual disability, we extended our investigations to assess the potential role of this CNV in other neurodevelopmental and psychiatric phenotypes. Given the genetic and clinical overlap observed between ASD and schizophrenia,3 we looked for this CNV in two large existing schizophrenia samples: GAIN and the SGENE consortium. We obtained and extracted intensities from the raw data, based on the Affymetrix 6.0 array platform, of 2167 schizophrenia cases and 2418 controls from GAIN48 (dbGaP; accession number phs000021.v), where we identified one female with the deletion among the cases and no deletions among controls. Additionally, three deletions (two females and one male) out of 4,173 cases were present in the SGENE collection, assessed via Illumina SNP microarrays (317K, 370K, 550K, 1M) as described above, whereas none of the 40,658 controls carried this imbalance. Again, via the Cochran-Mantel-Haenszel model to estimate the risk for the combined schizophrenia follow-up sample, we demonstrated a high risk of schizophrenia associated with deletion 17q12 (Table 2; OR ∞, 1.25-∞, p = 1.47 × 10−2).
Combined Discovery and Follow-up Samples
Overall, this deletion was present in 24 out of the 23,271 patients and in 0 out of the 52,448 controls. All the cytogenomic array platforms we used in this study (Figure S1) can detect CNVs of much smaller size than this 1.4 Mb deletion, so we do not expect false negative results among our samples. Likewise, none of the deletions we identified appear to be mosaic, as we anticipated, given that the mechanism yielding these imbalances is most probably nonhomologous allelic recombination, a well-known meiotic recombination event, as opposed to a mitotic or somatic event.
We did not have information on the ethnicity for all the individuals from the ISCA sample; hence, we could not apply the Cochran-Mantel-Haenszel correction to the pooled discovery and follow-up samples. However, we did calculate the uncorrected risk for this combined sample, which was highly significant (Table 2; OR ∞, 13.58-∞, p = 5 × 10−13). We believe this is a useful estimate, and not probably explained by population differences, for several reasons including the absence of this CNV from a very large control population, the fact that most of the deletions for which we had parental data were de novo, and the strong penetrance of the deletion on the phenotype of the patients who have it, albeit with variable expressivity.
Discussion
Initially believed to be one of the few genomic disorders that spared the central nervous system,46 the 17q12 deletion has been shown to be associated with intellectual disability or ASD in a few case series.47,49 Moreover, a recent study on patients with ADHD (MIM 143465) identified one case with the same 17q12 deletion.50 By analyzing large populations, we now show that this CNV confers a statistically significant high risk for ASD and schizophrenia. This is important because the 17q12 deletion is among the 10 most frequent pathogenic recurrent genomic deletions identified in children with unexplained neurodevelopmental impairments (E.B.K. et al., unpublished data), with a frequency of 1 in 875 in our clinical discovery sample and 1 in 1254 in our follow-up sample. Additionally, this CNV may increase risk for additional psychiatric conditions such as bipolar disorder. The 17q12 deletion was de novo in all but one of the cases for whom parents were available for testing; in the only case in which the deletion was inherited, the mother with the deletion was affected with bipolar disorder, diabetes, and kidney cysts. Taken together with the absence of this CNV from the 52,448 controls we assessed (Table 2), this demonstrates the high penetrance of the 17q12 deletion and its etiologic role on the phenotype of these patients.
The 17q12 deletion adds to the growing list of recurrent CNVs associated with ASD and schizophrenia (Table 3). Both conditions are characterized by a wide phenotypic and genetic heterogeneity; however, the same genetic anomalies are being identified across these different but related neurodevelopmental diagnoses.3,4 This suggests that the disruption of the core developmental processes of the central nervous system can give rise to a wide range of clinical manifestations, where the final outcome is probably modulated by the genetic background of each individual as well as environmental factors. Interestingly, the 17q12 genomic region overlaps a well-established ASD linkage peak;51–54 furthermore, an autism association study55 has implicated genes in this same interval on 17q (Figure 1). Within our discovery ISCA sample, six of the nine patients we assessed in detail had autistic features or autism, and we identified two patients with the deletion among the independent ASD and neurodevelopmental samples we analyzed. These observations, along with an additional report of three patients with the 17q12 deletion and ASD,49 further strengthen the hypothesis of a major risk factor for ASD at 17q12. Although none of the patients with the CNV from the ISCA discovery sample had a diagnosis of schizophrenia (probably because of their young age), we identified four individuals with the 17q12 deletion within the two schizophrenia samples we assessed. Unlike ASD,56 cytogenomic array testing is not currently considered a standard of care for patients with schizophrenia. However, this and previous studies show that pathogenic CNVs that would account for the phenotype of these patients can be identified in individuals with schizophrenia.57,58 Independently from the statistical significance at the population level, the presence of one of these pathogenic CNVs in a single affected individual is highly relevant on a clinical basis and could impact management, as is the case of patients with 22q11 deletions initially ascertained for autism or schizophrenia.
Table 3.
Recurrent CNVs Identified across ASD and Schizophrenia
| Genomic Region | Position (Mb)a | Size (Mb)a | Number of Genesb | CNV | References |
|---|---|---|---|---|---|
| 1q21.1 | chr1:144,963,732-145,864,377 | 0.9 | 7 | del | 11,12 |
| 3q29 | chr3:197,244,288-198,830,238 | 1.6 | 21 | del | 13,14 |
| 15q13.3 | chr15:28,698,632-30,234,007 | 1.5 | 6 | del | 12,15,16 |
| 16p11.2 | chr16:29,557,553-30,107,434 | 0.5 | 25 | dup | 18,19 |
| 16p13.11 | chr16:15,421,876-16,200,195 | 0.8 | 7 | dup | 20,21 |
| 17q12 | chr17:31,893,783-33,277,865 | 1.4 | 15 | del | this study; 49 |
| 22q11.2 | chr22:17,412,646-19,797,314 | 2.4 | 41 | del | 12,17 |
Size and position are calculated in the hg18 genome assembly and exclude the DNA sequence from flanking segmental duplications.
The number of genes in each region is based on RefSeq coding genes.
It is unclear why the 17q12 deletion has not been detected previously in large neuropsychiatric samples assessed for CNVs.59–61 One reason could be the presence of a renal phenotype in several of the patients with this CNV, which may have caused their exclusion from the research studies and led clinicians to seek a syndromic cause for the behavioral impairments. Alternatively, the population we assessed might be more severely affected than most ASD and schizophrenia samples, which may exclude patients with intellectual disability.
In addition to the neurocognitive and behavioral involvement and a mild but characteristic facial phenotype, our patients shared several other phenotypic features; macrocephaly or relative macrocephaly and dolichocephaly were seen across most of the them, suggesting that 17q12 deletions, along with PTEN (MIM 601728) mutations, should be considered in the differential diagnosis of patients with macrocephaly and ASD, even though the degree of macrocephaly in the latter is more severe.62 We also observed mild to moderate intellectual disability in most of our patients. Although we did find renal anomalies in our sample, the severity was variable and generally milder than in patients with the same deletion reported previously; most of our patients had normal renal function but they may be too young to have developed a substantial impairment. In a similar fashion, only the eldest patient (37 years old) had diabetes mellitus, but careful monitoring of glucose levels for all affected patients will be important as they age, given their genetic predisposition. We found malformations of the reproductive system in two of our patients, including urethral stenosis in a male (Case 4) and uterus didelphis (Case 1), as previously described.63–66 Finally, one of the patients had short stature, which has been observed in other patients with the 17q12 deletion.47
The co-occurrence of multiple, apparently unrelated clinical findings in patients with the 17q12 deletion is suggestive of a contiguous gene syndrome.67 Under this model, major phenotypic features may be due to haploinsufficiency of two or more different genes within the deletion. HNF1B, previously known as TCF2 (transcription factor 2), is responsible for the renal structural abnormalities and female reproductive tract malformations,64 as shown by the fact that mutations in this gene cause RCAD, also referred to as maturity-onset diabetes of the young, type 5 (MODY5).68 There are no reported neurocognitive or psychiatric phenotypes in patients with RCAD resulting from point mutations in HNF1B, although this may not have been assessed in a systematic fashion. It will be very important to gather data on a series of patients with RCAD because of point mutations versus deletion 17q12 to determine the risk of neurocognitive and psychiatric disorders in these two groups. If patients with HNF1B mutations do not have neurocognitive/psychiatric impairments, then a different gene within the deleted interval may account for this component of the phenotype. Point mutations in this other gene may also occur in patients with idiopathic ASD or schizophrenia who do not have the renal and endocrine phenotype of HNF1B haploinsufficiency. It is noteworthy that not all the patients we ascertained with this CNV resulting from developmental delay or ASD had a diagnosis of RCAD. However, it is important to refer these patients for evaluation of renal and endocrine function.
Among the other 14 genes within the deletion, LHX1 is an interesting candidate to explain the neurocognitive phenotype. As a potential transcriptional regulator that plays a role in the differentiation of neural cells and the transcriptional control of axonal guidance,69 it is expressed in the brain during early development.69 Additionally, an Lhx1 knockout mouse model shows that Lhx1 is an essential regulator of the vertebrate head organizer.70 However, LHX1 has yet to be associated with disease in humans, and no single gene deletions, disruptions, or mutations causing haploinsufficiency have been detected. Future studies are therefore necessary to identify the gene or genes responsible for the neurobehavioral phenotype in patients with the 17q12 deletion.
In conclusion, the current data show that deletion 17q12 is a recurrent pathogenic CNV that confers a very high risk for ASD, schizophrenia, or neurodevelopmental disorders. The clinical phenotype of patients with this deletion, which include macrocephaly, characteristic facial features, renal cysts, diabetes, neurocognitive impairment, ASD, and schizophrenia, are suggestive of a contiguous gene syndrome that extends beyond RCAD. These observations suggest that haploinsufficiency of 1 of the 15 genes in the deleted interval has a very high risk of diverse neurocognitive or psychiatric disorders.
Acknowledgments
We would like to thank all the individuals and families for their participation in this study. We also thank D. Kunig, B. Bunke, D.J. Cutler, M.K. Rudd, M.R. Rossi, M. Hegde, C.T. Stauss, A. Moreno De Luca, and J.F. Cubells for technical and editorial assistance. This work was funded in part by NIH grants MH074090 (D.H.L. and C.L.M.), HD064525 (D.H.L. and C.L.M.), MH080583 (J.G.M.), MH080129 (S.T.W.), and MH071425 (K. Stefansson). The SGENE project was sponsored by EU grant LSHM-CT-2006-037761 (Project SGENE).
Funding support for the genome-wide association of schizophrenia study was provided by the NIMH, and the genotyping of samples was provided through GAIN; this data set was obtained from dbGaP through accession number phs000021.v. Samples and associated phenotype data for the genome-wide association of schizophrenia study were provided by the Molecular Genetics of Schizophrenia Collaboration (PI: P.V. Gejman, Evanston Northwestern Healthcare [ENH] and Northwestern University, Evanston, Illinois, USA).
We are grateful to the SSC Genetics Committee and the principal investigators (A. Beaudet, R. Bernier, J. Constantino, E. Cook, E. Fombonne, D. Geschwind, D. Grice, A. Klin, D.H.L., C. Lord, C.L.M., D. Martin, R. Maxim, J. Miles, O. Ousley, B. Peterson, J. Piggot, C. Saulnier, M. State, W. Stone, J. Sutcliffe, C. Walsh, E. Wijsman). We appreciate obtaining access to phenotypic and genetic data on SFARI Base. Approved researchers can obtain the SSC population data set described in this study by applying online at the SFARI website.
Contributor Information
Daniel Moreno-De-Luca, Email: daniel.morenodeluca@emory.edu.
David H. Ledbetter, Email: david.ledbetter@emory.edu.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
CNVision, http://www.yale.edu/state/Homepage.htm
Database of Genomic Variants (DGV), http://projects.tcag.ca/variation/
Database of Genomic Structural Variation (dbVar), http://www.ncbi.nlm.nih.gov/dbvar
Database of Genotype and Phenotype (dbGaP), http://www.ncbi.nlm.nih.gov/gap
International Standards for Cytogenomic Arrays Consortium, https://isca.genetics.emory.edu
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
R Project for Statistical Computing, http://www.r-project.org/
SGENE consortium, http://SGENE.eu
Simons Foundation Autism Research Initiative (SFARI) Simplex Collection (SSC), https://base.sfari.org
Accession Numbers
The dbVar accession number for the new data set reported in this paper (ISCA consortium) is NSTD37.
References
- 1.Autism and Developmental Disabilities Monitoring Network Surveillance Year 2006 Principal InvestigatorsCenters for Disease Control and Prevention (CDC) Prevalence of autism spectrum disorders - Autism and Developmental Disabilities Monitoring Network, United States, 2006. MMWR Surveill. Summ. 2009;58:1–20. [PubMed] [Google Scholar]
- 2.Messias E.L., Chen C.Y., Eaton W.W. Epidemiology of schizophrenia: Review of findings and myths. Psychiatr. Clin. North Am. 2007;30:323–338. doi: 10.1016/j.psc.2007.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carroll L.S., Owen M.J. Genetic overlap between autism, schizophrenia and bipolar disorder. Genome Med. 2009;1:102. doi: 10.1186/gm102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cantor R.M., Geschwind D.H. Schizophrenia: Genome, interrupted. Neuron. 2008;58:165–167. doi: 10.1016/j.neuron.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 5.Abrahams B.S., Geschwind D.H. Advances in autism genetics: On the threshold of a new neurobiology. Nat. Rev. Genet. 2008;9:341–355. doi: 10.1038/nrg2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kirov G. The role of copy number variation in schizophrenia. Expert Rev. Neurother. 2010;10:25–32. doi: 10.1586/ern.09.133. [DOI] [PubMed] [Google Scholar]
- 7.Raymond F.L., Tarpey P. The genetics of mental retardation. Hum. Mol. Genet. 2006;15(Spec No 2, Spec No 2):R110–R116. doi: 10.1093/hmg/ddl189. [DOI] [PubMed] [Google Scholar]
- 8.Martin C.L., Ledbetter D.H. Autism and cytogenetic abnormalities: Solving autism one chromosome at a time. Curr. Psychiatry Rep. 2007;9:141–147. doi: 10.1007/s11920-007-0084-9. [DOI] [PubMed] [Google Scholar]
- 9.Addington A.M., Rapoport J.L. The genetics of childhood-onset schizophrenia: When madness strikes the prepubescent. Curr. Psychiatry Rep. 2009;11:156–161. doi: 10.1007/s11920-009-0024-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rudd M.K., Keene J., Bunke B., Kaminsky E.B., Adam M.P., Mulle J.G., Ledbetter D.H., Martin C.L. Segmental duplications mediate novel, clinically relevant chromosome rearrangements. Hum. Mol. Genet. 2009;18:2957–2962. doi: 10.1093/hmg/ddp233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brunetti-Pierri N., Berg J.S., Scaglia F., Belmont J., Bacino C.A., Sahoo T., Lalani S.R., Graham B., Lee B., Shinawi M. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat. Genet. 2008;40:1466–1471. doi: 10.1038/ng.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.International Schizophrenia Consortium Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Willatt L., Cox J., Barber J., Cabanas E.D., Collins A., Donnai D., FitzPatrick D.R., Maher E., Martin H., Parnau J. 3q29 microdeletion syndrome: Clinical and molecular characterization of a new syndrome. Am. J. Hum. Genet. 2005;77:154–160. doi: 10.1086/431653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mulle J.G., Dodd A.F., McGrath J.A., Wolyniec P.S., Mitchell A.A., Shetty A.C., Sobreira N.L., Valle D., Rudd M.K., Satten G. Microdeletions of 3q29 confer high risk for schizophrenia. Am. J. Hum. Genet. 2010;87:229–236. doi: 10.1016/j.ajhg.2010.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ben-Shachar S., Lanpher B., German J.R., Qasaymeh M., Potocki L., Nagamani S.C., Franco L.M., Malphrus A., Bottenfield G.W., Spence J.E. Microdeletion 15q13.3: A locus with incomplete penetrance for autism, mental retardation, and psychiatric disorders. J. Med. Genet. 2009;46:382–388. doi: 10.1136/jmg.2008.064378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stefansson H., Rujescu D., Cichon S., Pietiläinen O.P., Ingason A., Steinberg S., Fossdal R., Sigurdsson E., Sigmundsson T., Buizer-Voskamp J.E., GROUP Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Antshel K.M., Aneja A., Strunge L., Peebles J., Fremont W.P., Stallone K., Abdulsabur N., Higgins A.M., Shprintzen R.J., Kates W.R. Autistic spectrum disorders in velo-cardio facial syndrome (22q11.2 deletion) J. Autism Dev. Disord. 2007;37:1776–1786. doi: 10.1007/s10803-006-0308-6. [DOI] [PubMed] [Google Scholar]
- 18.Weiss L.A., Shen Y., Korn J.M., Arking D.E., Miller D.T., Fossdal R., Saemundsen E., Stefansson H., Ferreira M.A., Green T., Autism Consortium Association between microdeletion and microduplication at 16p11.2 and autism. N. Engl. J. Med. 2008;358:667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- 19.McCarthy S.E., Makarov V., Kirov G., Addington A.M., McClellan J., Yoon S., Perkins D.O., Dickel D.E., Kusenda M., Krastoshevsky O., Wellcome Trust Case Control Consortium Microduplications of 16p11.2 are associated with schizophrenia. Nat. Genet. 2009;41:1223–1227. doi: 10.1038/ng.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ingason A., Rujescu D., Cichon S., Sigurdsson E., Sigmundsson T., Pietilainen O.P., Buizer-Voskamp J.E., Strengman E., Francks C., Muglia P. Copy number variations of chromosome 16p13.1 region associated with schizophrenia. Mol. Psychiatry. 2009 doi: 10.1038/mp.2009.101. in press. Published online September 29, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ullmann R., Turner G., Kirchhoff M., Chen W., Tonge B., Rosenberg C., Field M., Vianna-Morgante A.M., Christie L., Krepischi-Santos A.C. Array CGH identifies reciprocal 16p13.1 duplications and deletions that predispose to autism and/or mental retardation. Hum. Mutat. 2007;28:674–682. doi: 10.1002/humu.20546. [DOI] [PubMed] [Google Scholar]
- 22.Miller D.T., Adam M.P., Aradhya S., Biesecker L.G., Brothman A.R., Carter N.P., Church D.M., Crolla J.A., Eichler E.E., Epstein C.J. Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 2010;86:749–764. doi: 10.1016/j.ajhg.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baldwin E.L., Lee J.Y., Blake D.M., Bunke B.P., Alexander C.R., Kogan A.L., Ledbetter D.H., Martin C.L. Enhanced detection of clinically relevant genomic imbalances using a targeted plus whole genome oligonucleotide microarray. Genet. Med. 2008;10:415–429. doi: 10.1097/GIM.0b013e318177015c. [DOI] [PubMed] [Google Scholar]
- 24.Spitzer R.L., Endicott J., Robins E. Research diagnostic criteria: Rationale and reliability. Arch. Gen. Psychiatry. 1978;35:773–782. doi: 10.1001/archpsyc.1978.01770300115013. [DOI] [PubMed] [Google Scholar]
- 25.Spitzer R., Endicott J. New York State Psychiatric Institute; New York: 1977. The Schedule for Affective Disorders and Schizophrenia, Lifetime Version. [Google Scholar]
- 26.Stefansson H., Rye D.B., Hicks A., Petursson H., Ingason A., Thorgeirsson T.E., Palsson S., Sigmundsson T., Sigurdsson A.P., Eiriksdottir I. A genetic risk factor for periodic limb movements in sleep. N. Engl. J. Med. 2007;357:639–647. doi: 10.1056/NEJMoa072743. [DOI] [PubMed] [Google Scholar]
- 27.Aromaa A., Koskinen S. National Public Health Institute, Finland; Helsinki, Finland: 2004. Health and Functional Capacity in Finland. Baseline Results of the Health 2000 Health Examination Survey. Publications of the National Public Health Institute Helsinki. [Google Scholar]
- 28.Pirkola S.P., Isometsä E., Suvisaari J., Aro H., Joukamaa M., Poikolainen K., Koskinen S., Aromaa A., Lönnqvist J.K. DSM-IV mood-, anxiety- and alcohol use disorders and their comorbidity in the Finnish general population—Results from the Health 2000 Study. Soc. Psychiatry Psychiatr. Epidemiol. 2005;40:1–10. doi: 10.1007/s00127-005-0848-7. [DOI] [PubMed] [Google Scholar]
- 29.Ekelund J., Lichtermann D., Hovatta I., Ellonen P., Suvisaari J., Terwilliger J.D., Juvonen H., Varilo T., Arajärvi R., Kokko-Sahin M.L. Genome-wide scan for schizophrenia in the Finnish population: Evidence for a locus on chromosome 7q22. Hum. Mol. Genet. 2000;9:1049–1057. doi: 10.1093/hmg/9.7.1049. [DOI] [PubMed] [Google Scholar]
- 30.Hovatta I., Terwilliger J.D., Lichtermann D., Mäkikyrö T., Suvisaari J., Peltonen L., Lönnqvist J. Schizophrenia in the genetic isolate of Finland. Am. J. Med. Genet. 1997;74:353–360. [PubMed] [Google Scholar]
- 31.Paunio T., Ekelund J., Varilo T., Parker A., Hovatta I., Turunen J.A., Rinard K., Foti A., Terwilliger J.D., Juvonen H. Genome-wide scan in a nationwide study sample of schizophrenia families in Finland reveals susceptibility loci on chromosomes 2q and 5q. Hum. Mol. Genet. 2001;10:3037–3048. doi: 10.1093/hmg/10.26.3037. [DOI] [PubMed] [Google Scholar]
- 32.McGuffin P., Farmer A., Harvey I. A polydiagnostic application of operational criteria in studies of psychotic illness. Development and reliability of the OPCRIT system. Arch. Gen. Psychiatry. 1991;48:764–770. doi: 10.1001/archpsyc.1991.01810320088015. [DOI] [PubMed] [Google Scholar]
- 33.Rosa A., Fañanás L., van Os J., Ribchester T., Davies N., Arias B., McDonald A., Murray R.M. Further evidence that congenital dermatoglyphic abnormalities are associated with psychosis: A twin study. Schizophr. Bull. 2002;28:697–701. doi: 10.1093/oxfordjournals.schbul.a006973. [DOI] [PubMed] [Google Scholar]
- 34.Toulopoulou T., Rabe-Hesketh S., King H., Murray R.M., Morris R.G. Episodic memory in schizophrenic patients and their relatives. Schizophr. Res. 2003;63:261–271. doi: 10.1016/s0920-9964(02)00324-9. [DOI] [PubMed] [Google Scholar]
- 35.Boydell J., Dean K., Dutta R., Giouroukou E., Fearon P., Murray R. A comparison of symptoms and family history in schizophrenia with and without prior cannabis use: Implications for the concept of cannabis psychosis. Schizophr. Res. 2007;93:203–210. doi: 10.1016/j.schres.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 36.World Health Organization. (1994). Schedules for Clinical Assessment in Neuropsychiatry (SCAN) Manual. (World Health Organization).
- 37.First M., Spitzer R.L., Gibbon M., Williams J.B. Biometrics Research; New York: 1994. Structured Clinical Interview for Axis I DSM-IV Disorders. [Google Scholar]
- 38.Stefansson H., Ophoff R.A., Steinberg S., Andreassen O.A., Cichon S., Rujescu D., Werge T., Pietiläinen O.P., Mors O., Mortensen P.B., Genetic Risk and Outcome in Psychosis (GROUP) Common variants conferring risk of schizophrenia. Nature. 2009;460:744–747. doi: 10.1038/nature08186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang K., Li M., Hadley D., Liu R., Glessner J., Grant S.F., Hakonarson H., Bucan M. PennCNV: An integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665–1674. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Colella S., Yau C., Taylor J.M., Mirza G., Butler H., Clouston P., Bassett A.S., Seller A., Holmes C.C., Ragoussis J. QuantiSNP: An Objective Bayes Hidden-Markov Model to detect and accurately map copy number variation using SNP genotyping data. Nucleic Acids Res. 2007;35:2013–2025. doi: 10.1093/nar/gkm076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mantel N., Haenszel W. Statistical aspects of the analysis of data from retrospective studies of disease. J. Natl. Cancer Inst. 1959;22:719–748. [PubMed] [Google Scholar]
- 43.Shaikh T.H., Gai X., Perin J.C., Glessner J.T., Xie H., Murphy K., O'Hara R., Casalunovo T., Conlin L.K., D'Arcy M. High-resolution mapping and analysis of copy number variations in the human genome: A data resource for clinical and research applications. Genome Res. 2009;19:1682–1690. doi: 10.1101/gr.083501.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Itsara A., Cooper G.M., Baker C., Girirajan S., Li J., Absher D., Krauss R.M., Myers R.M., Ridker P.M., Chasman D.I. Population analysis of large copy number variants and hotspots of human genetic disease. Am. J. Hum. Genet. 2009;84:148–161. doi: 10.1016/j.ajhg.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Iafrate A.J., Feuk L., Rivera M.N., Listewnik M.L., Donahoe P.K., Qi Y., Scherer S.W., Lee C. Detection of large-scale variation in the human genome. Nat. Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 46.Mefford H.C., Clauin S., Sharp A.J., Moller R.S., Ullmann R., Kapur R., Pinkel D., Cooper G.M., Ventura M., Ropers H.H. Recurrent reciprocal genomic rearrangements of 17q12 are associated with renal disease, diabetes, and epilepsy. Am. J. Hum. Genet. 2007;81:1057–1069. doi: 10.1086/522591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nagamani S.C., Erez A., Shen J., Li C., Roeder E., Cox S., Karaviti L., Pearson M., Kang S.H., Sahoo T. Clinical spectrum associated with recurrent genomic rearrangements in chromosome 17q12. Eur. J. Hum. Genet. 2009;18:278–284. doi: 10.1038/ejhg.2009.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shi J., Levinson D.F., Duan J., Sanders A.R., Zheng Y., Pe'er I., Dudbridge F., Holmans P.A., Whittemore A.S., Mowry B.J. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature. 2009;460:753–757. doi: 10.1038/nature08192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Loirat C., Bellanne-Chantelot C., Husson I., Deschenes G., Guigonis V., Chabane N. Autism in three patients with cystic or hyperechogenic kidneys and chromosome 17q12 deletion. Nephrol. Dial. Transplant. 2010;25:3430–3433. doi: 10.1093/ndt/gfq380. [DOI] [PubMed] [Google Scholar]
- 50.Elia J., Gai X., Xie H.M., Perin J.C., Geiger E., Glessner J.T., D'arcy M., deBerardinis R., Frackelton E., Kim C. Rare structural variants found in attention-deficit hyperactivity disorder are preferentially associated with neurodevelopmental genes. Mol. Psychiatry. 2010;15:637–646. doi: 10.1038/mp.2009.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McCauley J.L., Li C., Jiang L., Olson L.M., Crockett G., Gainer K., Folstein S.E., Haines J.L., Sutcliffe J.S. Genome-wide and Ordered-Subset linkage analyses provide support for autism loci on 17q and 19p with evidence of phenotypic and interlocus genetic correlates. BMC Med. Genet. 2005;6:1. doi: 10.1186/1471-2350-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.International Molecular Genetic Study of Autism Consortium (IMGSAC) A genomewide screen for autism: Strong evidence for linkage to chromosomes 2q, 7q, and 16p. Am. J. Hum. Genet. 2001;69:570–581. doi: 10.1086/323264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yonan A.L., Alarcón M., Cheng R., Magnusson P.K., Spence S.J., Palmer A.A., Grunn A., Juo S.H., Terwilliger J.D., Liu J. A genomewide screen of 345 families for autism-susceptibility loci. Am. J. Hum. Genet. 2003;73:886–897. doi: 10.1086/378778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stone J.L., Merriman B., Cantor R.M., Yonan A.L., Gilliam T.C., Geschwind D.H., Nelson S.F. Evidence for sex-specific risk alleles in autism spectrum disorder. Am. J. Hum. Genet. 2004;75:1117–1123. doi: 10.1086/426034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stone J.L., Merriman B., Cantor R.M., Geschwind D.H., Nelson S.F. High density SNP association study of a major autism linkage region on chromosome 17. Hum. Mol. Genet. 2007;16:704–715. doi: 10.1093/hmg/ddm015. [DOI] [PubMed] [Google Scholar]
- 56.Shen Y., Dies K.A., Holm I.A., Bridgemohan C., Sobeih M.M., Caronna E.B., Miller K.J., Frazier J.A., Silverstein I., Picker J., Autism Consortium Clinical Genetics/DNA Diagnostics Collaboration Clinical genetic testing for patients with autism spectrum disorders. Pediatrics. 2010;125:e727–e735. doi: 10.1542/peds.2009-1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bassett A.S., Costain G., Alan Fung W.L., Russell K.J., Pierce L., Kapadia R., Carter R.F., Chow E.W., Forsythe P.J. Clinically detectable copy number variations in a Canadian catchment population of schizophrenia. J. Psychiatr. Res. 2010 doi: 10.1016/j.jpsychires.2010.06.013. in press. Published online July 17, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bassett A.S., Scherer S.W., Brzustowicz L.M. Copy number variations in schizophrenia: Critical review and new perspectives on concepts of genetics and disease. Am. J. Psychiatry. 2010;167:899–914. doi: 10.1176/appi.ajp.2009.09071016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Glessner J.T., Wang K., Cai G., Korvatska O., Kim C.E., Wood S., Zhang H., Estes A., Brune C.W., Bradfield J.P. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pinto D., Pagnamenta A.T., Klei L., Anney R., Merico D., Regan R., Conroy J., Magalhaes T.R., Correia C., Abrahams B.S. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Glessner J.T., Reilly M.P., Kim C.E., Takahashi N., Albano A., Hou C., Bradfield J.P., Zhang H., Sleiman P.M., Flory J.H. Strong synaptic transmission impact by copy number variations in schizophrenia. Proc. Natl. Acad. Sci. USA. 2010;107:10584–10589. doi: 10.1073/pnas.1000274107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Buxbaum J.D., Cai G., Chaste P., Nygren G., Goldsmith J., Reichert J., Anckarsäter H., Rastam M., Smith C.J., Silverman J.M. Mutation screening of the PTEN gene in patients with autism spectrum disorders and macrocephaly. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2007;144B:484–491. doi: 10.1002/ajmg.b.30493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Müller D., Klopocki E., Neumann L.M., Mundlos S., Taupitz M., Schulze I., Ropers H.H., Querfeld U., Ullmann R. A complex phenotype with cystic renal disease. Kidney Int. 2006;70:1656–1660. doi: 10.1038/sj.ki.5001746. [DOI] [PubMed] [Google Scholar]
- 64.Bingham C., Ellard S., Cole T.R., Jones K.E., Allen L.I., Goodship J.A., Goodship T.H., Bakalinova-Pugh D., Russell G.I., Woolf A.S. Solitary functioning kidney and diverse genital tract malformations associated with hepatocyte nuclear factor-1beta mutations. Kidney Int. 2002;61:1243–1251. doi: 10.1046/j.1523-1755.2002.00272.x. [DOI] [PubMed] [Google Scholar]
- 65.Lindner T.H., Njolstad P.R., Horikawa Y., Bostad L., Bell G.I., Sovik O. A novel syndrome of diabetes mellitus, renal dysfunction and genital malformation associated with a partial deletion of the pseudo-POU domain of hepatocyte nuclear factor-1beta. Hum. Mol. Genet. 1999;8:2001–2008. doi: 10.1093/hmg/8.11.2001. [DOI] [PubMed] [Google Scholar]
- 66.Cheroki C., Krepischi-Santos A.C., Szuhai K., Brenner V., Kim C.A., Otto P.A., Rosenberg C. Genomic imbalances associated with mullerian aplasia. J. Med. Genet. 2008;45:228–232. doi: 10.1136/jmg.2007.051839. [DOI] [PubMed] [Google Scholar]
- 67.Schmickel R.D. Contiguous gene syndromes: A component of recognizable syndromes. J. Pediatr. 1986;109:231–241. doi: 10.1016/s0022-3476(86)80377-8. [DOI] [PubMed] [Google Scholar]
- 68.Bellanné-Chantelot C., Clauin S., Chauveau D., Collin P., Daumont M., Douillard C., Dubois-Laforgue D., Dusselier L., Gautier J.F., Jadoul M. Large genomic rearrangements in the hepatocyte nuclear factor-1beta (TCF2) gene are the most frequent cause of maturity-onset diabetes of the young type 5. Diabetes. 2005;54:3126–3132. doi: 10.2337/diabetes.54.11.3126. [DOI] [PubMed] [Google Scholar]
- 69.Avraham O., Hadas Y., Vald L., Zisman S., Schejter A., Visel A., Klar A. Transcriptional control of axonal guidance and sorting in dorsal interneurons by the Lim-HD proteins Lhx9 and Lhx1. Neural Dev. 2009;4:21. doi: 10.1186/1749-8104-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shawlot W., Behringer R.R. Requirement for Lim1 in head-organizer function. Nature. 1995;374:425–430. doi: 10.1038/374425a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.