

Abstract
Intellectual disability (ID) affects 1%–3% of the general population. We recently reported on a family with autosomal-recessive mental retardation with anterior maxillary protrusion and strabismus (MRAMS) syndrome. One of the reported patients with ID did not have dysmorphic features but did have temporal lobe epilepsy and psychosis. We report on the identification of a truncating mutation in the SOBP that is responsible for causing both syndromic and nonsyndromic ID in the same family. The protein encoded by the SOBP, sine oculis binding protein ortholog, is a nuclear zinc finger protein. In mice, Sobp (also known as Jxc1) is critical for patterning of the organ of Corti; one of our patients has a subclinical cochlear hearing loss but no gross cochlear abnormalities. In situ RNA expression studies in postnatal mouse brain showed strong expression in the limbic system at the time interval of active synaptogenesis. The limbic system regulates learning, memory, and affective behavior, but limbic circuitry expression of other genes mutated in ID is unusual. By comparing the protein content of the +/jc to jc/jc mice brains with the use of proteomics, we detected 24 proteins with greater than 1.5-fold differences in expression, including two interacting proteins, dynamin and pacsin1. This study shows mutated SOBP involvement in syndromic and nonsyndromic ID with psychosis in humans.
Main Text
Intellectual disability (ID) affects 1%–3% of the general population.1,2 Syndromic ID is characterized by ID accompanied by major physical abnormalities, dysmorphism, or neurological abnormalities, whereas nonsyndromic ID is characterized by cognitive impairment without any additional features. Progress in the identification of genes associated with ID has highlighted the fact that the distinction between syndromic and nonsyndromic ID appears to be blurred, because mutations in some genes can result in both syndromic and nonsyndromic forms of cognitive impaiment.3,4 To date, mutations in only six genes have been identified to cause nonsyndromic autosomal-recessive ID—these are PRSS12 (MIM 606709), CRBN (MIM 609262), CC2D1A (MIM 610055), GRIK2 (MIM 138244), TUSC3 (MIM 601385), and TRAPPC9 (MIM 611966).5–10
We recently reported on a syndrome characterized by autosomal-recessive ID, dysmorphic features, and strabismus, which we have named the MRAMS syndrome (for mental retardation, anterior maxillary protrusion, and strabismus).11 Interestingly, one of the seven reported patients with ID did not have dysmorphic features but did have temporal lobe epilepsy and psychosis. Esotropia was a common finding in this family and was present in 6 out of 7 individuals; it was resolved with conservative treatment in all but one patient. Patient IV-9 (Figure 1A) was diagnosed with esotropia and amblyopia in the left eye at the age of 6 years. Vision was reduced to 6/18 in the left eye. Cycloplegic refraction revealed hypermetropia of +4.0 diopters in the right eye and +4.5 diopters in the left eye. Glasses with full cycloplegic correction were prescribed and patching of the right eye was initiated. Vision was normalized, but the esotropia persisted; therefore, strabismus surgery was performed. Brainstem auditory evoked response examination in patient IV-1 showed mild cochlear hearing loss of 30–40 dB. On the MRI of this patient, no gross cochlear abnormalities were observed. In patient IV-5, premaxillary prominence, dental crowding, and strabismus were absent, and this patient has a slightly higher level of functioning together with psychosis and seizures. At 11 years of age he developed seizures, and from 13 years onward he developed increasingly severe psychotic symptoms; he started talking to himself and would respond to auditory and visual hallucinations for several hours a day. At the same time, there appeared to be fragmentation of his thought processes. He became easily frustrated and exhibited recurrent aggressive outbursts. Psychiatric symptoms are known to be a part of several syndromes, including velo-cardio-facial syndrome (MIM 192430), fragile X syndrome (MIM 300624), MRX30 (MIM 300558), and others.
Figure 1.
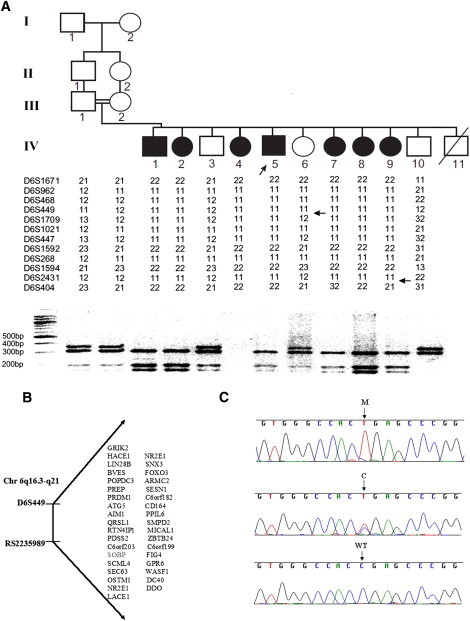
Gene Mapping and Mutation Identification
(A) Pedigree, haplotyping, and segregation of mutation in the family. The proband is indicated by an arrow. For mutation detection by restriction analysis, amplification of a 669 bp fragment from genomic DNA was performed with the use of primers A and B. The mutation introduces a Dde1 restriction site, which digests the 669 bp fragment into 310 bp, 195 bp, and164 bp fragments. The reaction products were separated by electrophoresis on 3% NuSieve/1% agarose gels.
(B) Candidate region on chromosome 6q16.3-q21, with the list of candidate genes.
(C) Sequence analysis in a patient, heterozygous carrier, and control individual. M, mutant allele; C, carrier; WT, homozygous wild-type. DNA from individual IV-4 was not available for mutation segregation analysis.
In order to identify the causative mutation in this family, after informed consent was obtained from all family members or their legal guardians (in accordance with a protocol approved and reviewed by the National Committee for Genetic Studies, Israel Ministry of Health), we genotyped 400 microsatellite markers from the ABI PRISM linkage mapping set, version 2.5 (Applied Biosystems, Foster City, CA, USA). All of the patients exhibited a large continuous segment of homozygosity on chromosomal region 6q21. A common homozygous disease-bearing haplotype was constructed; informative recombinations delineated a critical region of 9.8 Mb between the polymorphic markers D6S449 and D6S404 (Figure 1A). Individual IV-5, with nonsyndromic ID, showed the same disease-associated haplotype. Through SNP genotyping using Affymetrix Human Mapping 50k Xba 240 arrays on individuals IV-1, IV-2, IV-4, and IV-7, the candidate interval was reduced to the region between markers D6S449 and rs2235989. There was only one homozygosity region shared by all of the patients; there were no homozygosity regions shared by all individuals apart from individual IV-5 (Figure S1, available online). The two-point LOD score calculations were performed with the program SUPERLINK12 and yielded a Zmax score of 4.45 at recombination fraction (θ) 0.00 (Table S1). The candidate region contained 36 known or predicted genes (Figure 1B), as identified with the use of available databases (NCBI and UCSC Genome Browser, March 2006 version). All exons including exon-intron junctions of the following candidate genes were sequenced: GRIK2 (MIM 138244), LIN28B (MIM 611066), BVES (MIM 604577), POPDC3 (MIM 605824), PREP (MIM 600400), ATG5 (MIM 604261), RTN4IP1 (MIM 610502), PDSS2 (MIM 610564), SCML4, OSTM1(MIM 607649), NR2E1 (MIM 603849), SNX3 (MIM 605930), FOXO3A (MIM 602681), ARMC2, SESN1 (MIM 606103), CD164 (MIM 603356), LACE1, PPIL6, SMPD2 (MIM 603498), ZBTB24, C6ORF199, GPR6 (MIM 600553), WASF1 (MIM 605035), CDC40 (MIM 605585), and DDO (MIM 124450). No pathogenic sequence changes were found. Sequencing of SOBP was performed with the use of ten primer pairs (Table S2). In this gene, we identified a homozygous C>T change in nucleotide 1981 of exon 6 (c.1981C>T) (NM_018013.3) (Figure 1C), which creates an early stop codon that causes the truncation of 212 residues of the 873 amino acids of the protein (p.Arg661X). SOBP mutation screening was performed on genomic DNA by PCR amplification with primer pair 7 (Table S2). The mutation segregated with the disease (Figure 1A) and was not observed in 300 control chromosomes of Israeli Arab individuals. mRNA from the lymphoblasts of one of the patients was successfully amplified, indicating that mutant mRNA is not completely degraded (data not shown).
In order to determine the frequency of SOBP involvement, when mutated, in other families with autosomal-recessive ID, homozygosity at the 6q21 locus was tested by genotyping with the Affymetrix GeneChip Mapping 250K Array in 22 independent consanguineous patients presenting with isolated or mildly syndromic ID. However, no patient was found to be compatible with linkage to this locus.
The SOBP structure was described with the use of the UCSC (GRCh37/hg19, February 2009), NCBI (build 37.1, August 2009), and Ensembl (GRCh37, February 2009) genome browsers (Figure 2A).13 DNA and RNA binding motifs were predicted by the following servers: RNABindR, BindN, PPRint, and MEME. The human SOBP (sine oculis binding protein ortholog, also known as Jackson circler protein 1 [JXC1]; RefSeq NM_018013, GeneID: 55084) encodes a zinc finger protein that is located on human chromosome 6q21 and covers 171,192 bp of genomic DNA. The mRNA size is 6227 bp. The SOBP promoter consists of a distinct CpG island region; another CpG island region is clearly found in exon 6 and is predicted with high probability to be an additional promoter region. There are seven exons, the last of which is the untranslated region (UTR). Genomic conservation analysis shows that exons are highly conserved among vertebrates, whereas introns are conserved mainly within primates (Figure 2A). Transcript variants encode the main 873 amino acid protein (transcript 1) (NP_060483) and a shorter variant of 232 amino acids (transcript 2) (Figure 2A). It contains two FCS-type zinc fingers (MYM-type Zinc finger with FCS sequence motif, PF06467) and two proline-rich (PR) regions (IPR000694) (Figure 2B). There are many species with orthologs of the human protein. In the UCSC database, the chimp SOBP protein is a 668 residue protein, and the human is an 874 residue protein. The chimp protein divides into two parts. The first section is 1–409, with 99% identity to the human protein. Then there is a missing section in the chimp, found only in the human, and then comes another fragment (chimp 410–668; human 696–874) with 87% identity. Human SOBP is expressed in various tissues. There is a clear nuclear localization signal (NLS) at residues 9–24 and another putative NLS around residue 750. The motifs found in the protein are depicted in Figure 2B. Predictions for RNA and DNA binding motifs by different servers show that SOBP contains RNA binding motifs around and including residue 661 (Figure 2B). Interestingly, a search for DNA binding motifs using various bioinformatics databases did not show such motifs in SOBP. Truncation of SOBP after residue 661 causes elimination of a PR2 domain as well as possible NLS motifs and an RNA binding region, which may be important for the functioning of the protein. Protein-conservation analysis by the ConSeq server showed high sequence conservation and functionality of the C-terminal part of the protein; the N-terminal part is less conserved and has fewer functional residues (Figure S2).
Figure 2.
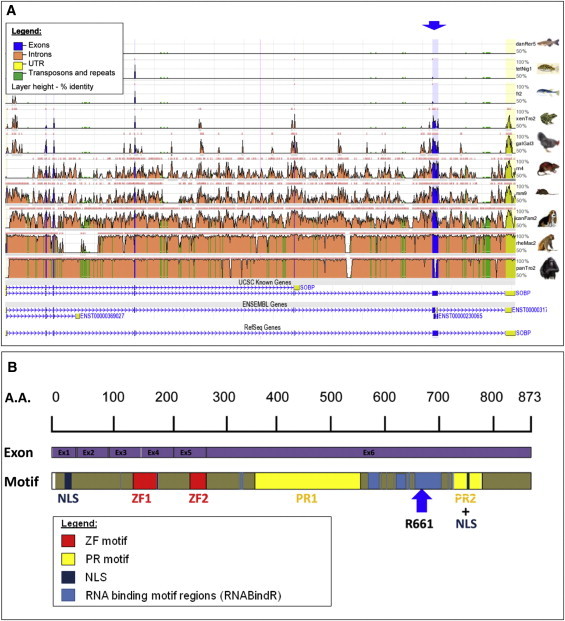
Bioinformatics Analysis of the Genomic Conservation of the SOBP and SOBP Domain and Motif Structure
(A) ECR browser analysis on human (hg18) was performed to show SOBP conservation between human and various species (danio, tetradon, fugu, Xenopus, gallus, rat, mouse, dog, rhesus, and chimp). Genomic conservation of exons (blue) and introns (orange), as well as the UTR (yellow) and repeats (green), is visualized. Layer height presents the percentage of identity (sequence conservation). Two different gene transcripts are shown at the bottom. The blue arrow shows a C>T change in nucleotide 1981 of exon 6 in SOBP.
(B) Domain properties for SOBP (A7XYQ1; SOBP_HUMAN) were obtained from UniProt. Two FCS-type zinc fingers (MYM-type zinc finger with FCS sequence motif, PF06467) and two proline-rich regions (IPR000694) are shown (red and yellow, respectively). NLS localization is also presented (navy). In addition, DNA and RNA binding motifs were predicted by the RNABindR server (light blue). The results from the BindN, PPRint, and MEME servers agree with those from the RNABindR server (data not shown). The blue arrow represents residue 661, in which the reported mutation occurred, causing truncation of SOBP.
In order to establish whether Sobp expression in the developing and adult brain could provide some insight into the cause of cognitive impairment, we performed a complete developmental characterization of Sobp mRNA expression in the murine brain by in situ hybridization. For in situ hybridization, plasmid templates for riboprobe synthesis were generated by subcloning a fragment of Sobp PCR amplified from IMAGE cDNA clone 10083890 containing a 493 bp sequence corresponding to nucleotide positions 21–514 of the Sobp coding sequence. In situ hybridization was performed as described by Tucker et al.14 Sopb was present at embryonic day (E) 14 throughout the developing brain, with intense staining in the cortical plate (Figure 3). During postnatal (P) development (P0, P10, and adult), Sobp was also expressed in all neurons, but a striking and intense expression pattern was especially evident in the limbic system, with the highest expression levels throughout layer V neurons in the cortex, the hippocampus, the pyriform cortex, the dorsomedial nucleus of the thalamus (Figures 3A and 3B), the amygdala, and the hypothalamus (Figure 3C). Cortical expression was strong throughout development, with no clear dorsoventral or rostrocaudal gradient, the highest levels at P10 in layers II/III and V and in the subplate (Figure 3D). Relatively strong expression was also observed in the mitral cells layer and anterior olfactory bulb (Figure 3B) and in the Purkinje cell layer in the cerebellum (not shown). We observed a striking and very specific expression of Sobp in the limbic system postnatally, corresponding to the time window of active synaptogenesis. It is known that on mouse P7 there is a period of significant synaptogenesis for neural-circuit formation and the expression of many genes encoding synaptic proteins and receptors.15 Another protein that has been found to be expressed in the limbic circuitry so strikingly is limbic system-associated membrane protein (LAMP).16 Because the circuit showing the highest Sobp expression regulates learning, memory, and affective behavior in humans, we suggest that disruption of SOBP in the limbic system during synaptogenesis leads to the abnormal cognition observed in the patients. The pathogenesis of strabismus in the patients is unclear. Because cortical Sobp expression is uniform, we cannot explain the origin of the strabismus on the basis of dynamic changes of Sobp expression levels in the visual cortex. Regulation of eye convergence may be affected at levels other than the cortex. Interestingly, in Drosophila, ectopic expression of Sobp leads to structural aberrations in the adult eye.17
Figure 3.
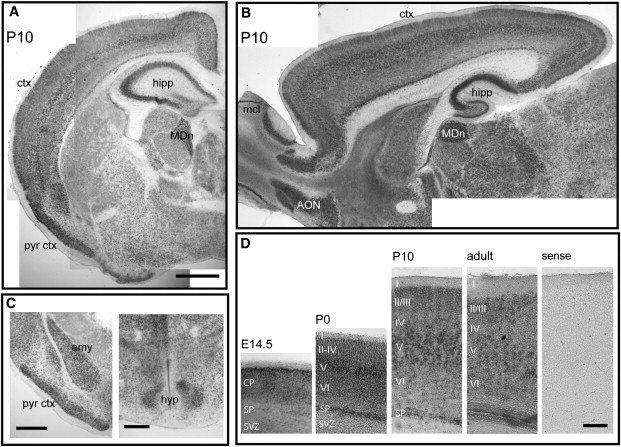
Mouse Sobp mRNA Is Highly Expressed in the Limbic System
(A and B) In situ hybridization on coronal (A) and parasagittal (B) sections of P10 murine brains show that Sobp is expressed throughout the brain, with the strongest expression in the cortex (ctx), especially in layer V, the hippocampus (hipp), the piriform cortex (pyr ctx), the mediodorsal nucleus of the thalamus (MDn), the anterior olfactory nucleus (AON), and the mitral cell layer in the olfactory bulb (mcl). Scale bars represent 500 μm.
(C) Strong Sobp expression is also observed in the amygdala (amy) and the hypothalamus (hyp). Scale bars represent 100 μm.
(D) Cortical expression is strong throughout development, with the highest levels of expression in layers II/III and V and in the subplate (SP). SVZ, subventricular zone. Scale bar represents 100 μm.
In mice, Jxc1/Sobp is critical for cochlear growth, cell fate, and patterning of the organ of Corti.18,19 Mice mutants also exhibit erratic circling behavior and have no vestibular evoked potentials. In contrast to mutant mice, one of our patients has a subclinical cochlear hearing loss of 30–40 dB but does not show any vestibular abnormalities or gross cochlear abnormalities. In order to gain some insight into the cause of cognitive impairment, we compared the protein content of the +/jc to jc/jc mice brains by using proteomics. For the proteomics analysis, proteins from brain tissue from each of +/jc and jc/jc mice were extracted and 300 mg of protein lysate was used for the analyses. Two-dimensional gel electrophoresis, spot identification, and mass spectrometry were performed by Applied Biomics. Proteins with greater than 1.3-fold differences between +/jc and jc/jc samples were identified by mass spectrometry. Protein lists were analyzed with the bioinformatics tool DAVID. Gene Ontology (GO) characterization of the proteins was performed by the tools GOTM, DAVID, and PANTHER. Functional annotation was performed with the use of mouse brain genes as background (MGI database). We detected 24 differentially expressed proteins with greater than 1.5-fold differences between +/jc and jc/jc samples (Table 1). Although these results should be further investigated and validated in additional studies, there are several interesting observations. Two of the differentially expressed proteins, pacsin1 and dynamin, may be involved the same cellular pathway—the rat ortholog of pacsin1 was identified as a synaptic protein that interacts with dynamin.20 Dynamin is a neuron-specific guanosine triphosphatase that is important in endocytosis at the fission step of nascent clathrin-coated vesicles from the plasma membrane.21 In mice lacking dynamin, synaptic vesicle endocytosis is severely impaired during strong exogenous stimulation but resumes efficiently when the stimulus is terminated, suggesting that dynamin is necessary during high levels of neuronal activity.22 In the rat brain, downregulation of dynamin in cultured hippocampal embryonic neurons leads to impairment of the formation of minor and axon-like processes in a dose-dependent manner.23 Another differentially expressed protein, parvalbumin, encoded by pvalb, belongs to the family of calcium-binding proteins. Parvalbumin-positive neurons are observed in the hippocampal cells; they constitute an abundant subpopulation of cells that express GABA.24 During the development of the rat neocortex and hippocampus, Pvalb mRNA expression is visible after P6.25 As with Sobp, by P10 Pvalb expression is detected mainly in layer V of the neocortex and in the hippocampus; by P14 there is a marked increase of expression in the entire neocortex and in the hippocampus. Significant functional annotations of differentially expressed proteins, with mouse brain genes used as background, were for axon (GO: 0030424) and neuron (GO: 0043005) projection, axon guidance, synaptic vesicle traffic, 5HT3- and 5HT4-type receptor-mediated processes, the alpha and beta3 adrenergic receptors, the cortocotropin releasing factor, and sugar metabolism (results not shown).
Table 1.
Proteins with Greater than 1.5-fold Differences in Expression
| Mus musculus Protein Name | NCBI Reference Sequence | Ratio of +/jc to jc/jc |
|---|---|---|
| Dynamin | 487851 | 1.55 |
| Pacsin1 | 148690599 | 1.59 |
| Dihydropyrimidinase-like 2 | 40254595 | −1.86 |
| Syntaxin binding protein 1 | 17225417 | −1.82 |
| Collapsin response mediator protein 1 isoform 2 | 40068507 | −1.52 |
| Solute carrier family 25 (mitochondrial carrier, Aralar), member 12 | 27369581 | −1.51 |
| Enolase 3, beta muscle, isoform CRA_a | 148680653 | 2.69 |
| Unnamed protein product | 12845061 | 2.77 |
| Acyl-CoA thioesterase 7 | 19923052 | −1.56 |
| Tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, epsilon polypeptide | 31981925 | −1.51 |
| Synaptosomal-associated protein 25 | 6755588 | −1.62 |
| NmrA-like family domain containing 1 | 24431937 | −1.64 |
| Actin related protein 2/3 complex, subunit 2 | 112363072 | −1.58 |
| Phosphoglycerate mutase 1 | 114326546 | −1.52 |
| Carbonic anhydrase III | 31982861 | 3.72 |
| ATP synthase, H+ transporting, mitochondrial F0 complex, subunit d | 21313679 | −3.79 |
| Hypothetical protein LOC66469 | 27754130 | 1.66 |
| Growth hormone | 6679997 | −5.96 |
| ATP synthase, H+ transporting, mitochondrial F1 complex, delta subunit, isoform CRA_a | 148699643 | −1.57 |
| Unnamed protein product | 74150634 | −1.96 |
| Parvalbumin | 53819 | 1.88 |
| PREDICTED: similar to expressed in non-metastatic cells 1, protein (NM23A) (nucleoside diphosphate) | 73963227 | 3.23 |
| Beta-globin | 156257681 | 1.56 |
| Heat-responsive protein 12 | 40807498 | −2.06 |
In summary, we describe a gene involved in human cognition, SOBP, that shows the highest expression in the limbic system postnatally and that, when mutated, causes syndromic and nonsyndromic autosomal-recessive ID.
Acknowledgments
This study was supported by the Israeli Ministry of Health Chief Scientist Foundation (grant no. 3-4963) and the Israeli Science Foundation (grant no. 558/09). The authors thank Gabrielle J. Halpern for her help with editing of the manuscript and the members of the family for their cooperation.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Database for Annotation, Visualization and Integrated Discovery (DAVID), http://david.abcc.ncifcrf.gov/home.jsp
ECR browser, http://ecrbrowser.dcode.org/xB.php?db=hg18&location=chr6:107918010-108089206
ESLPred, http://www.imtech.res.in/raghava/eslpred/index.html
Homozygosity Mapper, http://www.homozygositymapper.org/
Multiple Em for Motif Elicitation (MEME), http://meme.sdsc.edu/meme4_4_0/cgi-bin/meme.cgi
Mouse Genome Informatics (MGI) database, http://www.informatics.jax.org/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
PANTHER, http://www.pantherdb.org
Predict NLS, http://cubic.bioc.columbia.edu/services/predictNLS/
RNABindR, http://bindr2.gdcb.iastate.edu/RNABindR/
UniProt server, http://www.uniprot.org/
References
- 1.McLaren J., Bryson S.E. Review of recent epidemiological studies of mental retardation: prevalence, associated disorders, and etiology. Am. J. Ment. Retard. 1987;92:243–254. [PubMed] [Google Scholar]
- 2.Leonard H., Wen X. The epidemiology of mental retardation: challenges and opportunities in the new millennium. Ment. Retard. Dev. Disabil. Res. Rev. 2002;8:117–134. doi: 10.1002/mrdd.10031. [DOI] [PubMed] [Google Scholar]
- 3.Frints S.G., Froyen G., Marynen P., Fryns J.P. X-linked mental retardation: vanishing boundaries between non-specific (MRX) and syndromic (MRXS) forms. Clin. Genet. 2002;62:423–432. doi: 10.1034/j.1399-0004.2002.620601.x. [DOI] [PubMed] [Google Scholar]
- 4.Kleefstra T., Hamel B.C. X-linked mental retardation: further lumping, splitting and emerging phenotypes. Clin. Genet. 2005;67:451–467. doi: 10.1111/j.1399-0004.2005.00434.x. [DOI] [PubMed] [Google Scholar]
- 5.Basel-Vanagaite L. Genetics of autosomal recessive non-syndromic mental retardation: recent advances. Clin. Genet. 2007;72:167–174. doi: 10.1111/j.1399-0004.2007.00881.x. [DOI] [PubMed] [Google Scholar]
- 6.Garshasbi M., Hadavi V., Habibi H., Kahrizi K., Kariminejad R., Behjati F., Tzschach A., Najmabadi H., Ropers H.H., Kuss A.W. A defect in the TUSC3 gene is associated with autosomal recessive mental retardation. Am. J. Hum. Genet. 2008;82:1158–1164. doi: 10.1016/j.ajhg.2008.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Molinari F., Foulquier F., Tarpey P.S., Morelle W., Boissel S., Teague J., Edkins S., Futreal P.A., Stratton M.R., Turner G. Oligosaccharyltransferase-subunit mutations in nonsyndromic mental retardation. Am. J. Hum. Genet. 2008;82:1150–1157. doi: 10.1016/j.ajhg.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mir A., Kaufman L., Noor A., Motazacker M.M., Jamil T., Azam M., Kahrizi K., Rafiq M.A., Weksberg R., Nasr T. Identification of mutations in TRAPPC9, which encodes the NIK- and IKK-beta-binding protein, in nonsyndromic autosomal-recessive mental retardation. Am. J. Hum. Genet. 2009;85:909–915. doi: 10.1016/j.ajhg.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mochida G.H., Mahajnah M., Hill A.D., Basel-Vanagaite L., Gleason D., Hill R.S., Bodell A., Crosier M., Straussberg R., Walsh C.A. A truncating mutation of TRAPPC9 is associated with autosomal-recessive intellectual disability and postnatal microcephaly. Am. J. Hum. Genet. 2009;85:897–902. doi: 10.1016/j.ajhg.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Philippe O., Rio M., Carioux A., Plaza J.M., Guigue P., Molinari F., Boddaert N., Bole-Feysot C., Nitschke P., Smahi A. Combination of linkage mapping and microarray-expression analysis identifies NF-kappaB signaling defect as a cause of autosomal-recessive mental retardation. Am. J. Hum. Genet. 2009;85:903–908. doi: 10.1016/j.ajhg.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Basel-Vanagaite L., Rainshtein L., Inbar D., Gothelf D., Hennekam R., Straussberg R. Autosomal recessive mental retardation syndrome with anterior maxillary protrusion and strabismus: MRAMS syndrome. Am. J. Med. Genet. A. 2007;143A:1687–1691. doi: 10.1002/ajmg.a.31810. [DOI] [PubMed] [Google Scholar]
- 12.Fishelson M., Geiger D. Exact genetic linkage computations for general pedigrees. Bioinformatics. 2002;18(Suppl 1):S189–S198. doi: 10.1093/bioinformatics/18.suppl_1.s189. [DOI] [PubMed] [Google Scholar]
- 13.Ovcharenko I., Nobrega M.A., Loots G.G., Stubbs L. ECR Browser: a tool for visualizing and accessing data from comparisons of multiple vertebrate genomes. Nucleic Acids Res. 2004;32(Web Server issue):W280–W286. doi: 10.1093/nar/gkh355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tucker E.S., Segall S., Gopalakrishna D., Wu Y., Vernon M., Polleux F., Lamantia A.S. Molecular specification and patterning of progenitor cells in the lateral and medial ganglionic eminences. J. Neurosci. 2008;28:9504–9518. doi: 10.1523/JNEUROSCI.2341-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Han X., Wu X., Chung W.Y., Li T., Nekrutenko A., Altman N.S., Chen G., Ma H. Transcriptome of embryonic and neonatal mouse cortex by high-throughput RNA sequencing. Proc. Natl. Acad. Sci. USA. 2009;106:12741–12746. doi: 10.1073/pnas.0902417106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pimenta A.F., Reinoso B.S., Levitt P. Expression of the mRNAs encoding the limbic system-associated membrane protein (LAMP): II. Fetal rat brain. J. Comp. Neurol. 1996;375:289–302. doi: 10.1002/(SICI)1096-9861(19961111)375:2<289::AID-CNE8>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 17.Kenyon K.L., Li D.J., Clouser C., Tran S., Pignoni F. Fly SIX-type homeodomain proteins Sine oculis and Optix partner with different cofactors during eye development. Dev. Dyn. 2005;234:497–504. doi: 10.1002/dvdy.20442. [DOI] [PubMed] [Google Scholar]
- 18.Calderon A., Derr A., Stagner B.B., Johnson K.R., Martin G., Noben-Trauth K. Cochlear developmental defect and background-dependent hearing thresholds in the Jackson circler (jc) mutant mouse. Hear. Res. 2006;221:44–58. doi: 10.1016/j.heares.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 19.Chen Z., Montcouquiol M., Calderon R., Jenkins N.A., Copeland N.G., Kelley M.W., Noben-Trauth K. Jxc1/Sobp, encoding a nuclear zinc finger protein, is critical for cochlear growth, cell fate, and patterning of the organ of corti. J. Neurosci. 2008;28:6633–6641. doi: 10.1523/JNEUROSCI.1280-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qualmann B., Roos J., DiGregorio P.J., Kelly R.B. Syndapin I, a synaptic dynamin-binding protein that associates with the neural Wiskott-Aldrich syndrome protein. Mol. Biol. Cell. 1999;10:501–513. doi: 10.1091/mbc.10.2.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takei K., Slepnev V.I., Haucke V., De Camilli P. Functional partnership between amphiphysin and dynamin in clathrin-mediated endocytosis. Nat. Cell Biol. 1999;1:33–39. doi: 10.1038/9004. [DOI] [PubMed] [Google Scholar]
- 22.Ferguson S.M., Brasnjo G., Hayashi M., Wölfel M., Collesi C., Giovedi S., Raimondi A., Gong L.W., Ariel P., Paradise S. A selective activity-dependent requirement for dynamin 1 in synaptic vesicle endocytosis. Science. 2007;316:570–574. doi: 10.1126/science.1140621. [DOI] [PubMed] [Google Scholar]
- 23.Torre E., McNiven M.A., Urrutia R. Dynamin 1 antisense oligonucleotide treatment prevents neurite formation in cultured hippocampal neurons. J. Biol. Chem. 1994;269:32411–32417. [PubMed] [Google Scholar]
- 24.Sauer J.-F., Bartos M. Recruitment of early postnatal parvalbumin-positive hippocampal interneurons by GABAergic excitation. J. Neurosci. 2010;30:110–115. doi: 10.1523/JNEUROSCI.4125-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Lecea L., del Río J.A., Soriano E. Developmental expression of parvalbumin mRNA in the cerebral cortex and hippocampus of the rat. Brain Res. Mol. Brain Res. 1995;32:1–13. doi: 10.1016/0169-328x(95)00056-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.