

Abstract
In human mitochondria, polyadenylation of mRNA, undertaken by the nuclear-encoded mitochondrial poly(A) RNA polymerase, is essential for maintaining mitochondrial gene expression. Our molecular investigation of an autosomal-recessive spastic ataxia with optic atrophy, present among the Old Order Amish, identified a mutation of MTPAP associated with the disease phenotype. When subjected to poly(A) tail-length assays, mitochondrial mRNAs from affected individuals were shown to have severely truncated poly(A) tails. Although defective mitochondrial DNA maintenance underlies a well-described group of clinical disorders, our findings reveal a defect of mitochondrial mRNA maturation associated with human disease and imply that this disease mechanism should be considered in other complex neurodegenerative disorders.
Main Text
In the current study, we investigated an extensive Old Order Amish family in which multiple children were affected by a slowly progressive autosomal-recessive neurodegenerative condition, the key features of which are cerebellar ataxia (limb and truncal), spastic paraparesis (increased lower limb tone with brisk knee jerks and extensor plantars, but no weakness), dysarthria (mixture of cerebellar and spastic), optic atrophy, learning difficulties, and, in some, emotional liability. The clinical features found in the affected individuals are shown in detail in Table 1. Speech and walking are usually delayed and abnormal from the onset. The two younger cases (IX-6 and IX-7) have predominantly cerebellar gait and speech disturbance. In the four older cases (IX-2 to IX-5), tongue movements are slow and spastic, jaw jerk is brisk, and two have increased tone in the upper limbs. Their parents report a clear progressive decline in function, although all are still independently mobile with a spastic ataxic gait, with no obvious intellectual deterioration, needing only mild assistance for most self-care. The most severely ataxic is IX-5, whose oromandibular coordination has become so poor that, since the age of 16, she has had to use a straw for drinking. In addition to the parents' accounts, the relative severity of the clinical picture in the older individuals compared to the much younger IX-6 and IX-7 also supports the impression that this is a progressive condition. Diminished or absent upper limb and ankle reflexes in older cases, which were said to have been present in pediatric assessments, suggest additional progressive lower motor neuron involvement. Older individuals attended school until the age of 14 (the traditional limit of Amish education), but special tuition was required from early on. Individual IX-3 had a full IQ estimate at age 12 of only 47 (verbal 61, performance 45). All patients live with their parents, none are married, and older individuals either attend a special workshop or help with basic household tasks.
Table 1.
Summary of Clinical Features of Affected Cases
| Patient | IX-2 | IX-3 | IX-4 | IX-5 | IX-6 | IX-7 |
|---|---|---|---|---|---|---|
| Age when seen (years) | 27 | 26 | 25 | 18 | 2 | 6 |
| Age at which walked (months) | 12 (tiptoed until age 3) | 16 | 11 (with falls) | 12 | 24 (abnormal) | 18 (with falls) |
| Age at which talked (years) | 3 | 2 | 1 | >1 | 2 | 2 |
| Speech normal at onset? | N | N | Y | N | N | N |
| Optic atrophy | Y | Y | Y | Y | NK | NK |
| Nystagmus | N | Y (horizontal) | Y (vertical) | N | N | N |
| Emotional liability | Y | N | Y | Y | N | Y |
| Dysarthria | Y | Y | Y | Y | Y | Y |
| Tone in upper limbs | Increased | Normal | Increased | Normal | Normal | Normal |
| Tone in lower limbs | Increased | Increased | Increased | Increased | Normal | Normal |
| Power in limbs | Normal | Normal | Normal | Normal | Normal | Normal |
| Reflexes: biceps | + | + | − | − | ++ | + |
| Reflexes: supinator | + | + | − | − | ++ | + |
| Reflexes: triceps | + | + | − | − | ++ | + |
| Reflexes: knee | +++ | +++ | − | +++ | +++ | +++ |
| Reflexes: ankle | − | − | − | − | ++ | ++ |
| Plantars | Mute | Extensor | Extensor | Extensor | Extensor | Extensor |
| Limb ataxia | Y | Y | Y | Y | N | N |
| Gait ataxia | Y | Y | Y | Y | Y | Y |
The following abbreviations are used: Y, yes; N, no; NK, not known; reflexes: −, absent; +, diminished; ++, normal; +++, pathologically brisk.
Assuming that a founder mutation was responsible, we undertook a genome-wide microarray scan with DNA extracted from blood samples of family members obtained with informed consent and following approved institutional review board protocols. The genome-wide screen was performed with Affymetrix 250K SNP NspI arrays (Geneservice) by using DNA from the four affected cases initially available (IX-2, IX-3, IX-4, IX-5; Figure 1A). Homozygosity analysis identified a single autozygous region of 6.5 Mb on chromosome 10p11.23, delimited by markers rs1144522 and rs910967, likely to correspond to the disease locus; no other notable regions of autozygosity were observed. Autozygosity across this interval was corroborated by microsatellite marker analysis, determined by PCR amplification, and subsequent analysis by polyacrylamide gel electrophoresis, visualized by silver staining via standard methodologies (Figure 1B). Multipoint LOD scores were calculated across the microsatellite markers with Simwalk version 2.91,1,2 revealing a LODmax score of 7.64, assuming an autosomal-recessive mode of inheritance, complete penetrance, and a disease gene frequency of 0.0001%. The putative disease locus is predicted to contain 46 genes, of which 7 are hypothetical and 15 are pseudogenes. While screening the remaining 24 genes, a nonsynonymous base change was identified in the nuclear-encoded mitochondrial poly(A) RNA polymerase gene MTPAP (c.1432A>G; Figure 1C). The c.1432A>G variant in exon 9 of PAPD1 creates a BamHI restriction endonuclease recognition site. Exon 9 PCR products generated from control and patient samples were subsequently digested with one unit of BamHI and were visualized on agarose gels. This sequence variant cosegregated with the disease phenotype and was not detected in 600 control chromosomes of unaffected individuals of European ancestry. Analysis of 200 control chromosomes from unaffected individuals from the same Ohio Amish community identified a single heterozygous carrier of the sequence variant, which is not unexpected in this endogamous community, in which founder mutations are commonly associated with rare inherited diseases in multiple families.3–5 The resultant predicted p.N478D substitution occurs within an N-P-F-E sequence (Figure 1D) located within one of the most highly conserved regions of the molecule in all species examined.6
Figure 1.
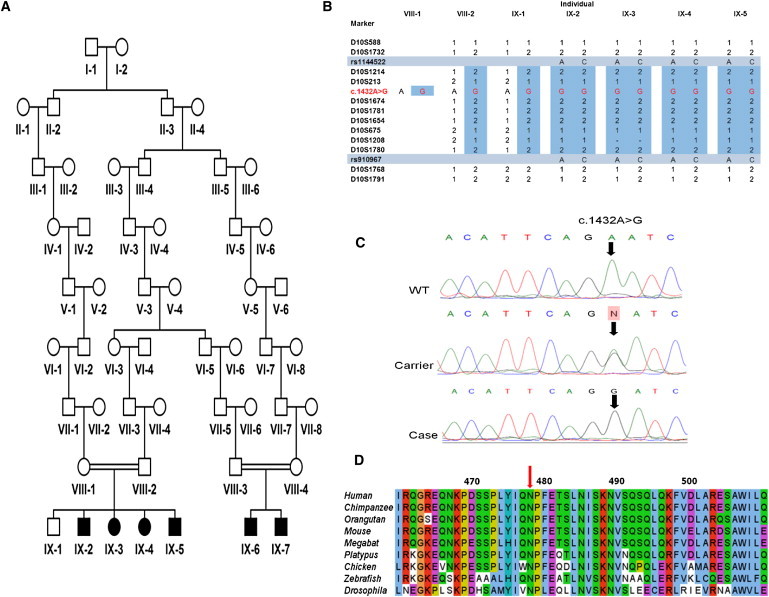
Identification of MTPAP Mutation in Amish Pedigree
(A) Pedigree diagram of Amish family.
(B) Marker genotypings across the region of homozygosity identified by whole-genome SNP analysis. Initial genome-wide screen was undertaken in individuals IX-2, IX-3, IX-4, and IX-5 for which samples were available at the time. The region of autozygosity defined was delimited by SNPs rs1144522 and rs910967. The mutation (NC_000010.10: g.30602855T>C; NM_018109.3: c.1432A>G) is shown in red. All affected individuals, including IX-6 and IX-7, were subsequently found to be homozygous for the c.1432A>G variant, whereas parental samples and the unaffected sibling were carriers.
(C) Electropherograms showing MTPAP exon 9 sequence encompassing the NM_018109.3: c.1432A>G variant in a wild-type (WT) control, a heterozygous carrier parent, and a homozygous affected individual.
(D) ClustalW2 alignment of the region encompassing the PAP/25A-associated domain of MTPAP from various species. Amino acid altered by the c.1432A>G substitution (p.N478D) is indicated with the red arrow.
In order to confirm that the c.1432A>G alteration is pathogenic and to determine its functional consequence, we undertook mitochondrial poly(A) tail (MPAT) assays of representative mitochondrial-encoded transcripts (RNA14 and MTCO1 [MIM 516030]) of the Amish family with the putative mutation. These assays allowed us to visualize the heterogeneous poly(A) tails of the various species generated by mitochondrial poly(A) RNA polymerase (mtPAP). RNA (2.5 μg) was extracted from whole blood (PAXgene Blood RNA kit, QIAGEN) according to the manufacturer's instructions. Linker primers7 were ligated with NEB T4 RNA ligase. All subsequent steps were exactly as described in Temperley et al.,7 including primer sequences for RNA14 and anti-ligation primer. For MTCO1, first-round PCR primer was 5′-CATATTCATCGGCGTAAATC-3′ and labeled second-round PCR primer was 5′-CAACCCCATGGCCTCCA-3′. PCR profiles were as described.7
Gels were exposed to PhosphorImage screens and ImageQuant 5.2 software used for visualization and densitometric analyses. Relative fraction of oligoadenylated species was calculated as oligo/(oligo + poly) and presented as a percentage. Oligoadenylated species were defined as >10 nt extension from the deadenylated species and were measured against the size markers. For polyadenylated species, RNA14 was defined as 30–50 nt extensions, and MTCO1 was defined as 29–50 nt extensions. As shown in Figure 2, oligoadenylated species were present in all samples, consistent with previous reports in which mtPAP had been depleted by small interfering RNA treatment.6,8 Polyadenylation in both carrier parents appeared to be normal, but the MPAT assay revealed a profound effect on polyadenylation associated with homozygosity of the sequence variant in all affected cases. Whereas the percentage of oligoadenylated (>10 nt) versus polyadenylated (>30 nt extension) RNA14 transcript was less than 10% in the unaffected parent carriers (Figure 2, bottom, lanes 1 and 2), a dramatic reversal in this ratio was apparent in all patients homozygous for the c.1432A>G mutation (87%–93%, lanes 3–6). A second transcript, MTCO1, was also assessed (Figure 2, top), with similar results (unaffected parents 23%–40%, affected siblings 80%–92%). Together with the genetic data, these findings strongly suggest that the p.N478D mtPAP is responsible for the condition in this family.
Figure 2.
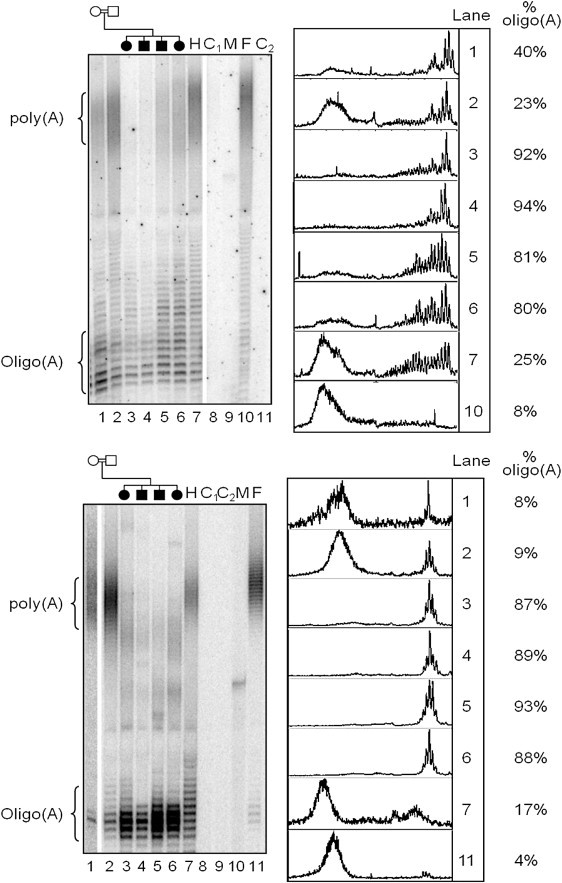
Poly(A) Status of Representative Mitochondrial mRNAs from Patient and Control Samples Available
RNA (2.5 μg) from each individual was ligated to linker and subjected to the mitochondrial poly(A) tail-length assay as described in Temperley et al.7 Radiolabeled products were separated through a 10% denaturing polyacrylamide gel, and lengths were calculated by comparison against size markers (P32 end-labeled synthetic oligomers of 20 and 75 nt). Oligoadenylated (<10 nt extension from processed transcript) or polyadenylated (35–50 nt extension RNA14; 29–50 nt MTCO1) species were quantified following densitometric scans (ImageQuant software). Lane 1 denotes the mother, lane 2 denotes the father, and lanes 3–6 denote the affected children. The following abbreviations are used: H, HEK293T control cell line; C1 and C2, negative amplification controls from the first and second rounds of PCR; M, size marker; F, control fibroblast cell line. Top: MTCO1 transcript; bottom: RNA14 transcript.
What is the function of polyadenylation in the mammalian mitochondrion? The answer is complex and, to date, has not been fully resolved. Terminal 3′ oligoadenylation of mitochondrial mRNAs is clearly essential, because processing of several mitochondrial open reading frames from the primary polycistronic transcript leaves a truncated terminal codon that requires adenylation for completion.9 Polyadenylation of mRNA is, however, crucial for maintaining global mRNA expression in mammalian mitochondria.8,10,11 Depletion of mtPAP clearly causes instability of some transcripts,8 but not others.6,10,11 Indeed, enzymatic removal of the poly(A) tail from mitochondrial transcripts in intact cells can actually promote stability of a subset of mRNAs.11 In contrast, occlusion of the poly(A) extensions clearly leads to a decrease in translation efficiency.11 Although the exact role or roles of mitochondrial mRNA polyadenylation remain unclear, depletion of mtPAP has a profound effect on mitochondrial function8 and is evidenced by the severe neurodegenerative disorder found in this family.
Inherited mitochondrial disorders commonly affect postmitotic tissues, such as muscle and the central and peripheral nervous system, and are widely acknowledged as an important cause of neurological disease, with cerebellar ataxia a frequent feature and occasionally the main one (e.g., the mitochondrial recessive ataxia syndrome). Adult-onset mitochondrial diseases are typically caused by mutations within the mitochondrial genome, whereas mutations in nuclear-encoded mitochondrial proteins usually present with more severe childhood phenotypes and multisystem disorders, in addition to neurological involvement (reviewed in Rahman and Hanna12). These childhood disorders are often associated with instability of the mitochondrial genome, resulting in mtDNA deletions (e.g., POLG [MIM 174763], POLG2 [MIM 604983], ANT1 [MIM 103220], PEO1 [MIM 606075], OPA1 [MIM 605290], TYMP [MIM 131222]) or mtDNA depletion (DGUOK [MIM 601465], SUCLA2 [MIM 603921], SUCLG1 [MIM 611224], RRM2B [MIM 604712], MPV17 [MIM 137960], TK2 [MIM 188250]).
In contrast, one category of mitochondrial disorders that is becoming increasingly diagnosed is that of nuclear gene mutations that cause mitochondrial protein synthesis disorders with no evidence of mtDNA involvement. This highly heterogeneous group mostly shares a combined disorder of respiratory chain complexes. To date, the underlying genetic defects encode mt-tRNA modifying and aminoacylating enzymes (PUS1, TRMU, DARS2, RARS2, YARS2), an mRNA stability factor and translational activator (LRPPRC, TACO1), translation factors (GFM1, TUFM, TSFM, C12orf65), components of the mitochondrial ribosome (MRPS16/22), and factors that mediate mitochondrial ribosome assembly (SPG7, AFG3L2). Clinical presentations are diverse, but, other than the mt-tRNA modifying defects, almost all patients have neurological deficits. Most strikingly, mutations in two components of the mitochondrial mAAA protease that are essential for mitochondrial ribosome maturation, paraplegin and AFG3-like protein, cause hereditary spastic paraplegia (SPG713) and cerebellar ataxia (SCA2814), respectively. Both presentations are similar to the clinical disorder that we have identified in patients with mtPAP defects. The SPG7 phenotype can include cerebellar ataxia and optic atrophy in addition to spastic paraparesis,15 and SCA28 patients have pyramidal signs in addition to ataxia.16 Similarly, Friedreich's ataxia, the most common autosomal-recessive ataxia, may display a comparable phenotype to the patients described here with spasticity, areflexia, and optic atrophy. The mutated protein, frataxin, is also a mitochondrial protein, and the latest evidence suggests that it is involved in synthesis of iron-sulfur clusters whose diverse functions include electron transfer in the mitochondrial respiratory complex.17 Although a number of diverse molecular mechanisms lead to autosomal-recessive ataxias, our findings provide evidence of a new mitochondrial abnormality that may lead to a complex neurodegenerative disorder with ataxia as the prominent component.
Acknowledgments
This work was supported by the Birth Defects Foundation (UK)/Newlife Foundation for Disabled Children. We would like to thank the Amish families for participating in this study. Sequencing was done in the Medical Biomics (St. George's, University of London). R.N.L. and Z.M.A.C.-L. would like to thank The Wellcome Trust (074454/Z/04/Z) and the Biotechnology and Biological Sciences Research Council (BB/F011520/1) for continuing support.
Web Resources
The URLs for data presented herein are as follows:
ClustalW2, http://www.ebi.ac.uk/Tools/clustalw2
NCBI National Center for Biotechnology Information, http://www.ncbi.nlm.nih.gov/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
UCSC Genome Browser, http://genome.ucsc.edu/
Accession Numbers
The GenBank accession numbers for PAPD1 are NM_018109.3 and NP_060579.3.
References
- 1.Sobel E., Lange K. Descent graphs in pedigree analysis: Applications to haplotyping, location scores, and marker-sharing statistics. Am. J. Hum. Genet. 1996;58:1323–1337. [PMC free article] [PubMed] [Google Scholar]
- 2.Sobel E., Sengul H., Weeks D.E. Multipoint estimation of identity-by-descent probabilities at arbitrary positions among marker loci on general pedigrees. Hum. Hered. 2001;52:121–131. doi: 10.1159/000053366. [DOI] [PubMed] [Google Scholar]
- 3.Patel H., Cross H., Proukakis C., Hershberger R., Bork P., Ciccarelli F.D., Patton M.A., McKusick V.A., Crosby A.H. SPG20 is mutated in Troyer syndrome, an hereditary spastic paraplegia. Nat. Genet. 2002;31:347–348. doi: 10.1038/ng937. [DOI] [PubMed] [Google Scholar]
- 4.Simpson M.A., Cross H., Proukakis C., Pryde A., Hershberger R., Chatonnet A., Patton M.A., Crosby A.H. Maspardin is mutated in mast syndrome, a complicated form of hereditary spastic paraplegia associated with dementia. Am. J. Hum. Genet. 2003;73:1147–1156. doi: 10.1086/379522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simpson M.A., Cross H., Proukakis C., Priestman D.A., Neville D.C., Reinkensmeier G., Wang H., Wiznitzer M., Gurtz K., Verganelaki A. Infantile-onset symptomatic epilepsy syndrome caused by a homozygous loss-of-function mutation of GM3 synthase. Nat. Genet. 2004;36:1225–1229. doi: 10.1038/ng1460. [DOI] [PubMed] [Google Scholar]
- 6.Tomecki R., Dmochowska A., Gewartowski K., Dziembowski A., Stepien P.P. Identification of a novel human nuclear-encoded mitochondrial poly(A) polymerase. Nucleic Acids Res. 2004;32:6001–6014. doi: 10.1093/nar/gkh923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Temperley R.J., Seneca S.H., Tonska K., Bartnik E., Bindoff L.A., Lightowlers R.N., Chrzanowska-Lightowlers Z.M. Investigation of a pathogenic mtDNA microdeletion reveals a translation-dependent deadenylation decay pathway in human mitochondria. Hum. Mol. Genet. 2003;12:2341–2348. doi: 10.1093/hmg/ddg238. [DOI] [PubMed] [Google Scholar]
- 8.Nagaike T., Suzuki T., Katoh T., Ueda T. Human mitochondrial mRNAs are stabilized with polyadenylation regulated by mitochondria-specific poly(A) polymerase and polynucleotide phosphorylase. J. Biol. Chem. 2005;280:19721–19727. doi: 10.1074/jbc.M500804200. [DOI] [PubMed] [Google Scholar]
- 9.Ojala D., Crews S., Montoya J., Gelfand R., Attardi G. A small polyadenylated RNA (7 S RNA), containing a putative ribosome attachment site, maps near the origin of human mitochondrial DNA replication. J. Mol. Biol. 1981;150:303–314. doi: 10.1016/0022-2836(81)90454-x. [DOI] [PubMed] [Google Scholar]
- 10.Slomovic S., Schuster G. Stable PNPase RNAi silencing: Its effect on the processing and adenylation of human mitochondrial RNA. RNA. 2008;14:310–323. doi: 10.1261/rna.697308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wydro M., Bobrowicz A., Temperley R.J., Lightowlers R.N., Chrzanowska-Lightowlers Z.M. Targeting of the cytosolic poly(A) binding protein PABPC1 to mitochondria causes mitochondrial translation inhibition. Nucleic Acids Res. 2010;38:3732–3742. doi: 10.1093/nar/gkq068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rahman S., Hanna M.G. Diagnosis and therapy in neuromuscular disorders: Diagnosis and new treatments in mitochondrial diseases. J. Neurol. Neurosurg. Psychiatry. 2009;80:943–953. doi: 10.1136/jnnp.2008.158279. [DOI] [PubMed] [Google Scholar]
- 13.Casari G., De Fusco M., Ciarmatori S., Zeviani M., Mora M., Fernandez P., De Michele G., Filla A., Cocozza S., Marconi R. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell. 1998;93:973–983. doi: 10.1016/s0092-8674(00)81203-9. [DOI] [PubMed] [Google Scholar]
- 14.Di Bella D., Lazzaro F., Brusco A., Plumari M., Battaglia G., Pastore A., Finardi A., Cagnoli C., Tempia F., Frontali M. Mutations in the mitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28. Nat. Genet. 2010;42:313–321. doi: 10.1038/ng.544. [DOI] [PubMed] [Google Scholar]
- 15.Wilkinson P.A., Crosby A.H., Turner C., Bradley L.J., Ginsberg L., Wood N.W., Schapira A.H., Warner T.T. A clinical, genetic and biochemical study of SPG7 mutations in hereditary spastic paraplegia. Brain. 2004;127:973–980. doi: 10.1093/brain/awh125. [DOI] [PubMed] [Google Scholar]
- 16.Mariotti C., Brusco A., Di Bella D., Cagnoli C., Seri M., Gellera C., Di Donato S., Taroni F. Spinocerebellar ataxia type 28: A novel autosomal dominant cerebellar ataxia characterized by slow progression and ophthalmoparesis. Cerebellum. 2008;7:184–188. doi: 10.1007/s12311-008-0053-9. [DOI] [PubMed] [Google Scholar]
- 17.Stemmler T.L., Lesuisse E., Pain D., Dancis A. Frataxin and mitochondrial FeS cluster biogenesis. J. Biol. Chem. 2010;285:26737–26743. doi: 10.1074/jbc.R110.118679. [DOI] [PMC free article] [PubMed] [Google Scholar]