

Abstract
Mutations in smooth muscle cell (SMC)-specific isoforms of α-actin and β-myosin heavy chain, two major components of the SMC contractile unit, cause familial thoracic aortic aneurysms leading to acute aortic dissections (FTAAD). To investigate whether mutations in the kinase that controls SMC contractile function (myosin light chain kinase [MYLK]) cause FTAAD, we sequenced MYLK by using DNA from 193 affected probands from unrelated FTAAD families. One nonsense and four missense variants were identified in MYLK and were not present in matched controls. Two variants, p.R1480X (c.4438C>T) and p.S1759P (c.5275T>C), segregated with aortic dissections in two families with a maximum LOD score of 2.1, providing evidence of linkage of these rare variants to the disease (p = 0.0009). Both families demonstrated a similar phenotype characterized by presentation with an acute aortic dissection with little to no enlargement of the aorta. The p.R1480X mutation leads to a truncated protein lacking the kinase and calmodulin binding domains, and p.S1759P alters amino acids in the α-helix of the calmodulin binding sequence, which disrupts kinase binding to calmodulin and reduces kinase activity in vitro. Furthermore, mice with SMC-specific knockdown of Mylk demonstrate altered gene expression and pathology consistent with medial degeneration of the aorta. Thus, genetic and functional studies support the conclusion that heterozygous loss-of-function mutations in MYLK are associated with aortic dissections.
Main Text
Myosin light chain kinase (MLCK [MIM 600922]), encoded by MYLK, is a ubiquitously expressed kinase whose only known target of phosphorylation is the 20 kDa regulatory light chain (RLC [MIM 160781]) of smooth and nonmuscle myosin II. RLC phosphorylation increases actin-activated myosin II ATPase activity and regulates many cellular actin-myosin II cytoskeleton-mediated functions, such as secretion, endocytosis, cytokinesis, cell spreading and migration, cytoskeletal clustering of integrins at focal adhesions, and stress-fiber formation.1 MLCK is highly expressed in smooth muscle cells (SMCs), where phosphorylation of RLC by MLCK initiates the physiologic contraction of smooth muscle within hollow organs, and targeted deletion of MLCK in mouse SMCs results in markedly reduced RLC phosphorylation, as well as arterial hypotension, reduced gut motility, and urinary dysfunction.2 In arteries, the SMC myogenic response to mechanical load occurs as a result of the stretch activation of cation channels, triggering an influx of calcium that binds to calmodulin (CaM) encoded by CALM1 (MIM 114180).3,4 The association of the calcium/CaM complex to MLCK activates the kinase, leading to RLC phosphorylation and SMC contractile shortening.5,6 MLCK is also expressed in other muscle cells: skeletal and cardiac muscle express tissue-specific isoforms of MLCK, with MYLK2 (MIM 606566) predominantly expressed in skeletal muscle cells and MYLK3 (MIM 612147) expressed in cardiac muscle cells.7–9
The necessity of maintaining proper SMC contractile function in the ascending aorta throughout a lifetime is suggested by the identification of heterozygous mutations in genes encoding the SMC-specific isoforms of α-actin or β-myosin heavy chain (ACTA2 and MYH11) in approximately 15% of families with inherited adult-onset thoracic aortic disease (MIM 611788 and MIM 132900, respectively).10–12 Approximately 20% of patients with thoracic aortic disease have a family history of the disease, and the disease is typically inherited in an autosomal-dominant manner with decreased penetrance and variable expression. The phenotype is usually characterized by progressive enlargement of the ascending aorta, which predisposes those affected to acute dissections. Acute aortic dissections are a common cause of sudden death that can be prevented if the aorta is surgically repaired prior to dissection.13 However, in a subset of families, little to no enlargement of the ascending thoracic aorta is present prior to the dissection, making clinical decisions that weigh risk of dissection against the risks of surgery extremely difficult for family members unless a predisposing gene mutation is identified. Given the pivotal role of MLCK, CaM, and RLC in regulating SMC contractile function, we sequenced MYLK (NM_053025.3) and CALM1 (NM_006888.4), along with the light chains expressed in SMCs MYL6 (MIM 609931), MYL6B (MIM 609930), and MYL9 (MIM 609905), using DNA of 94 affected probands from unrelated families with two or more members with thoracic aortic aneurysms or aortic dissections (TAAD) in whom the causative mutation was unknown (the study was approved by the Committee for the Protection of Human Subjects at the University of Texas Health Science Center at Houston and informed consent was obtained from study participants).10 We sought to identify rare genetic variants leading to nonsynonymous amino acid changes or disrupting splice donor or acceptor sites in these genes. Although five variants leading to nonsynonymous amino acid changes were identified in MYLK, only one variant, c.5275T>C (p.S1759P), in family TAA026, was absent in 188 ethnically matched controls. In addition, this variant segregated with aortic disease in family TAA026 (Figures 1A and B). No variants identified in the other genes fulfilled these criteria. MYLK was sequenced in DNA from an additional 99 probands with familial TAAD, and another alteration that met the described criteria, c.4438C>T (p.R1480X), in family TAA400, was identified. Three additional variants were identified in MYLK—c.5260G>A (p.A1754T) in TAA225, c.3637G>A (p.V1213M) in TAA445, and c.4195G>A (p.E1399K) in TAA043—that were not present in 188 ethnically matched controls (controls of European descent were used for comparison, with the exception of Hispanic controls used for TAA225), but additional affected family members were not available for segregation analysis.
Figure 1.

Identification of MYLK as a Causative Gene Leading to Familial TAAD
(A) This panel shows the two families with MYLK mutations, p.S1759P and p.R1480X, which segregate with TAAD. The disease status and mutation status of individuals are indicated in the figure key. Current age or age at death for affected individuals and the current age for unaffected individuals are indicated under the individual ID.
(B) MYLK encodes three products from independent promoters: long form, short form, and telokin. The alterations in black indicate polymorphisms that are present in controls. The alterations in blue are absent in ethnically matched controls, but segregation with disease could not be tested. These changes do not affect conserved amino acids and do not disrupt the kinase domain. The alterations shown in red are predicted to affect kinase activity.
(C) Three-dimensional structures of the MLCK kinase domain and the CaM-binding sequence are presented in green and yellow, respectively. The remainder of the structure is shown in gray. The CaM-binding sequence forms a basic α-helix secondary structure containing Alaline-1754 and Serine-1759, shown in red.
MLCK p.S1759P segregated with aortic disease in family TAA026 with a LOD score of 0.3, and p.R1480X segregated in TAA400 with a LOD score of 1.2.10 Because aortic dissections can cause sudden death, two TAA400 family members who died suddenly of unknown causes were also included, which raised the LOD score to 1.8. Because a candidate-gene approach was used to identify these rare genetic variants rather than a genome-wide search, the combined LOD score of 2.1 provided significant evidence of linkage of the genotype to the disease (p = 0.0009).
MYLK encodes three gene products expressed from separate promoters, with two isoforms containing the catalytic and CaM-binding domains (the 220 kDa long form and the 130 kDa short form, respectively) and a third, small, noncatalytic protein product called telokin (Figure 1B).1 Telokin is a 17 kDa protein that affects calcium sensitivity of contraction, primarily in intestinal smooth muscle. Two identified genetic variants, p.V1213M and p.E1399K, lie outside the kinase domain and are not predicted to disrupt kinase activity or telokin expression. The p.R1480X mutation leads to either nonsense-mediated decay of the message or a truncated protein missing the kinase and CaM binding domains and is therefore predicted to disrupt kinase activity but not to disturb telokin expression. The missense alterations p.A1754T and p.S1759P disrupt amino acids in the α-helix of the CaM-binding sequence (Figure 1C). The p.S1759P alteration was particularly interesting because phosphorylation of this serine in MLCK disrupts CaM binding, thereby desensitizing MLCK to activation by calcium/CaM.14 On the basis of this information, we predicted that these last two alterations lead to loss of MLCK function by altering CaM binding.
To test this hypothesis, we initially confirmed that the 130 kDa short form of MLCK is the expressed isoform in human and mouse aortic tissue, as previously described (Figure 2A).15 To assess the effect of the missense alterations on MLCK function, the 130 kDa form of wild-type MLCK along with the p.S1759P and p.A1754T MLCK mutants were expressed in COS7 cells with an N-terminal Flag tag. Immunoblot analysis using anti-MLCK and anti-Flag indicated that endogenous expression of MLCK was minimal in transfected COS7 cells, and protein levels increased significantly with transfection of the MLCK constructs (Figure 2B).16 Immunoprecipitation with anti-Flag in the presence of calcium demonstrated that the wild-type MLCK binds to endogenous CaM in the presence of Ca2+, but the binding was abolished in the p.S1759P mutant MLCK and decreased in the p.A1754T mutant MLCK (Figure 2C). For further analysis of the kinase activity and CaM binding, the wild-type and mutant MLCK were purified, and the kinase activity (Vmax) and binding to CaM (KCaM) were determined for both. MLCK mutants p.S1759P and p.A1754T showed significant reduction in kinase activity, with a 6-fold reduction for p.S1759P (Figure 2D). MLCK mutants p.S1759P and p.A1754T also, respectively, required 7- and 4-fold increases in CaM concentration for half-maximal activation as compared to wild-type kinase, indicating reduced binding affinity to CaM (Figure 2E). The genetic-segregation and functional studies support the conclusion that p.S1759P and p.R1480X are causative alterations for FTAAD. On the other hand, additional studies are needed to confirm whether p.A1754T, p.V1213M, or p.E1399K lead to aortic disease.
Figure 2.
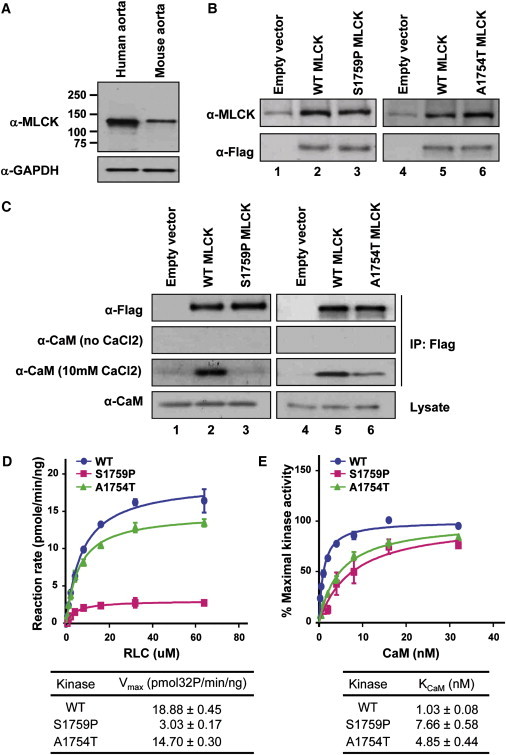
Expression of MLCK in Aorta and Assessment of Kinase Activity in MYLK Mutants
(A) Long-form and short-form MLCK were examined in human and mouse aortas. Only a 130 kDa protein band was detected in extracts from aortic tissues. Positions of molecular-weight markers are indicated at the side of the panel.
(B) COS7 cells were transfected with an empty vector (lane 1) or with vectors expressing Flag-tagged, wild-type short-form MLCK (lane 2), or S1759P short-form MLCK (lane 3). After 48 hr, whole cell lysates were collected for detection of target proteins by immunoblotting with anti-MLCK and anti-Flag. In contrast to lane 1, Flag-tagged short-form MLCK proteins were detected in lanes 2 and 3. Additional COS-7 cells were transfected with empty vectors (lane 4) or with vectors expressing Flag-tagged, wild-type short-form MLCK (lane 5) or A1754T short-form MLCK (lane 6). The target proteins were also detected with anti-MLCK and anti-Flag.
(C) Binding of CaM to MLCK mutants. COS7 cell lysates expressing empty vectors (lane 1), wild-type short-form MLCK (lane 2), and S1759P short-form MLCK (lane 3) were pulled down with the use of anti-Flag. Immunoprecipitates were analyzed by immunoblotting with the use of anti-CaM. In the presence of 10 mM CaCl2, CaM was coimmunoprecipitated with wild-type short-form MLCK but not with S1759P short-form MLCK. In the absence of CaCl2, no α-CaM signal was detected. Additional COS7 cell lysates expressing empty vectors (lane 4), wild-type short-form MLCK (lane 5), and A1754T short-form MLCK (lane 6) were pulled down with the use of anti-Flag and blotted with anti-CaM. The CaM band in lane 5 is more strongly detected than the band in lane 6 in the presence of 10 mM CaCl2.
(D) Assessment of kinase activity of wild-type and mutant MLCK proteins. The rate of 32P incorporation into RLC was measured. The maximal activities of WT MLCK (blue circle), A1754T MLCK (green triangle), and S1759P MLCK (red square) were obtained at different RLC concentrations. The data points represented the mean ± standard error of three or more determinations. The data were fit to the Michaelis-Menten equation for calculation of the Vmax values.
(E) CaM activation of wild-type and mutant MLCK proteins. The relative percentage of maximal kinase activity of WT MLCK (blue circle), A1754T MLCK (green triangle), and S1759P MLCK (red square) was plotted versus various CaM concentrations. The data were presented as mean ± standard error from three experiments. The data were fit to the Michaelis-Menten equation to obtain the KCaM value.
All affected members of families TAA026 and TAA400 similarly presented with acute aortic dissections at variable ages at onset (Table S1 available online), but the most notable feature of these events was that acute aortic dissections occurred with little to no aortic enlargement. For example, one individual (Figure 1A; TAA026, II:2) presented at the age of 43 years with chest pain but no evidence of dissection and minimal ascending aortic enlargement on aortic imaging (4.0 cm, BSA 1.55 m2), although she was found to have a focal acute aortic dissection at the time of surgical repair.17 With acute aortic dissections as the only clinical marker of the presence of the defective gene in an individual, it is likely that there are many other family members with the defective gene who are phenotypically normal. Examination of ascending aortic tissue available from two members of family TAA026 indicated medial degeneration of the aorta, characterized by increased proteoglycan deposition and mild elastic fiber thinning and fragmentation, along with a significant increase in the presence of small arteries in the medial layer of the aorta (Figure 3). Interestingly, a similar increase of arteries in the medial layer has been described in a patient with a MYH11 mutation.18 Other clinical complications reported by individuals harboring MYLK mutations involved the gastrointestinal tract and included diverticulosis, polyps, duodenal ulcers, adenocarcinoma of the colon, and irritable bowel syndrome.
Figure 3.

Aortic Pathology from Two Patients with the MLCK mutation p.S1759P, TAA026:II:2 and TAA026:II:3, and an Unaffected Control
(A) Movat staining of aortas from patients shows medial degeneration of the aorta, as shown by increased proteoglycan deposition (blue, arrows) and elastic-fiber fragmentation and loss (black). Von Willebrand factor (marker of endothelial cells) immunostaining of the aorta (red) was performed to demonstrate the increased vascularity in the medial layer in TAA026:II2 and TAA026:II3 (arrows) as compared to the control aorta.
(B) Quantification of the number of arteries in the aortic media from patients with MYLK mutations and controls. The average number of arteries per field from the patients' aortic media is significantly higher than that in the control aortas. Data are expressed as mean ± standard error of the mean, and p values are indicated.
Conventional deletion of Mylk in mice leads to embryonic and perinatal lethality, whereas inducible, SMC-specific deletion leads to reduced gut motility, airway constriction, reduced blood pressure, and urinary dysfunction.2,19,20 Although the mice survive only 17–20 days after induction as a result of the gut hypomotility, arterial hypotension occurs. Interestingly, missense mutations in ACTA2 predispose humans to thoracic aortic disease but when deleted in mice also cause hypotension without reported aneurysm formation.10,21
To determine whether deficiency of MLCK in mice leads to aortic medial degeneration, we harvested aortas from mice with tamoxifen-induced and SMC-specific knockout of Mylk. Mylkf/f mice were bred to homozygosity and crossed with a transgenic mouse line expressing tamoxifen-activated Cre under the control of the smooth-muscle myosin heavy chain promoter.2,22 This promoter yields a more efficient Cre-mediated recombination in vascular SMCs than the SM22 promoter used in earlier studies. MLCK expression in knockout aortas was reduced to 33.5% ± 12.4% of that in controls 13 days after initiation of tamoxifen injections (n = 4 per group, p < 0.01), and immunoblot analysis showed that MLCK content in the aortas in the tamoxifen-treated mice was 53.4% ± 24.2% of that in controls (n = 3 per group, p = 0.04). Aortas were harvested for histology at day 13 before any overt signs of morbidity in the mice. Previously studied mouse models of thoracic aortic disease have determined that increased proteoglycan deposition can be the initial pathogenic abnormality observed in the progression of aortic disease.23 Although aortic dilation was not present after 13 days, increased pools of proteoglycans were found in the aortic media in the aortas of the tamoxifen-treated mice compared with controls, along with increased expression of lumican and decorin (Figure 4). Increased collagen staining in the adventitial layer and increased type III collagen (COL3A1 [MIM 120180]) expression were also indentified, findings similar to adventitial changes observed with aortic aneurysm formation with angiotensin II infusion in mice.24,25 Although elastic fibers were not degraded in the aortic media, expression of MMP2 (MIM 120360), an elastin-degrading metalloproteinase that has been shown to be increased in ascending aortic aneurysms and in mouse models of aneurysm formation, was also increased in the aortas of the mice.26–28
Figure 4.
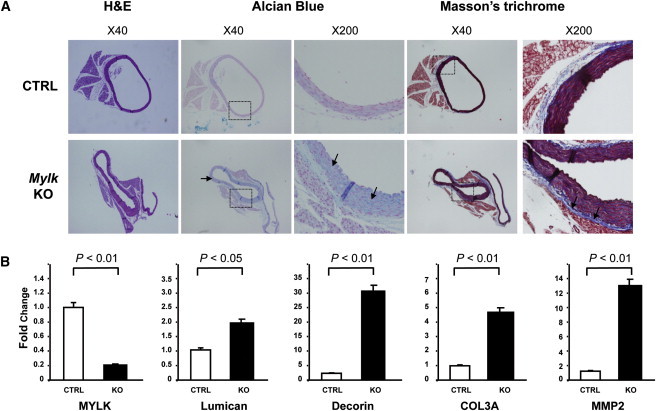
Pathology of the Ascending Aorta with Smooth-Muscle-Specific Deficiency of MLCK in Mice
Mylkf/f mice were bred to homozygosity and crossed with a transgenic mouse line expressing tamoxifen-activated Cre under the control of the smooth-muscle myosin heavy chain promoter.2MlckSMKO male mice 12–13 weeks old and littermate controls harboring the Cre transgene in mixed 129/B6 backgrounds were used for this study.
(A) Paraffin-embedded ascending aortic tissues from control (n = 3) and SM-Mylk-KO (n = 4) mice were sectioned and stained for proteoglycans and collagen. The upper panels are from the control mice (CTRL), and the lower panels are from the tamoxifen-treated Sm-Mylk-KO mice (KO). The specific stains and magnifications are indicted above the panels. Alcian blue staining indicates increased proteoglycan accumulation (blue) in the SM-Mylk-KO mice compared with controls (arrows). Masson's trichrome staining indicates increased collagen fibers (blue) in the adventitia (arrows) in the SM-MYLK-KO mice compared with controls.
(B) Quantitative PCR was used to determine changes in expression levels of MYLK, MMP2, lumican, decorin, and type III collagen. Expression analysis was performed with the use of RNA extracted from ascending aortas of control (n = 4) and Sm-Mylk-KO (n = 4) mice. The analysis shows decreased MYLK expression, whereas MMP2 messages increased 13-fold, lumican messages increased 2-fold, decorin messages increased 32-fold, and the type III procollagen (COL3A1) messages increased 5-fold. Gene-expression levels are standardized to GAPDH messages. The relative expression values were determined via the ΔΔCt method, and assays were performed in triplicate. Data are expressed as mean ± standard deviation.
In summary, a candidate-gene approach to identifying genes for familial thoracic aortic aneurysms and aortic dissections, followed by linkage, structural, and functional analyses, indicates that heterozygous loss-of-function mutations in MYLK cause dissections of the thoracic aorta. In contrast, sequencing of other genes encoding proteins involved in regulating SMC contraction, including MYL6, MYL6B, MYL9, and CALM1, failed to identify any causative mutations. Given the ubiquitous expression of MYLK and its role in many cellular processes involving actin-myosin molecular motors, the absence of other phenotypic manifestations suggests that half of normal MLCK activity does not disrupt type II myosin motors in the majority of cells to an extent that a phenotype is manifested. This fact may reflect that normal cellular molecular motors can function effectively with only half of normal MLCK activity. Although SMCs have an abundance of total CaM, the Ca2+/CaM complexes available to activate MLCK are limiting, which would exacerbate physiological effects resulting from reduced amounts of MLCK.5 Alternatively, other kinases proposed to phosphorylate RLC may compensate for the decreased MLCK activity in other cells, such as integrin-linked kinase or zipper-interacting protein kinase.29 It is not yet clear whether phosphorylation of RLC by these other Ca2+-independent kinases may significantly affect SMC contraction.2,20 The phenotype of MYLK mutations specifically involves the ascending thoracic aorta, the arterial segment exposed to the highest biomechanical force from pulsatile blood flow, suggesting that haploinsufficiency of MLCK decreases SMC contractile function to the extent that the aorta cannot withstand a lifetime of biomechanical forces.
Acknowledgments
The authors are grateful to the families and their physicians involved in this study. The following sources provided funding for these studies: National Institutes of Health (R21 HL091509 [D.M.M.], P50HL083794-01 [D.M.M.], RO1 HL62594 [D.M.M.], HL29043 [J.T.S.], UL1 RR024148), the Moss Heart Fund (J.T.S.), the Fouad A. and Val Imm Bashour Distinguished Chair in Physiology (J.T.S.), the Vivian L. Smith Foundation (D.M.M.), the TexGen Foundation (D.M.M.), and the Doris Duke Charitable Trust (D.M.M.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
National Center for Biotechnology Information, http://www.ncbi.nlm.nih.gov/
Ensembl Genome Browser, http://www.ensembl.org/index.html
Online Mendelian Inheritance in Man, http://www.ncbi.nlm.nih.gov/omim/
PyMOL, http://www.pymol.org/
References
- 1.Kamm K.E., Stull J.T. Dedicated myosin light chain kinases with diverse cellular functions. J. Biol. Chem. 2001;276:4527–4530. doi: 10.1074/jbc.R000028200. [DOI] [PubMed] [Google Scholar]
- 2.He W.Q., Peng Y.J., Zhang W.C., Lv N., Tang J., Chen C., Zhang C.H., Gao S., Chen H.Q., Zhi G. Myosin light chain kinase is central to smooth muscle contraction and required for gastrointestinal motility in mice. Gastroenterology. 2008;135:610–620. doi: 10.1053/j.gastro.2008.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schubert R., Lidington D., Bolz S.S. The emerging role of Ca2+ sensitivity regulation in promoting myogenic vasoconstriction. Cardiovasc. Res. 2008;77:8–18. doi: 10.1016/j.cardiores.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 4.Welsh D.G., Nelson M.T., Eckman D.M., Brayden J.E. Swelling-activated cation channels mediate depolarization of rat cerebrovascular smooth muscle by hyposmolarity and intravascular pressure. J. Physiol. 2000;527:139–148. doi: 10.1111/j.1469-7793.2000.t01-1-00139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Isotani E., Zhi G., Lau K.S., Huang J., Mizuno Y., Persechini A., Geguchadze R., Kamm K.E., Stull J.T. Real-time evaluation of myosin light chain kinase activation in smooth muscle tissues from a transgenic calmodulin-biosensor mouse. Proc. Natl. Acad. Sci. USA. 2004;101:6279–6284. doi: 10.1073/pnas.0308742101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ding H.L., Ryder J.W., Stull J.T., Kamm K.E. Signaling processes for initiating smooth muscle contraction upon neural stimulation. J. Biol. Chem. 2009;284:15541–15548. doi: 10.1074/jbc.M900888200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shmueli O., Horn-Saban S., Chalifa-Caspi V., Shmoish M., Ophir R., Benjamin-Rodrig H., Safran M., Domany E., Lancet D. GeneNote: whole genome expression profiles in normal human tissues. C. R. Biol. 2003;326:1067–1072. doi: 10.1016/j.crvi.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 8.Chan J.Y., Takeda M., Briggs L.E., Graham M.L., Lu J.T., Horikoshi N., Weinberg E.O., Aoki H., Sato N., Chien K.R., Kasahara H. Identification of cardiac-specific myosin light chain kinase. Circ. Res. 2008;102:571–580. doi: 10.1161/CIRCRESAHA.107.161687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhi G., Ryder J.W., Huang J., Ding P., Chen Y., Zhao Y., Kamm K.E., Stull J.T. Myosin light chain kinase and myosin phosphorylation effect frequency-dependent potentiation of skeletal muscle contraction. Proc. Natl. Acad. Sci. USA. 2005;102:17519–17524. doi: 10.1073/pnas.0506846102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo D.C., Pannu H., Tran-Fadulu V., Papke C.L., Yu R.K., Avidan N., Bourgeois S., Estrera A.L., Safi H.J., Sparks E. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat. Genet. 2007;39:1488–1493. doi: 10.1038/ng.2007.6. [DOI] [PubMed] [Google Scholar]
- 11.Zhu L., Vranckx R., Khau Van Kien P., Lalande A., Boisset N., Mathieu F., Wegman M., Glancy L., Gasc J.M., Brunotte F. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat. Genet. 2006;38:343–349. doi: 10.1038/ng1721. [DOI] [PubMed] [Google Scholar]
- 12.Milewicz D.M., Guo D.C., Tran-Fadulu V., Lafont A.L., Papke C.L., Inamoto S., Kwartler C.S., Pannu H. Genetic basis of thoracic aortic aneurysms and dissections: focus on smooth muscle cell contractile dysfunction. Annu. Rev. Genomics Hum. Genet. 2008;9:283–302. doi: 10.1146/annurev.genom.8.080706.092303. [DOI] [PubMed] [Google Scholar]
- 13.Hiratzka L.F., Bakris G.L., Beckman J.A., Bersin R.M., Carr V.F., Casey D.E., Jr., Eagle K.A., Hermann L.K., Isselbacher E.M., Kazerooni E.A., American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. American Association for Thoracic Surgery. American College of Radiology. American Stroke Association. Society of Cardiovascular Anesthesiologists. Society for Cardiovascular Angiography and Interventions. Society of Interventional Radiology. Society of Thoracic Surgeons. Society for Vascular Medicine 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with Thoracic Aortic Disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation. 2010;121:e266–e369. doi: 10.1161/CIR.0b013e3181d4739e. [DOI] [PubMed] [Google Scholar]
- 14.Goeckeler Z.M., Masaracchia R.A., Zeng Q., Chew T.L., Gallagher P., Wysolmerski R.B. Phosphorylation of myosin light chain kinase by p21-activated kinase PAK2. J. Biol. Chem. 2000;275:18366–18374. doi: 10.1074/jbc.M001339200. [DOI] [PubMed] [Google Scholar]
- 15.Herring B.P., El-Mounayri O., Gallagher P.J., Yin F., Zhou J. Regulation of myosin light chain kinase and telokin expression in smooth muscle tissues. Am. J. Physiol. Cell Physiol. 2006;291:C817–C827. doi: 10.1152/ajpcell.00198.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fitzsimons D.P., Herring B.P., Stull J.T., Gallagher P.J. Identification of basic residues involved in activation and calmodulin binding of rabbit smooth muscle myosin light chain kinase. J. Biol. Chem. 1992;267:23903–23909. [PMC free article] [PubMed] [Google Scholar]
- 17.Elefteriades J.A., Tranquilli M., Darr U., Cardon J., Zhu B.Q., Barrett P. Symptoms plus family history trump size in thoracic aortic aneurysm. Ann. Thorac. Surg. 2005;80:1098–1100. doi: 10.1016/j.athoracsur.2004.02.130. [DOI] [PubMed] [Google Scholar]
- 18.Pannu H., Tran-Fadulu V., Papke C.L., Scherer S., Liu Y., Presley C., Guo D., Estrera A.L., Safi H.J., Brasier A.R. MYH11 mutations result in a distinct vascular pathology driven by insulin-like growth factor 1 and angiotensin II. Hum. Mol. Genet. 2007;16:2453–2462. doi: 10.1093/hmg/ddm201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Somlyo A.V., Wang H., Choudhury N., Khromov A.S., Majesky M., Owens G.K., Somlyo A.P. Myosin light chain kinase knockout. J. Muscle Res. Cell Motil. 2004;25:241–242. doi: 10.1023/b:jure.0000038362.84697.c0. [DOI] [PubMed] [Google Scholar]
- 20.Zhang W.C., Peng Y.J., Zhang G.S., He W.Q., Qiao Y.N., Dong Y.Y., Gao Y.Q., Chen C., Zhang C.H., Li W. Myosin light chain kinase is necessary for tonic airway smooth muscle contraction. J. Biol. Chem. 2010;285:5522–5531. doi: 10.1074/jbc.M109.062836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schildmeyer L.A., Braun R., Taffet G., Debiasi M., Burns A.E., Bradley A., Schwartz R.J. Impaired vascular contractility and blood pressure homeostasis in the smooth muscle alpha-actin null mouse. FASEB J. 2000;14:2213–2220. doi: 10.1096/fj.99-0927com. [DOI] [PubMed] [Google Scholar]
- 22.Wirth A., Benyó Z., Lukasova M., Leutgeb B., Wettschureck N., Gorbey S., Orsy P., Horváth B., Maser-Gluth C., Greiner E. G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat. Med. 2008;14:64–68. doi: 10.1038/nm1666. [DOI] [PubMed] [Google Scholar]
- 23.Hassane S., Claij N., Lantinga-van Leeuwen I.S., Van Munsteren J.C., Van Lent N., Hanemaaijer R., Breuning M.H., Peters D.J., DeRuiter M.C. Pathogenic sequence for dissecting aneurysm formation in a hypomorphic polycystic kidney disease 1 mouse model. Arterioscler. Thromb. Vasc. Biol. 2007;27:2177–2183. doi: 10.1161/ATVBAHA.107.149252. [DOI] [PubMed] [Google Scholar]
- 24.Tieu B.C., Lee C., Sun H., Lejeune W., Recinos A., 3rd, Ju X., Spratt H., Guo D.C., Milewicz D., Tilton R.G., Brasier A.R. An adventitial IL-6/MCP1 amplification loop accelerates macrophage-mediated vascular inflammation leading to aortic dissection in mice. J. Clin. Invest. 2009;119:3637–3651. doi: 10.1172/JCI38308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daugherty A., Rateri D.L., Charo I.F., Owens A.P., Howatt D.A., Cassis L.A. Angiotensin II infusion promotes ascending aortic aneurysms: attenuation by CCR2 deficiency in apoE-/- mice. Clin. Sci. (Lond.) 2010;118:681–689. doi: 10.1042/CS20090372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lesauskaite V., Tanganelli P., Sassi C., Neri E., Diciolla F., Ivanoviene L., Epistolato M.C., Lalinga A.V., Alessandrini C., Spina D. Smooth muscle cells of the media in the dilatative pathology of ascending thoracic aorta: morphology, immunoreactivity for osteopontin, matrix metalloproteinases, and their inhibitors. Hum. Pathol. 2001;32:1003–1011. doi: 10.1053/hupa.2001.27107. [DOI] [PubMed] [Google Scholar]
- 27.Fedak P.W., de Sa M.P., Verma S., Nili N., Kazemian P., Butany J., Strauss B.H., Weisel R.D., David T.E. Vascular matrix remodeling in patients with bicuspid aortic valve malformations: implications for aortic dilatation. J. Thorac. Cardiovasc. Surg. 2003;126:797–806. doi: 10.1016/s0022-5223(03)00398-2. [DOI] [PubMed] [Google Scholar]
- 28.Ailawadi G., Moehle C.W., Pei H., Walton S.P., Yang Z., Kron I.L., Lau C.L., Owens G.K. Smooth muscle phenotypic modulation is an early event in aortic aneurysms. J. Thorac. Cardiovasc. Surg. 2009;138:1392–1399. doi: 10.1016/j.jtcvs.2009.07.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murthy K.S. Signaling for contraction and relaxation in smooth muscle of the gut. Annu. Rev. Physiol. 2006;68:345–374. doi: 10.1146/annurev.physiol.68.040504.094707. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.