


Abstract
In order to support the structural genomic initiatives, both by rapidly classifying newly determined structures and by suggesting suitable targets for structure determination, we have recently developed several new protocols for classifying structures in the CATH domain database (http://www.biochem.ucl.ac.uk/bsm/cath). These aim to increase the speed of classification of new structures using fast algorithms for structure comparison (GRATH) and to improve the sensitivity in recognising distant structural relatives by incorporating sequence information from relatives in the genomes (DomainFinder). In order to ensure the integrity of the database given the expected increase in data, the CATH Protein Family Database (CATH-PFDB), which currently includes 25 320 structural domains and a further 160 000 sequence relatives has now been installed in a relational ORACLE database. This was essential for developing more rigorous validation procedures and for allowing efficient querying of the database, particularly for genome analysis. The associated Dictionary of Homologous Superfamilies [Bray,J.E., Todd,A.E., Pearl,F.M.G., Thornton,J.M. and Orengo,C.A. (2000) Protein Eng., 13, 153–165], which provides multiple structural alignments and functional information to assist in assigning new relatives, has also been expanded recently and now includes information for 903 homologous superfamilies. In order to improve coverage of known structures, preliminary classification levels are now provided for new structures at interim stages in the classification protocol. Since a large proportion of new structures can be rapidly classified using profile-based sequence analysis [e.g. PSI-BLAST: Altschul,S.F., Madden,T.L., Schaffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Nucleic Acids Res., 25, 3389–3402], this provides preliminary classification for easily recognisable homologues, which in the latest release of CATH (version 1.7) represented nearly three-quarters of the non-identical structures.
INTRODUCTION
Although the protein structure database (RCSB) (1), has expanded significantly over the last year from 10 714 to 13 400 chains, we can expect even more substantial increases in the coming years due to the international structural genomic initiatives. Several projects in the United States, Germany, France and Japan already aim to increase the number of structures solved annually by using robotic approaches to cloning and crystallisation and there have also been major improvements in synchrotron technology for crystallography.
POPULATION OF THE CURRENT CATH PROTEIN FAMILY DATABASE (CATH-PFDB)
CATH is a hierarchical classification in which structures are assigned to evolutionary families (S) and superfamilies (H) on the basis of sequence, structure and functional similarity and to fold groups (T) on the basis of similarity in the spatial arrangements and connectivity of the secondary structures. Additionally, the architecture level (A) provides a description of the orientations of secondary structures regardless of connectivity, with the top level in the hierarchy simply grouping structures according to protein class (C). Table 1 shows the population of structures at different levels in the hierarchy. Recently, CATH was expanded to include 160 000 sequence relatives from the genomes identified using profile-based sequence comparison methods (2,3). Conservative thresholds were employed to minimise incorrect assignments (DomainFinder; F.M.G.Pearl, D.Lee, J.E.Bray, D.W.A.Buchan, A.J.Shepherd and C.A.Orengo, manuscript in preparation). This protocol has now been extended to identify more putative homologues (D.W.A.Buchan, F.M.G.Pearl, D.Lee, I.Sillitoe and C.A.Orengo, manuscript in preparation), allowing further matches to be tentatively assigned to structural families within CATH. These are not formerly integrated into the database but are maintained as gene neighbour lists for each non-identical structure in CATH. They can be viewed in a related web resource (Gene-3D) currently being developed.
Table 1. The population for each class, at each level in the CATH hierarchy.
| Class |
Architecture |
Fold |
Homologous superfamily |
Sequence
family |
S95 groups |
S99 groups |
Total number of entries |
| 1 (mainly alpha) | 5 | 173 | 269 | 508 | 888 | 1853 | 4981 |
| 2 (mainly beta) | 18 | 106 | 218 | 530 | 1391 | 2492 | 7282 |
| 3 (alpha/beta) | 13 | 250 | 471 | 1036 | 1798 | 3725 | 10586 |
| 4 (few secondary structures) | N/A | 63 | 77 | 94 | 176 | 287 | 705 |
| 5 (multi-domain) | N/A | N/A | N/A | 577 | 1180 | 2594 | 5821 |
| 6 (single-domain PSI-BLAST clusters) | N/A | N/A | 369 | 501 | 640 | 867 | 1767 |
| 7 (chain-wise PSI-BLAST clusters) | N/A | N/A | N/A | 239 | 274 | 483 | 909 |
| 8 (sequence family clusters) | N/A | N/A | N/A | 77 | 88 | 142 | 234 |
IMPLICATIONS FOR STRUCTURAL GENOME INITIATIVES
Figure 1 shows the increase in the number of sequence families (≥35% identity) in the CATH database, obtained by identifying gene sequence relatives for each structural class in CATH. It can be seen from Figure 1 that only a small proportion of these families (<15%) contain a structural relative from which a reliable homology model can be built. Many of these families belong to highly populated fold groups within the database (4) and the data suggest that within these broad fold groups, representative sequences should be selected from each diverse sequence family for determination in the structural genome initiatives. This will enable models to be built across the superfamily, providing more reliable structural data for many more sequence relatives than is currently possible.
Figure 1.
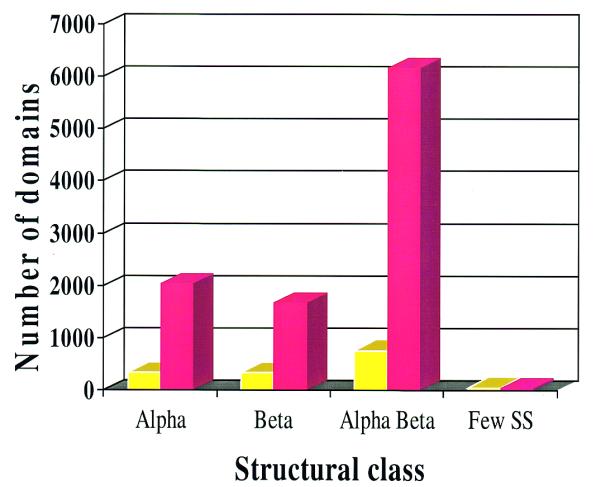
Distribution of sequence families with close structural relatives (>35% identity) in the CATH-PFDB database on a class-wise basis are shown in yellow. Sequence families with more distant structural relatives (<35% identity) are shown in red.
Another approach to identifying potential targets for structure determination is to focus on a particular organism of biological interest (e.g. a microbial pathogen). In this context, the gene neighbour lists generated by the PSI-BLAST (2) and IMPALA (3) searches used for generating the CATH-PFDB can also be used to assign structural annotation to ORFs within partial or complete genomes. Such data will shortly be available on the CATH Gene-3D web site and can be used to eliminate ORFs for which structural data has already been assigned, allowing research groups to focus on the remaining ORFs when selecting suitable targets for structure determination.
IMPROVED DETECTION OF HOMOLOGUES USING SEQUENCE-BASED APPROACHES
The recognition of evolutionary relatives is one of the most important stages in the classification of new structures. This level in the hierarchy is of particular interest to biologists as functional data can often be suggested for a new structure, depending on the degree of similarity to other relatives in the family to which it has been assigned. Several recent analyses (5; A.E.Todd, C.A.Orengo and J.M.Thornton, manuscript submitted) have shown that for a reasonable degree of sequence similarity (≥40% identity) there is a high probability (≥96%) that relatives will all exhibit similar functions.
In order to improve the recognition of distant homologues for classification in CATH, sequence profiles have been generated for representatives from each gene sequence S60 family (≥60% identity), in the extended CATH-PFDB (currently 9900 profiles). These profiles, which were generated using IMPALA software (3), were added to the library of 2200 profiles previously generated for all the non-identical protein structures in CATH. The expansion of the profile library in this way improves the recognition of distant homologues by considerably broadening the region of sequence ‘space’ currently represented by the CATH-IMPALA profiles (F.M.G.Pearl et al., manuscript in preparation).
IMPROVED CLASSIFICATION PROTOCOL FOR THE CATH DATABASE
The recent improvements in iterative profile-based search methods (6) has meant that a large proportion of structural homologues can be rapidly classified in CATH, using these sequence-based approaches, without the need for the much slower structure-based comparisons. To reflect these developments, the CATH classification protocol has recently been revised. Figure 2 shows the various stages in the classification of new structures. Preliminary sequence clustering using a Needleman and Wunsch algorithm (HOMOL; 7) is followed by scanning all the non-identical structures against the CATH-IMPALA profiles. Any matches indicating putative homologues are subsequently checked by the structure comparison method SSAP (8,9) and where validated are added to their homologous superfamily. Information within the Dictionary of Homologous Superfamilies (10) is updated to reflect these new relatives. Any sequences unmatched are checked for domain boundaries using a consensus suite of programs (DBS; 11) and the HOMOL and IMPALA stages are then rerun using the domain sequences.
Figure 2.

Flow chart describing the improved update procedure. New sequences are first compared pairwise, by sequence, with each other and all the entries in the database (HOMOL). Those that have not been identified as a sequence match are then compared using IMPALA. If a homologue is found, the structure is compared structurally with all the members of the homologous superfamily (SSAPH) and the DHS family data updated. Those that are unmatched 7 are assigned domain boundaries using DBS (11). The resulting single domains 6 are again compared by pairwise and sequence profile methods. If no homologous relative is found, a fast structural comparison program (GRATH) is used to compare the domain with all sequence families within the CATH database. If a similar fold is detected CORA and ConAlign is used to compare the structure with representatives from all the top scoring fold groups to assign homology. If GRATH does not find a significant hit then structural templates CORA (12) are used to find the correct fold. Finally pairwise SSAPs against the database are run on any remaining structures. If there is no significant fold match the architecture is assigned manually.
For sequences still unmatched, a fast structural pre-screen is used to check for fold similarities to any domain in CATH. This uses a vector-based approach to search for similarities in secondary structure orientations (GRATH; A.P.Harrison, F.M.G.Pean, T.Slidel, C.A.Orengo and J.M.Thornton, manuscript in preparation). Any matches to a particular fold group are scanned against a library of structural templates for all the homologous superfamilies adopting that fold (CORA; 12) to identify the particular superfamily to which the structure belongs. A contact-based comparison program (CONALIGN; I.Sillitoe and C.A.Orengo, manuscript in preparation) is used to suggest putative homologues on the basis of conserved contacts. These are then manually validated by checking available functional information. Structures returning no hits by GRATH are scanned against a library of CORA templates for all the fold groups in CATH. Again any matches are scanned against templates for the homologous superfamilies within that fold group to check for potential homologues. Any structures failing to match a fold group must be scanned against all the structures within their class, using SSAP. These SSAP scans can be many orders of magnitude slower than any of the previous steps in the classification, dependant on the size of the protein.
Encouragingly, Figure 3 shows that for the 6040 new structural domains classified in CATH during the last year, 66% could be classified using simple pairwise methods (HOMOL), and a further 10% could be recognised using the IMPALA CATH-PFDB profiles. GRATH matched 17% of the remaining structures and the CORA templates enabled a further 9% to be identified. This meant that only 7% of the structures required the computationally expensive SSAP scans. Finally architectures are manually assigned for the unique structures. This represented 5% of the structures classified last year.
Figure 3.
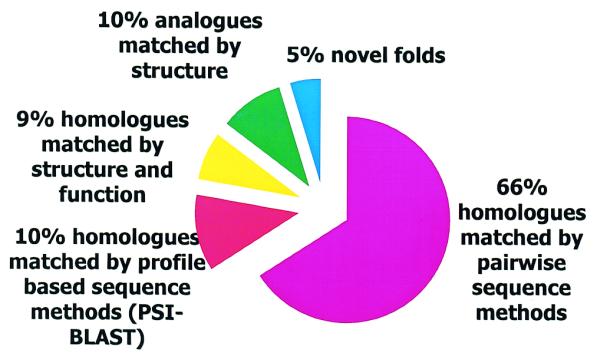
Pie chart describing the proportion of 6040 domains classified in CATH in the last 12 months, which have been recognised using different classification procedures. Eighty-four percent of the structural domains were homologous to an entry in the database: 66% of new domains were classified as homologues using pairwise sequence methods (shown in pink), 10% were classified as homologues using PSI-BLAST, using SSAP to validate the matches (shown in red) and 9% were classified as homologues using CORA and ConAlign (shown in yellow). A further 10% of structural domains were classified at the fold level using SSAP and GRATH (shown in green). Five percent were novel folds (shown in blue).
PORTING CATH INTO AN ORACLE DATABASE
Previous releases of CATH have been implemented on top of a collection of flat files. We are now re-implementing the CATH database using the Oracle8i relational database management system in order to gain the benefits such a system brings. In particular, use of a database management system enables us to ensure the integrity of the data while at the same time providing more flexible views of that data together with powerful querying facilities.
In designing the database scheme, a key consideration has been that the design should be flexible enough to readily accommodate new sources of derived structural, functional and relationship data. While object-oriented database systems can model the complex interrelationships characteristic of such data, we do not believe that current object-oriented database products would be able to support the flexible views required with the query performance needed.
Our approach has been to use conventional Oracle 8i relational tables to represent not only the data as would be expected in a flat file representation, but also to explicitly represent the interrelationships implicit in a flat file representation. As an example, lipocalin (CATH id# 2.40.128.20) is represented as a single row in a relational table with column values for the C, A, T and H identifiers 2, 40, 128 and 20, respectively, but additionally the relationship between each CATH subfamily and its parent family is also stored in a separate ‘metalevel’ table with internal identifiers representing the CATH families. Use of materialized views enables the flat file table representation to be efficiently generated from the underlying metalevel representation.
This explicit representation of otherwise implicit relationships gives rise to a number of advantages. First, the metalevel tables can be used to store additional information about a relationship including the period for which it is valid, so enabling historical ‘version’ information to be stored, as well as the degree of certainty with which the relationship is believed to be true, such as the position of domain boundaries. Secondly, the relationships between data can be themselves searched enabling the question ‘how is protein A related to protein B?’ to be answered. Thirdly, the use of internal identifiers in the metalevel tables ensures the relationships within the database are modelled independently of the external identifiers, which may be expected to change over time.
IMPROVEMENTS TO THE CATH SERVER
The CATH server, which allows the user to search CATH with a newly-determined structure or set of coordinates, has recently been improved by incorporating the fast secondary structure-based GRATH search algorithm. Recent trials suggest this method identifies related folds within the top 10 matches, with a 97% coverage rate in all structural classes. The current protocol takes the top 50 matches and then performs the slower SSAP comparison to provide alignments and superpositions for significant matches. In addition, a SSAP server has also been set up to allow the user to align pairs of structures. Again alignments are provided and superpositions, generated using the SSAP alignment, can be viewed in RASMOL.
CONTENT OF THE CURRENT RELEASE AND INTERIM CLASSIFICATION LEVELS
Table 1 shows the population of families, superfamilies, folds, architectures and classes currently held within the CATH database. There are 23 553 fully-classified domains. In addition, preliminary classifications using sequence-based approaches have been performed for a further 2901 structures, which have been assigned to chain-based sequence families (class 8), chain-based superfamilies (class 7) and domain-based superfamilies (class 6).
ACCESSING CATH
The CATH database can be accessed at http://www.biochem.ucl.ac.uk/bsm/cath. The web interface may be browsed, or alternatively searched with PDB codes. There is also a facility for keyword searches. The ftp site ftp://ftp.biochem.ucl.ac.uk/pub/cathdata enables the complete classification (including interim data) and domain definitions to be downloaded. There are links directly to PDBsum (13) which gives summary information for each of the structures within the database. The Dictionary of Homologous Superfamilies can be directly accessed at http://www.biochem.ucl.ac.uk/bsm/dhs.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Drs Roman Laskowski, Andrew Martin and David Jones for use of their computer programs. We acknowledge the support of the BBSRC and the MRC. A.E.T., J.E.B., D.W.A.B., G.A.R. and I.S. are in receipt of special BBSRC studentships. C.A.O, A.P.H. and D.L are supported by the MRC. We acknowledge the support from the BBSRC and MRC computing facilities.
References
- 1.Berman H.M., Westbrook,J., Feng,Z., Gilliland,G., Bhat,T.N., Weissig,H., Shindyalov,N., Bourne,P.E. (2000) The Protein Data Bank. Nucleic Acids Res., 28, 235–242. Updated article in this issue: Nucleic Acids Res. (2001), 29, 214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altschul S.F., Madden,T.L., Schaffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schaffer A.A, Wolf,Y.I., Ponting,C.P., Koonin,E.V., Avarind,L. and Altschul,S.F.(1999) IMPALA: matching a protein sequence against a collection of PSI-BLAST-constructed position-specific score matrices. Bioinformatics, 15, 1000–1011. [DOI] [PubMed]
- 4.Orengo C.A, Jones,D.T. and Thornton,J.M. (1994) Protein superfamilies and domain superfolds. Nature, 372, 631–634. [DOI] [PubMed] [Google Scholar]
- 5.Wilson C.A., Kreychman,J. and Gerstein,M. (2000) Assessing annotation transfer for genomics: quantifying the relationships between protein sequence, structure and function through traditional and probabilistic scores. J. Mol. Biol., 297, 233–249. [DOI] [PubMed] [Google Scholar]
- 6.Park J., Karplus,K., Barret,C., Hughey,R., Haussler,D., Hubbard,T. and Chothia,C. (1998) Sequence comparison using multiple sequences detect three times as many remote homologues as pairwise method. J. Mol. Biol., 284, 1201–1210. [DOI] [PubMed] [Google Scholar]
- 7.Orengo C.A., Michie,A.D., Jones,S., Jones,D.T., Swindells,M.B. and Thornton,J.M. (1997) CATH—A hierarchical classification of protein domain structures. Structure, 5, 1093–1108. [DOI] [PubMed] [Google Scholar]
- 8.Taylor W.R. and Orengo,C.A. (1989) Protein structure alignment J. Mol. Biol., 208, 1–22. [DOI] [PubMed] [Google Scholar]
- 9.Orengo C.A., Brown,N.P. and Taylor,W.R. (1992) Fast structure alignment for databank searching. Proteins: Struct. Func. Genet., 14, 139–167. [DOI] [PubMed] [Google Scholar]
- 10.Bray J.E., Todd,A.E., Pean,F.M.G., Thornton,J.M. and Orengo,C.A. (2000) Protein Eng., 13, 153–165. [DOI] [PubMed] [Google Scholar]
- 11.Jones S, Stewart,M., Michie,A., Swindells,M.B., Orengo,C.A. and Thornton,J.M. (1998) Domain assignment for protein structures using a consensus approach: characterisation and analysis. Protein Sci., 7, 233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Orengo C.A. (1999) CORA- Topological fingerprints for protein structural families. Protein Sci., 8, 699–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laskowski R.A., Hutchinson,E.G., Michie,A.D., Wallace,A.C., Jones,M.L. and Thornton,J.M. (1997). PDBsum: A Web-based database of summaries and analyses of all PDB structures. Trends Biochem. Sci., 22, 488–490. [DOI] [PubMed] [Google Scholar]