

Abstract
In the title molecular salt, C6H9N2 +·C7H3N2O7 −, the 2-amino-5-methylpyridinium cation is essentially planar, with a maximum deviation of 0.023 (1) Å. There is an intramolecular O—H⋯O hydrogen bond in the 3,5-dinitrosalicylate anion, which generates an S(6) ring motif. In the crystal, the protonated N atom and the 2-amino group are hydrogen bonded to the carboxylate O atoms via a pair of N—H⋯O hydrogen bonds, forming an R 2 2(8) ring motif. Weak intermolecular C—H⋯O interactions help to further stabilize the crystal structure.
Related literature
For background to the chemistry of substituted pyridines, see: Pozharski et al. (1997 ▶); Katritzky et al. (1996 ▶); Nahringbauer & Kvick (1977 ▶). For 3,5-dinitrosalicylic acid, see: Hindawey et al. (1980 ▶); Issa et al. (1981 ▶). For details of hydrogen bonding, see: Jeffrey & Saenger (1991 ▶); Jeffrey (1997 ▶); Scheiner (1997 ▶). For hydrogen-bond motifs, see: Bernstein et al. (1995 ▶). For bond-length data, see: Allen et al. (1987 ▶). For the stability of the temperature controller used in the data collection, see: Cosier & Glazer (1986 ▶).
Experimental
Crystal data
C6H9N2 +·C7H3N2O7 −
M r = 336.27
Triclinic,
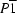
a = 5.8673 (7) Å
b = 8.0991 (9) Å
c = 15.2437 (17) Å
α = 86.844 (3)°
β = 84.252 (3)°
γ = 81.209 (3)°
V = 711.69 (14) Å3
Z = 2
Mo Kα radiation
μ = 0.13 mm−1
T = 100 K
0.29 × 0.14 × 0.08 mm
Data collection
Bruker APEX DUO CCD area-detector diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2009 ▶) T min = 0.963, T max = 0.990
12709 measured reflections
4947 independent reflections
3922 reflections with I > 2σ(I)
R int = 0.023
Refinement
R[F 2 > 2σ(F 2)] = 0.045
wR(F 2) = 0.150
S = 1.08
4947 reflections
219 parameters
H-atom parameters constrained
Δρmax = 0.51 e Å−3
Δρmin = −0.46 e Å−3
Data collection: APEX2 (Bruker, 2009 ▶); cell refinement: SAINT (Bruker, 2009 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXTL (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXTL; molecular graphics: SHELXTL; software used to prepare material for publication: SHELXTL and PLATON (Spek, 2009 ▶).
Supplementary Material
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536810014480/hb5405sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810014480/hb5405Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| O1—H1A⋯O7 | 0.82 | 1.66 | 2.4200 (13) | 152 |
| N1—H1⋯O6i | 0.86 | 1.82 | 2.6768 (14) | 174 |
| N2—H2A⋯O7i | 0.86 | 2.11 | 2.9668 (15) | 176 |
| N2—H2B⋯O1ii | 0.86 | 2.16 | 2.8468 (15) | 137 |
| N2—H2B⋯O2ii | 0.86 | 2.40 | 3.1723 (16) | 149 |
| C2—H2⋯O4iii | 0.93 | 2.49 | 3.4073 (16) | 169 |
| C4—H4⋯O3iv | 0.93 | 2.39 | 3.2371 (16) | 151 |
| C5—H5⋯O2ii | 0.93 | 2.44 | 3.2328 (17) | 143 |
Symmetry codes: (i) 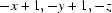 ; (ii)
; (ii) 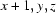 ; (iii)
; (iii) 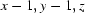 ; (iv)
; (iv) 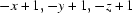 .
.
Acknowledgments
MH and HKF thank the Malaysian Government and Universiti Sains Malaysia for the Research University Golden Goose grant No. 1001/PFIZIK/811012. MH also thanks Universiti Sains Malaysia for a post-doctoral research fellowship.
supplementary crystallographic information
Comment
Pyridine and its derivatives play an important role in heterocyclic chemistry (Pozharski et al., 1997; Katritzky et al., 1996). They are often involved in hydrogen-bond interactions (Jeffrey & Saenger, 1991; Jeffrey, 1997; Scheiner, 1997). The nitro-substituted aromatic acid 3,5-dinitrosalicylic acid (DNSA) has proven potential for formation of proton-transfer compounds, particularly because of its acid strength (pKa = 2.18), its interactive ortho-related phenolic substituent group together with the nitro substituents which have potential for both π···π interactions as well as hydrogen-bonding interactions. A large number of both neutral and proton-transfer compounds of Lewis bases with DNSA, together with their IR spectra have been reported (Hindawey et al., 1980; Issa et al., 1981) in the literature. Since our aim is to study some interesting hydrogen bonding interactions, the crystal structure of the title compound is presented here.
The asymmetric unit (Fig. 1) contains one 2-amino-5-methylpyridinium cation and one 3,5-dinitrosalicylate anion. The proton transfer from the carboxyl group to atom N1 of 2-amino-5-methylpyridine resulted in the widening of C1—N1—C2 angle of the pyridinium ring to 123.48 (10)°, compared to the corresponding angle of 117.4° in neutral 2-amino-5-methylpyridine (Nahringbauer & Kvick, 1977). The 2-amino-5-methylpyridinium cation is essentially planar, with a maximum deviation of 0.023 (1) Å for atom N1. The bond lengths and angles are normal (Allen et al., 1987). The phenol oxygen atoms are bent slightly away from the mean plane of the benzene ring [torsion angle O1-C7-C8-C9 = 179.70 (12)°].
In the crystal packing (Fig. 2), the protonated N1 atom and the 2-amino group (N2) is hydrogen-bonded to the carboxylate oxygen atoms (O6 and O7) via a pair of intermolecular N1—H1···O6 and N2—H2A···O7 hydrogen bonds forming a ring motif R22(8) (Bernstein et al., 1995). There is an intramolecular O—H···O hydrogen bond in the 3,5-dinitrosalicylate anion, which generates an S(6) ring motif. The crystal structure is further stabilized by weak C—H···O (Table 1) hydrogen bonds.
Experimental
A hot methanol solution (20 ml) of 2-amino-5-methylpyridine (27 mg, Aldrich) and 3,5-dinitrosalicylic acid (58 mg, Merck) were mixed and warmed over a heating magnetic stirrer for a few minutes. The resulting solution was allowed to cool slowly at room temperature and yellow blocks of (I) appeared after a few days.
Refinement
All hydrogen atoms were positioned geometrically [C–H = 0.93 or 0.96 Å, N–H = 0.86 Å and O–H = 0.82 Å] and were refined using a riding model, with Uiso(H) = 1.2 Ueq(C, N) or 1.5 Ueq(O). The methyl H atoms were positioned geometrically [C–H = 0.96 Å] and were refined using a riding model, with Uiso(H) = 1.5Ueq(C). A rotating group model was used for the methyl group.
Figures
Fig. 1.

The asymmetric unit of (I). Displacement ellipsoids are drawn at the 50% probability level.
Fig. 2.

The crystal packing of (I), showing hydrogen-bonded (dashed lines) networks. H atoms are not involing the hydrogen bond interactions are omitted for clarity.
Crystal data
| C6H9N2+·C7H3N2O7− | Z = 2 |
| Mr = 336.27 | F(000) = 348 |
| Triclinic, P1 | Dx = 1.569 Mg m−3 |
| Hall symbol: -P 1 | Mo Kα radiation, λ = 0.71073 Å |
| a = 5.8673 (7) Å | Cell parameters from 5139 reflections |
| b = 8.0991 (9) Å | θ = 2.7–32.4° |
| c = 15.2437 (17) Å | µ = 0.13 mm−1 |
| α = 86.844 (3)° | T = 100 K |
| β = 84.252 (3)° | Block, yellow |
| γ = 81.209 (3)° | 0.29 × 0.14 × 0.08 mm |
| V = 711.69 (14) Å3 |
Data collection
| Bruker APEX DUO CCD area-detector diffractometer | 4947 independent reflections |
| Radiation source: fine-focus sealed tube | 3922 reflections with I > 2σ(I) |
| graphite | Rint = 0.023 |
| φ and ω scans | θmax = 32.5°, θmin = 1.3° |
| Absorption correction: multi-scan (SADABS; Bruker, 2009) | h = −8→8 |
| Tmin = 0.963, Tmax = 0.990 | k = −12→11 |
| 12709 measured reflections | l = −22→23 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.045 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.150 | H-atom parameters constrained |
| S = 1.08 | w = 1/[σ2(Fo2) + (0.0741P)2 + 0.3055P] where P = (Fo2 + 2Fc2)/3 |
| 4947 reflections | (Δ/σ)max < 0.001 |
| 219 parameters | Δρmax = 0.51 e Å−3 |
| 0 restraints | Δρmin = −0.46 e Å−3 |
Special details
| Experimental. The crystal was placed in the cold stream of an Oxford Cryosystems Cobra open-flow nitrogen cryostat (Cosier & Glazer, 1986) operating at 100.0 (1) K. |
| Geometry. All s.u.'s (except the s.u. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell s.u.'s are taken into account individually in the estimation of s.u.'s in distances, angles and torsion angles; correlations between s.u.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell s.u.'s is used for estimating s.u.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > 2σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| N1 | 0.50349 (18) | 0.28625 (13) | 0.24313 (7) | 0.0151 (2) | |
| H1 | 0.4877 | 0.2768 | 0.1882 | 0.018* | |
| N2 | 0.82364 (19) | 0.41676 (14) | 0.19955 (7) | 0.0176 (2) | |
| H2A | 0.8060 | 0.4015 | 0.1454 | 0.021* | |
| H2B | 0.9352 | 0.4662 | 0.2121 | 0.021* | |
| C1 | 0.6777 (2) | 0.36374 (15) | 0.26413 (8) | 0.0146 (2) | |
| C2 | 0.3509 (2) | 0.22205 (16) | 0.30452 (8) | 0.0162 (2) | |
| H2 | 0.2353 | 0.1692 | 0.2859 | 0.019* | |
| C3 | 0.3654 (2) | 0.23421 (16) | 0.39271 (8) | 0.0177 (2) | |
| C4 | 0.5424 (2) | 0.31915 (17) | 0.41660 (8) | 0.0196 (2) | |
| H4 | 0.5553 | 0.3318 | 0.4761 | 0.023* | |
| C5 | 0.6949 (2) | 0.38321 (16) | 0.35500 (8) | 0.0180 (2) | |
| H5 | 0.8088 | 0.4391 | 0.3726 | 0.022* | |
| C6 | 0.2014 (2) | 0.1628 (2) | 0.46135 (9) | 0.0252 (3) | |
| H6A | 0.0930 | 0.1115 | 0.4329 | 0.038* | |
| H6B | 0.1192 | 0.2508 | 0.4971 | 0.038* | |
| H6C | 0.2873 | 0.0806 | 0.4980 | 0.038* | |
| O1 | 0.17251 (16) | 0.61810 (13) | 0.14298 (6) | 0.01924 (19) | |
| H1A | 0.1638 | 0.6119 | 0.0899 | 0.029* | |
| O2 | 0.10528 (18) | 0.60967 (13) | 0.31694 (7) | 0.0243 (2) | |
| O3 | 0.28454 (18) | 0.76459 (15) | 0.38862 (6) | 0.0270 (2) | |
| O4 | 0.93341 (17) | 0.99542 (13) | 0.26211 (7) | 0.0237 (2) | |
| O5 | 1.00019 (18) | 1.02494 (14) | 0.11971 (8) | 0.0274 (2) | |
| O6 | 0.55546 (18) | 0.76372 (13) | −0.07527 (6) | 0.0222 (2) | |
| O7 | 0.26539 (17) | 0.63243 (12) | −0.01529 (6) | 0.01923 (19) | |
| N3 | 0.25086 (18) | 0.70325 (14) | 0.31952 (7) | 0.0166 (2) | |
| N4 | 0.89119 (18) | 0.97295 (13) | 0.18590 (8) | 0.0181 (2) | |
| C7 | 0.3389 (2) | 0.70035 (15) | 0.15497 (8) | 0.0136 (2) | |
| C8 | 0.3891 (2) | 0.74498 (15) | 0.23904 (8) | 0.0141 (2) | |
| C9 | 0.5696 (2) | 0.83248 (15) | 0.24908 (8) | 0.0152 (2) | |
| H9 | 0.6004 | 0.8588 | 0.3049 | 0.018* | |
| C10 | 0.7029 (2) | 0.87992 (15) | 0.17497 (8) | 0.0148 (2) | |
| C11 | 0.6613 (2) | 0.84379 (15) | 0.09005 (8) | 0.0154 (2) | |
| H11 | 0.7517 | 0.8788 | 0.0409 | 0.018* | |
| C12 | 0.4831 (2) | 0.75506 (14) | 0.08054 (7) | 0.0136 (2) | |
| C13 | 0.4365 (2) | 0.71642 (15) | −0.01015 (8) | 0.0160 (2) |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| N1 | 0.0161 (4) | 0.0167 (5) | 0.0128 (4) | −0.0032 (4) | −0.0022 (3) | −0.0010 (3) |
| N2 | 0.0184 (5) | 0.0215 (5) | 0.0143 (4) | −0.0086 (4) | −0.0004 (3) | −0.0001 (4) |
| C1 | 0.0161 (5) | 0.0138 (5) | 0.0138 (5) | −0.0018 (4) | −0.0014 (4) | −0.0003 (4) |
| C2 | 0.0144 (5) | 0.0164 (5) | 0.0180 (5) | −0.0032 (4) | −0.0014 (4) | −0.0009 (4) |
| C3 | 0.0183 (5) | 0.0184 (5) | 0.0156 (5) | −0.0022 (4) | 0.0005 (4) | 0.0009 (4) |
| C4 | 0.0238 (6) | 0.0234 (6) | 0.0123 (5) | −0.0048 (5) | −0.0033 (4) | −0.0007 (4) |
| C5 | 0.0200 (5) | 0.0205 (6) | 0.0149 (5) | −0.0058 (5) | −0.0039 (4) | −0.0005 (4) |
| C6 | 0.0225 (6) | 0.0321 (7) | 0.0206 (6) | −0.0078 (5) | 0.0030 (5) | 0.0045 (5) |
| O1 | 0.0192 (4) | 0.0257 (5) | 0.0154 (4) | −0.0105 (4) | −0.0028 (3) | −0.0014 (3) |
| O2 | 0.0271 (5) | 0.0276 (5) | 0.0202 (5) | −0.0141 (4) | 0.0037 (4) | −0.0011 (4) |
| O3 | 0.0285 (5) | 0.0422 (6) | 0.0122 (4) | −0.0098 (5) | −0.0019 (4) | −0.0062 (4) |
| O4 | 0.0210 (4) | 0.0230 (5) | 0.0296 (5) | −0.0032 (4) | −0.0110 (4) | −0.0068 (4) |
| O5 | 0.0235 (5) | 0.0269 (5) | 0.0337 (6) | −0.0121 (4) | −0.0010 (4) | 0.0021 (4) |
| O6 | 0.0286 (5) | 0.0266 (5) | 0.0127 (4) | −0.0099 (4) | 0.0000 (3) | −0.0007 (3) |
| O7 | 0.0215 (4) | 0.0242 (5) | 0.0142 (4) | −0.0094 (4) | −0.0031 (3) | −0.0008 (3) |
| N3 | 0.0163 (4) | 0.0197 (5) | 0.0134 (4) | −0.0017 (4) | −0.0011 (3) | −0.0005 (4) |
| N4 | 0.0144 (4) | 0.0149 (5) | 0.0261 (5) | −0.0027 (4) | −0.0048 (4) | −0.0025 (4) |
| C7 | 0.0133 (5) | 0.0139 (5) | 0.0135 (5) | −0.0018 (4) | −0.0015 (4) | −0.0009 (4) |
| C8 | 0.0137 (5) | 0.0158 (5) | 0.0128 (5) | −0.0021 (4) | −0.0006 (4) | −0.0009 (4) |
| C9 | 0.0145 (5) | 0.0151 (5) | 0.0162 (5) | −0.0007 (4) | −0.0038 (4) | −0.0030 (4) |
| C10 | 0.0125 (5) | 0.0132 (5) | 0.0195 (5) | −0.0031 (4) | −0.0028 (4) | −0.0017 (4) |
| C11 | 0.0148 (5) | 0.0140 (5) | 0.0171 (5) | −0.0019 (4) | −0.0007 (4) | −0.0010 (4) |
| C12 | 0.0147 (5) | 0.0144 (5) | 0.0119 (5) | −0.0029 (4) | −0.0014 (4) | −0.0008 (4) |
| C13 | 0.0187 (5) | 0.0162 (5) | 0.0136 (5) | −0.0028 (4) | −0.0028 (4) | −0.0014 (4) |
Geometric parameters (Å, °)
| N1—C1 | 1.3509 (15) | O1—H1A | 0.8200 |
| N1—C2 | 1.3668 (15) | O2—N3 | 1.2302 (14) |
| N1—H1 | 0.8600 | O3—N3 | 1.2348 (14) |
| N2—C1 | 1.3355 (15) | O4—N4 | 1.2404 (15) |
| N2—H2A | 0.8600 | O5—N4 | 1.2291 (15) |
| N2—H2B | 0.8600 | O6—C13 | 1.2331 (15) |
| C1—C5 | 1.4181 (16) | O7—C13 | 1.3067 (15) |
| C2—C3 | 1.3652 (17) | N3—C8 | 1.4546 (15) |
| C2—H2 | 0.9300 | N4—C10 | 1.4563 (15) |
| C3—C4 | 1.4164 (18) | C7—C8 | 1.4208 (16) |
| C3—C6 | 1.5019 (18) | C7—C12 | 1.4372 (16) |
| C4—C5 | 1.3681 (18) | C8—C9 | 1.3861 (16) |
| C4—H4 | 0.9300 | C9—C10 | 1.3794 (17) |
| C5—H5 | 0.9300 | C9—H9 | 0.9300 |
| C6—H6A | 0.9600 | C10—C11 | 1.3956 (17) |
| C6—H6B | 0.9600 | C11—C12 | 1.3793 (16) |
| C6—H6C | 0.9600 | C11—H11 | 0.9300 |
| O1—C7 | 1.2957 (14) | C12—C13 | 1.4943 (16) |
| C1—N1—C2 | 123.48 (10) | O2—N3—O3 | 122.24 (11) |
| C1—N1—H1 | 118.2 | O2—N3—C8 | 119.70 (10) |
| C2—N1—H1 | 118.3 | O3—N3—C8 | 118.06 (11) |
| C1—N2—H2A | 120.0 | O5—N4—O4 | 123.31 (11) |
| C1—N2—H2B | 120.0 | O5—N4—C10 | 118.76 (11) |
| H2A—N2—H2B | 120.0 | O4—N4—C10 | 117.93 (11) |
| N2—C1—N1 | 119.16 (11) | O1—C7—C8 | 123.94 (11) |
| N2—C1—C5 | 123.65 (11) | O1—C7—C12 | 120.07 (10) |
| N1—C1—C5 | 117.18 (11) | C8—C7—C12 | 115.98 (10) |
| C3—C2—N1 | 121.14 (11) | C9—C8—C7 | 122.15 (11) |
| C3—C2—H2 | 119.4 | C9—C8—N3 | 116.22 (10) |
| N1—C2—H2 | 119.4 | C7—C8—N3 | 121.63 (10) |
| C2—C3—C4 | 116.59 (11) | C10—C9—C8 | 118.97 (11) |
| C2—C3—C6 | 122.06 (12) | C10—C9—H9 | 120.5 |
| C4—C3—C6 | 121.34 (12) | C8—C9—H9 | 120.5 |
| C5—C4—C3 | 122.13 (11) | C9—C10—C11 | 122.23 (11) |
| C5—C4—H4 | 118.9 | C9—C10—N4 | 118.70 (11) |
| C3—C4—H4 | 118.9 | C11—C10—N4 | 119.06 (11) |
| C4—C5—C1 | 119.43 (11) | C12—C11—C10 | 118.54 (11) |
| C4—C5—H5 | 120.3 | C12—C11—H11 | 120.7 |
| C1—C5—H5 | 120.3 | C10—C11—H11 | 120.7 |
| C3—C6—H6A | 109.5 | C11—C12—C7 | 122.12 (10) |
| C3—C6—H6B | 109.5 | C11—C12—C13 | 118.93 (10) |
| H6A—C6—H6B | 109.5 | C7—C12—C13 | 118.95 (10) |
| C3—C6—H6C | 109.5 | O6—C13—O7 | 123.31 (11) |
| H6A—C6—H6C | 109.5 | O6—C13—C12 | 120.35 (11) |
| H6B—C6—H6C | 109.5 | O7—C13—C12 | 116.34 (10) |
| C7—O1—H1A | 109.5 | ||
| C2—N1—C1—N2 | 177.44 (11) | N3—C8—C9—C10 | −178.15 (10) |
| C2—N1—C1—C5 | −2.27 (17) | C8—C9—C10—C11 | 0.44 (18) |
| C1—N1—C2—C3 | 0.56 (19) | C8—C9—C10—N4 | 179.53 (11) |
| N1—C2—C3—C4 | 1.24 (18) | O5—N4—C10—C9 | −175.21 (11) |
| N1—C2—C3—C6 | −179.26 (12) | O4—N4—C10—C9 | 4.88 (17) |
| C2—C3—C4—C5 | −1.29 (19) | O5—N4—C10—C11 | 3.91 (17) |
| C6—C3—C4—C5 | 179.21 (13) | O4—N4—C10—C11 | −176.00 (11) |
| C3—C4—C5—C1 | −0.4 (2) | C9—C10—C11—C12 | −1.15 (18) |
| N2—C1—C5—C4 | −177.56 (12) | N4—C10—C11—C12 | 179.76 (11) |
| N1—C1—C5—C4 | 2.14 (18) | C10—C11—C12—C7 | 0.51 (18) |
| O1—C7—C8—C9 | 179.70 (12) | C10—C11—C12—C13 | 179.74 (11) |
| C12—C7—C8—C9 | −1.52 (17) | O1—C7—C12—C11 | 179.60 (11) |
| O1—C7—C8—N3 | −1.24 (19) | C8—C7—C12—C11 | 0.77 (17) |
| C12—C7—C8—N3 | 177.54 (10) | O1—C7—C12—C13 | 0.37 (17) |
| O2—N3—C8—C9 | −171.71 (11) | C8—C7—C12—C13 | −178.46 (10) |
| O3—N3—C8—C9 | 8.51 (17) | C11—C12—C13—O6 | −0.21 (18) |
| O2—N3—C8—C7 | 9.17 (18) | C7—C12—C13—O6 | 179.05 (11) |
| O3—N3—C8—C7 | −170.60 (11) | C11—C12—C13—O7 | −179.85 (11) |
| C7—C8—C9—C10 | 0.96 (18) | C7—C12—C13—O7 | −0.59 (16) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1—H1A···O7 | 0.82 | 1.66 | 2.4200 (13) | 152 |
| N1—H1···O6i | 0.86 | 1.82 | 2.6768 (14) | 174 |
| N2—H2A···O7i | 0.86 | 2.11 | 2.9668 (15) | 176 |
| N2—H2B···O1ii | 0.86 | 2.16 | 2.8468 (15) | 137 |
| N2—H2B···O2ii | 0.86 | 2.40 | 3.1723 (16) | 149 |
| C2—H2···O4iii | 0.93 | 2.49 | 3.4073 (16) | 169 |
| C4—H4···O3iv | 0.93 | 2.39 | 3.2371 (16) | 151 |
| C5—H5···O2ii | 0.93 | 2.44 | 3.2328 (17) | 143 |
Symmetry codes: (i) −x+1, −y+1, −z; (ii) x+1, y, z; (iii) x−1, y−1, z; (iv) −x+1, −y+1, −z+1.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: HB5405).
References
- Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor, R. (1987). J. Chem. Soc. Perkin Trans. 2, pp. S1–19.
- Bernstein, J., Davis, R. E., Shimoni, L. & Chang, N.-L. (1995). Angew. Chem. Int. Ed. Engl.34, 1555–1573.
- Bruker (2009). APEX2, SAINT and SADABS Bruker AXS Inc., Madison, Wisconsin, USA.
- Cosier, J. & Glazer, A. M. (1986). J. Appl. Cryst.19, 105–107.
- Hindawey, A. M., Nasser, A. M. G., Issa, R. M. & Issa, Y. M. (1980). Indian J. Chem. Sect. A, 19, 615–619.
- Issa, Y. M., Hindawey, A. M., El-Kholy, A. E. & Issa, R. M. (1981). Gazz. Chim. Ital.111, 27–33.
- Jeffrey, G. A. (1997). An Introduction to Hydrogen Bonding. Oxford University Press.
- Jeffrey, G. A. & Saenger, W. (1991). Hydrogen Bonding in Biological Structures. Berlin: Springer.
- Katritzky, A. R., Rees, C. W. & Scriven, E. F. V. (1996). Comprehensive Heterocyclic Chemistry II. Oxford: Pergamon Press.
- Nahringbauer, I. & Kvick, Å. (1977). Acta Cryst. B33, 2902–2905.
- Pozharski, A. F., Soldatenkov, A. T. & Katritzky, A. R. (1997). Heterocycles in Life and Society. New York: Wiley.
- Scheiner, S. (1997). Hydrogen Bonding. A Theoretical Perspective. Oxford University Press.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536810014480/hb5405sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810014480/hb5405Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report