

Abstract
Cobblestone (type II) lissencephaly and mental retardation are characteristic features of a subset of congenital muscular dystrophies that include Walker–Warburg syndrome, muscle-eye-brain disease, and Fukuyama-type congenital muscular dystrophy. Although the majority of clinical cases are genetically undefined, several causative genes have been identified that encode known or putative glycosyltransferases in the biosynthetic pathway of dystroglycan. Here we test the effects of brain-specific deletion of dystroglycan, and show distinct functions for neuronal and glial dystroglycan. Deletion of dystroglycan in the whole brain produced glial/neuronal heterotopia resembling the cerebral cortex malformation in cobblestone lissencephaly. In wild-type mice, dystroglycan stabilizes the basement membrane of the glia limitans, thereby supporting the cortical infrastructure necessary for neuronal migration. This function depends on extracellular dystroglycan interactions, since the cerebral cortex developed normally in transgenic mice that lack the dystroglycan intracellular domain. Also, forebrain histogenesis was preserved in mice with neuron-specific deletion of dystroglycan, but hippocampal long-term potentiation was blunted, as is also the case in the Largemyd mouse, in which dystroglycan glycosylation is disrupted. Our findings provide genetic evidence that neuronal dystroglycan plays a role in synaptic plasticity and that glial dystroglycan is involved in forebrain development. Differences in dystroglycan glycosylation in distinct cell types of the CNS may contribute to the diversity of dystroglycan function in the CNS, as well as to the broad clinical spectrum of type II lissencephalies.
Introduction
The laminar development of the cerebral cortex is dependent on the proliferation of neuronal precursors, scaffolding of radial glia, interactions between glia and neurons, and the presence of external localization cues (Diaz and Gleeson, 2009). Genetic mutations that impair these processes are associated with cognitive impairment and epilepsy, as well as structural malformations (Guerrini and Carrozzo, 2001; Ross and Walsh, 2001; Mochida and Walsh, 2004; Guerrini et al., 2008). A subset of congenital muscular dystrophies (CMDs), including Fukuyama-type congenital muscular dystrophy (FCMD), muscle-eye-brain disease (MEB), and Walker–Warburg syndrome (WWS), feature brain defects that include cobblestone (type II) lissencephaly (Muntoni and Voit, 2004). In contrast to type I (classical) lissencephaly, which is characterized by a four-layer cerebral cortex, cobblestone lissencephaly is characterized by glial/neuronal heterotopia that disrupt the laminar organization of the cerebral cortex (Olson and Walsh, 2002).
The genes linked to CMD-associated cobblestone lissencephaly encode known or putative glycosyltransferases, and muscle biopsies show that α-dystroglycan is hypoglycosylated in patients (Kobayashi et al., 1998; Yoshida et al., 2001; Beltrán-Valero de Bernabé et al., 2002, 2004; Barresi et al., 2004; van Reeuwijk et al., 2005, 2007; Godfrey et al., 2007; Clement et al., 2008). Dystroglycan is composed of α and β subunits that are noncovalently associated (Ibraghimov-Beskrovnaya et al., 1992, 1993). The α subunit of dystroglycan (α-dystroglycan) is an extracellular protein that binds laminin and laminin G-like domains of perlecan, agrin, neurexin, and pikachurin (Smalheiser and Schwartz, 1987; Bowe et al., 1994; Gee et al., 1994; Talts et al., 1999; Sugita et al., 2001; Sato et al., 2008). The β subunit of dystroglycan (β-dystroglycan) contains a single transmembrane domain, and its cytoplasmic tail binds dystrophin (Ervasti and Campbell, 1993). Dystroglycan forms a transmembrane link between the extracellular matrix and the intracellular actin cytoskeleton through its interactions with laminin and dystrophin. In skeletal muscle, this link provides structural integrity to the sarcolemma (Ervasti and Campbell, 1993). In addition, the cytoplasmic domain of β-dystroglycan is associated with rapsyn and the Ras/MAPK signaling pathway through the adapter protein Grb2 (Yang et al., 1995; Oak et al., 2003; Moore and Winder, 2010).
Previous immunohistochemical and in situ hybridization studies indicate that dystroglycan is expressed in migrating neurons as well as in radially oriented glia during CNS development (Henion et al., 2003; Ohtsuka-Tsurumi et al., 2004). It is not known whether dystroglycan mediates neuron-glia interactions or response to external cues during neuron migration. Dystroglycan binds α- and β-neurexins (Sugita et al., 2001), and these interactions might mediate neuron-glia adhesions. Furthermore, dystroglycan is a receptor for laminin, perlecan, and agrin–extracellular matrix proteins that are important to the organization and development of both the PNS and CNS (Smalheiser and Schwartz, 1987; Bowe et al., 1994; Miner et al., 1998; Ruegg and Bixby, 1998; Costell et al., 1999; Talts et al., 1999; Occhi et al., 2005; Chen et al., 2009).
Here we provide genetic evidence that dystroglycan has distinct functions in glia and neurons of the adult and developing CNS. Dystroglycan stabilizes the glial limitans basement membrane during cerebral cortex development and, in contrast to its mode of operation in skeletal muscle, this function is independent of the β-dystroglycan cytoplasmic domain. Neuronal expression of α- and β-dystroglycan was dispensible for the laminar development of the cerebral cortex; however, deletion of dystroglycan in neurons blunted hippocampal long-term potentiation (LTP), indicating a novel role in synaptic plasticity.
Materials and Methods
Generation of dystroglycan-deficient mice.
The generation of the floxed-dystroglycan mouse strain was previously described (Cohn et al., 2002). Heterozygous floxed dystroglycan (Dag1lox/lox) mice were bred to hemizygous mice expressing GFAP-Cre (Zhuo et al., 2001; Moore et al., 2002), Nestin-Cre (Tronche et al., 1999; Graus-Porta et al., 2001), NEX-Cre (Goebbels et al., 2006), or Pcp2-Cre (Barski et al., 2000). Mice heterozygous for the floxed dystroglycan allele and hemizygous for the Cre transgene (Cre-Dag1 +/lox) were bred with mice homozygous for the floxed dystroglycan allele (Dag1lox/lox) to generate conditional dystroglycan-deficient mice (Cre-Dag1lox/lox). The generation of the truncated dystroglycan mutant mouse strain was previously described (Satz et al., 2009). The genotypes of the mice were determined by PCR analysis of tail DNA.
Biochemistry.
For immunoblotting, brains were solubilized in TBS containing 1% Triton X-100 and protease inhibitors. The solubilized fraction was incubated with WGA-agarose beads (Vector Labs) for 24 h. The beads were washed three times in TBS containing 0.1% Triton X-100 and protease inhibitors. The bound proteins were eluted with TBS containing 0.1% Triton X-100, protease inhibitors and 300 mm N-acetyl-glucosamine. Proteins were separated by SDS-PAGE on 3–15% gels, transferred to polyvinylidene fluoride membranes, and probed with antibodies to dystroglycan (Ervasti and Campbell, 1991; Duclos et al., 1998).
Histology and immunofluorescence.
For routine histology, adult mice 10–30 weeks of age were deeply anesthetized with ketamine and killed by intracardiac perfusion with 4% paraformaldehyde. Sections were stained with hematoxylin and eosin or with reticulin stain, and imaged on a Leica DMRXA microscope. For immunofluorescence analysis, brains were frozen in isopentane cooled by dry ice. Sections 8 μm in thickness were cut on a RM2135 cryostat (Leica) and stored at −80°C before use. The sections were fixed for 10 min in 2% paraformaldehyde and blocked for 30 min with 2% BSA and 0.3% Triton X-100 in PBS. After overnight incubation of the primary antibody and washes with PBS, sections were incubated with CY3- or FITC-conjugated secondary antibodies (Jackson Laboratories) or biotinylated secondary antibodies (Vector Labs) and streptavidin conjugated to FITC or CY3 (Jackson Laboratories). Images were acquired on DMRXA (Leica), BX41 (Olympus) and MRC600 (Bio-Rad) microscopes.
Antibodies.
The following antibodies were used. IIH6 (1:100) (Ervasti and Campbell, 1991), AP83 (1:100) (Duclos et al., 1998), anti-laminin (1:1000) (Sigma L9393), anti-dystrophin (1:100) (Duclos et al., 1998). IIH6 is a monoclonal antibody to the fully glycosylated species of α-dystroglycan (Ervasti and Campbell, 1991). AP83 is a rabbit polyclonal antibody raised against the C-terminal 15 aa of β-dystroglycan (Duclos et al., 1998).
Electrophysiology.
For electrophysiological recordings, the brain was removed and sectioned in ice-cold artificial CSF (ACSF) at pH 7.4 containing (in mm): 119 NaCl, 2.5 KCl, 2.5 CaCl2, 1.0 NaH2PO4, 1.3 MgSO4 26.2 NaHCO3, 11 glucose, bubbled with 95% O2 with 5% CO2. The slices were incubated in the same solution at 31°C for 1–5 h before the recordings.
Standard extracellular field potential recordings were performed at 28 ± 0.5°C. Field EPSPs (fEPSPs) were evoked in CA1 stratum radiatum by stimulation of Schaffer collaterals with a tungsten bipolar electrode (World Precision Instruments) placed at the border of CA3–CA1, and recorded using a ACSF-filled glass pipette (4–6 m) and an Axopatch 200A amplifier (Axon). Stimulation (100 μs) was delivered every 30 s by a stimulus isolation unit (Grass Instruments, SD9). Stimulus-response curves were obtained by plotting the stimulus voltages against the amplitudes of the fEPSPs. The voltage that induced 40–50% of maximal synaptic response was then applied to establish a stable baseline for at least 10 min before LTP was induced by a high-frequency stimulation (HFS; 100 Hz, 1 s) with the same voltage. The signal was measured by normalizing the fEPSP amplitude after HFS to the baseline fEPSP before HFS. Data were digitized (10 kHz), filtered at 1 kHz, and monitored online using PatchMachine (http://www.hoshi.org). The offline analysis was performed using IgorPro version 6.0 (WaveMetrics).
Results
Targeting strategy for Dag1 inactivation in mice
Insertion of a neomycin resistance cassette into the second exon of the dystroglycan gene resulted in loss of dystroglycan expression and subsequent embryonic lethality (Williamson et al., 1997). A conditional dystroglycan allele was generated by using homologous recombination to insert LoxP sites flanking the second exon of the dystroglycan gene, as previously described (Cohn et al., 2002). Mice with the floxed-dystroglycan allele are viable and the colony was maintained with homozygous expression of the floxed allele. Mice bearing the floxed-dystroglycan allele were intercrossed with Nestin-Cre (Tronche et al., 1999; Graus-Porta et al., 2001; Satz et al., 2009), GFAP-Cre (Zhuo et al., 2001; Moore et al., 2002; Satz et al., 2009), NEX-Cre (Goebbels et al., 2006), or Pcp2-Cre (Barski et al., 2000) mice. Homozygous floxed-dystroglycan (Dag1 lox/lox) mice were bred to mice hemizygous for Cre. Mice heterozygous for the floxed-dystroglycan allele and hemizygous for Cre (Cre+-Dag1 +/lox) were bred with mice homozygous for the floxed-dystroglycan allele (Dag1 lox/lox) to generate dystroglycan-deficient mice (Cre/DG-null). The generation of Nestin-Cre/DG-null (Satz et al., 2009) and GFAP-Cre/DG-null (Moore et al., 2002; Satz et al., 2009) mice was previously described.
Nestin-Cre inactivation of Dag1 in brain
In Nestin-Cre mice, the neuron-specific enhancer of the Nestin promoter drives expression of Cre recombinase in glia and neurons throughout the CNS, beginning by embryonic day 10.5 (Tronche et al., 1999; Graus-Porta et al., 2001). Nestin-Cre expression occurs earlier and more broadly in the brain than GFAP-Cre, which has been previously used to delete dystroglycan in brain (Zhuo et al., 2001; Moore et al., 2002). Maximal expression of GFAP-Cre occurs at embryonic day 14.5 and expression of the transgene is restricted to radial glia and astrocytes, silencing dystroglycan in these cells and a subset of neurons that are the progeny of radial glia (Zhuo et al., 2001).
Nestin-Cre/DG-null mice were born according to Mendelian ratio. To confirm the loss of dystroglycan in the mutant mice, brains were examined by immunoblotting and immunofluorescence analysis with antibodies to α- and β-dystroglycan. For immunoblotting, adult Nestin-Cre/DG-null and littermate control brains were dissected into brain regions and run separately on SDS-PAGE. A 120 kDa band corresponding to α-dystroglycan, and a 43 kDa band corresponding to β-dystroglycan were detected in all brain regions from control mice but not in those from Nestin-Cre/DG-null mice (Fig. 1 A). In addition, the antibody to α-dystroglycan detected additional high molecular-weight bands in WGA-enriched homogenates from the cerebral cortex, hippocampus, and cerebellum of wild-type mice. The high molecular-weight bands were absent in Nestin-Cre/DG-null mice.
Figure 1.
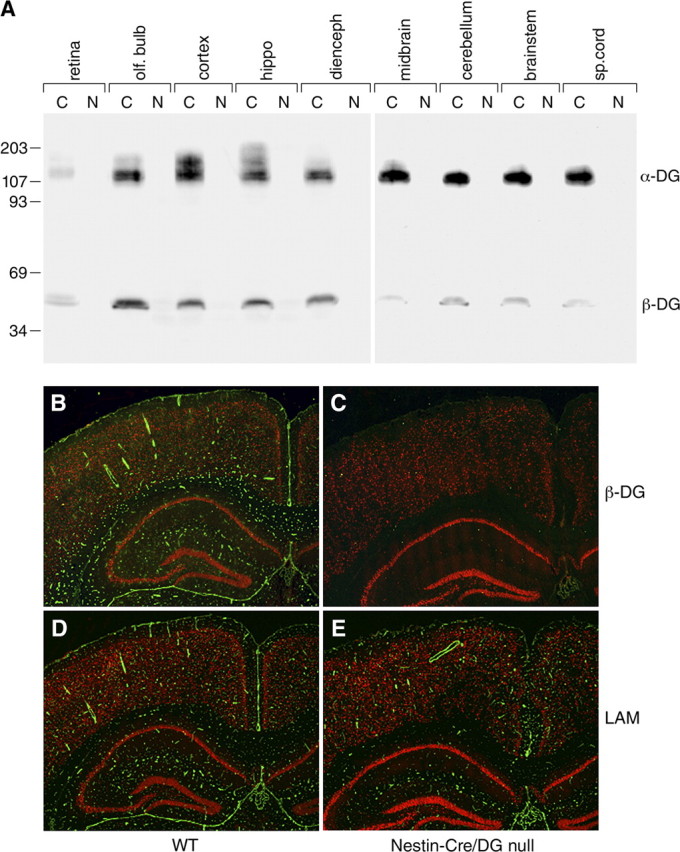
Loss of dystroglycan expression in Nestin-Cre/DG-null mouse brain. A, Western blot analysis of multiple brain regions from wild-type littermate control (C) and Nestin-Cre/DG-null (N) adult brain. B, C, Immunofluorescence detection of β-dystroglycan (green) in the cerebrum of wild-type littermate control (B) and Nestin-Cre/DG-null (C) mice. D, E, Immunofluorescence detection of laminin (green) in littermate control (D) and Nestin-Cre/DG-null (E) mice. Scale bars, 200 μm.
Immunofluorescence analysis revealed that in wild-type mice dystroglycan was present in glial foot processes abutting the glia limitans, in the intraparenchymal vasculature, and in the epithelium of the choroid plexus (Fig. 1 B). In the Nestin-Cre/DG-null mice, dystroglycan expression persisted in the choroid plexus, which does not express the Nestin-Cre transgene (Graus-Porta et al., 2001), but was undetected in other cell types in the brain (Fig. 1 C). Furthermore, laminin localization was discontinuous at the glia limitans but preserved at perivascular glial foot processes despite the loss of dystroglycan (Fig. 1 E). Dystroglycan persisted in Schwann cells of cranial nerves, vascular smooth muscle of cerebral arteries on the ventral surface of the brainstem (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material), and Schwann cells of spinal nerve roots (data not shown).
The Nestin-Cre transgene has been reported to drive recombination of floxed alleles beginning at embryonic day 10.5 (Tronche et al., 1999; Graus-Porta et al., 2001). PCR analysis on genomic DNA derived from heads [embryonic day 9.5 (E9.5)–E15.5] or dissected brains (E16.5–E18.5) from Nestin-Cre/DG-null embryos revealed that Dag1 recombination occurred from E9.5 through E18.5 (data not shown; see Moore et al., 2002, for methods). A wide range of adult tissues was also evaluated by Dag1 recombination-specific PCR in Nestin-Cre/DG-null mice, and in Nestin-Cre mice crossed with ROSA26 reporter mice (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). Nestin-Cre recombinase activity detected in non-CNS tissues (especially skeletal muscle, heart, and lung) was largely vascular (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material), except for a small minority of epithelial cells that expressed Nestin-Cre in kidney and liver (data not shown). To confirm the time of dystroglycan loss in the Nestin-Cre/DG-null mice, we immunostained cryosections of embryonic brain with antibodies to dystroglycan. At embryonic day 12.5, dystroglycan was detected in the glial foot processes and choroid plexus from both littermate control and Nestin-Cre/DG-null mice (data not shown). At embryonic day 13.5, dystroglycan expression was no longer detected in glial foot processes from Nestin-Cre/DG-null mice (supplemental Fig. 2A,B, available at www.jneurosci.org as supplemental material).
Brain malformation in Nestin-Cre/DG-null mice includes hydrocephalus, megalencephaly, and cortical dysplasia characteristic of cobblestone lissencephaly
Approximately 38% of the Nestin-Cre/DG-null mice developed domed skulls (n = 24) (Fig. 2), indicating that hydrocephalus was present before the fusion of the cranial sutures in some mice. Gross examination and histological analysis revealed that the lateral and third ventricles were dramatically enlarged (Fig. 2 D,E). Patent cerebral aqueducts were observed in serial sections (Fig. 2 G) of the affected brains. In all Nestin-Cre/DG-null mice, megalencephaly was readily recognizable at the time of brain dissection. The brain mass was 30% greater than that of littermate controls, even though the Nestin-Cre/DG-null mice were runted (supplemental Fig. 3, available at www.jneurosci.org as supplemental material); brain mass in mice with severe hydrocephalus was not evaluated. When viewed from the surface, Nestin-Cre/DG-null brains showed a less complex network of vascular branches than those of littermate controls, but small pial surface vessels may have been obscured by the presence of glial/neuronal heterotopia within the leptomeninges (supplemental Fig. 4B,D, available at www.jneurosci.org as supplemental material). Sections of the brains showed no apparent difference in the density of microvessels, but the radial orientation of the cortical vessels was disrupted in the dystroglycan-deficient mice (supplemental Fig. 4F, available at www.jneurosci.org as supplemental material).
Figure 2.

Brain-specific loss of dystrolgycan produces a high incidence of hydrocephalus. A–C, Photos of wild-type littermate (upper mouse in each panel) and Nestin-Cre/DG-null (lower mouse in each panel) mice that show the abnormally domed cranium of DG-null mice. D, Gross coronal section of Nestin-Cre/DG-null mouse with severe hydrocephalus. E, Hematoxylin- and eosin-stained coronal section of Nestin-Cre/DG-null mouse. The section roughly corresponds to the gross photo in D. F, Higher-magnification hematoxylin and eosin stain of Nestin-Cre/DG-null cerebral cortex midline. G, Hematoxylin- and eosin-stained section of the aqueduct of Sylvius of a Nestin-Cre/DG-null mouse midbrain. Scale bars, 100 μm.
Histological examination of Nestin-Cre/DG-null mice revealed defects in neuronal migration that resemble cobblestone lissencephaly (Fig. 3 B,D,F; supplemental Fig. 5B,C, available at www.jneurosci.org as supplemental material). The laminar organization of the cerebral cortex was disrupted, and extensive glial/neuronal heterotopia were present over the entire cerebral convexities; heterotopia extended into leptomeninges and obliterated the subarachnoid space. In the hippocampus, focal irregularities of the dentate gyrus granule cell layer were present in some animals (Fig. 3 H). In the cerebellum, the number of cerebellar lobules was preserved, but ectopic granule neurons were present at sites of fusion between adjacent cerebellar lobules and on the surface of cerebellar lobules (Fig. 3 J). Disruptions of the glia limitans accompanied by glial/neuronal heterotopia were observed along the dorsal surface of the brainstem, while extensive examination of spinal cord revealed no pathology of either the glia limitans or leptomeninges (supplemental Fig. 5G,I,K).
Figure 3.
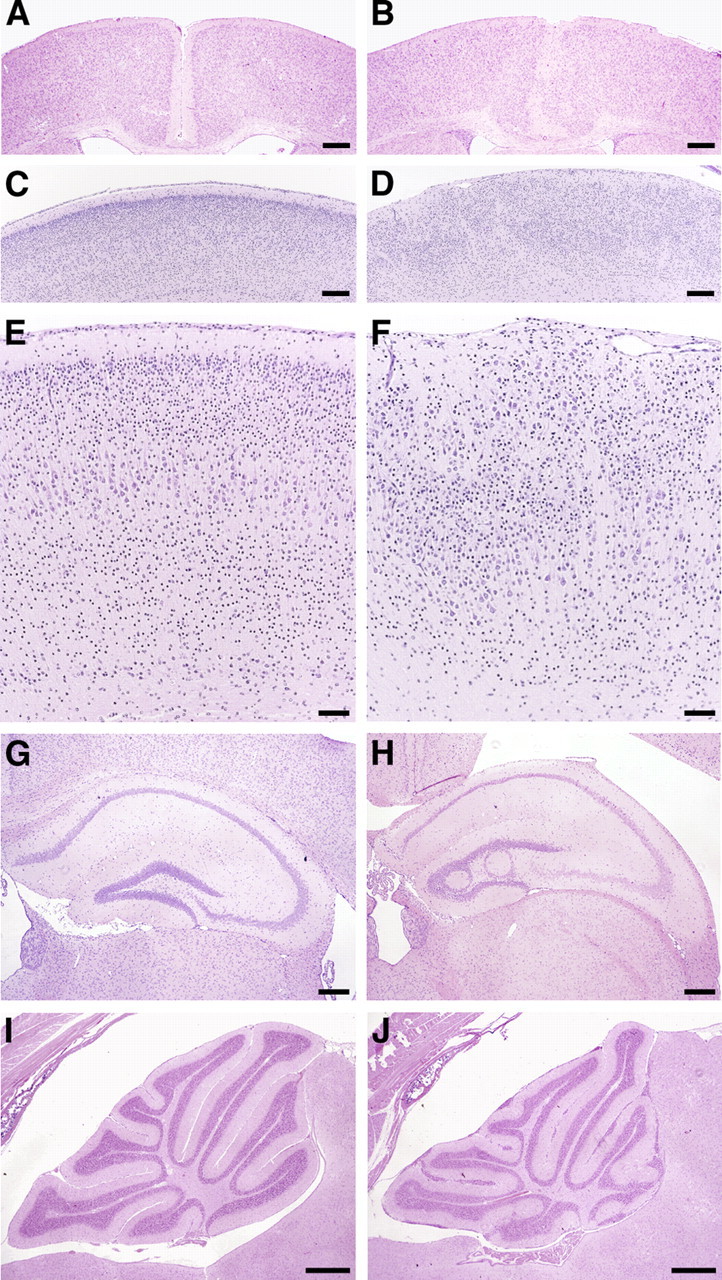
Neuron migration defects in adult Nestin-Cre/DG-null mouse brain. A–J, Hematoxylin- and eosin-stained sections of wild-type littermate control (A, C, E, G, I) and Nestin-Cre/DG-null (B, D, F, H, J) brains. B, Fused cerebral hemispheres in Nestin-Cre/DG-null brain. D, F, Dysplastic cerebral cortex far lateral to the midline in Nestin-Cre/DG-null brain. Heterotopic cells extend into layer I of the cortex and into the leptomeninges. H, Loops of ectopic granule neurons in the dentate gyrus of Nestin-Cre/DG-null hippocampus. J, Cerebellar granule cells that have failed to migrate from the lobular surfaces of Nestin-Cre/DG-null cerebellum. Fissures are fused in many regions. Scale bars: A–D, 250 μm; E, F, 100 μm; G, H, 250 μm; I, J, 500 μm.
The relationship between the loss of dystroglycan expression and defects in neuron migration was examined by comparing the histology of cerebral cortices from Nestin-Cre/DG-null and GFAP-Cre/DG-null mice. Light microscopy revealed disruptions of the glia limitans and glial/neuronal heterotopia on the first day that dystroglycan immunostaining was negative, and an increase in glial/neuronal heterotopia throughout embryonic development (Fig. 4 E–H; supplemental Fig. 2B, available at www.jneurosci.org as supplemental material). Ultrastructural evaluation revealed disruptions a day earlier (Fig. 4 B). Glial/neuronal heterotopias affected a larger area of the cerebral cortex in the Nestin-Cre/DG-null mice. Although the medial cortex was disrupted by glial/neuronal heterotopia in both mouse strains, the lateral cortex was affected only in the Nestin-Cre/DG-null animals (Fig. 5; supplemental Fig. 6, available at www.jneurosci.org as supplemental material). The spatial differences in pathology appear to have been caused by the late expression of GFAP-Cre, but may also have been a consequence of incomplete deletion of neuronal dystroglycan by GFAP-Cre expression. GFAP-Cre transgene expression is first detected in the dorsal and medial telencephalon at E13.5, although GFAP-Cre expression is present throughout the cerebral cortex by the time of birth (Zhuo et al., 2001).
Figure 4.
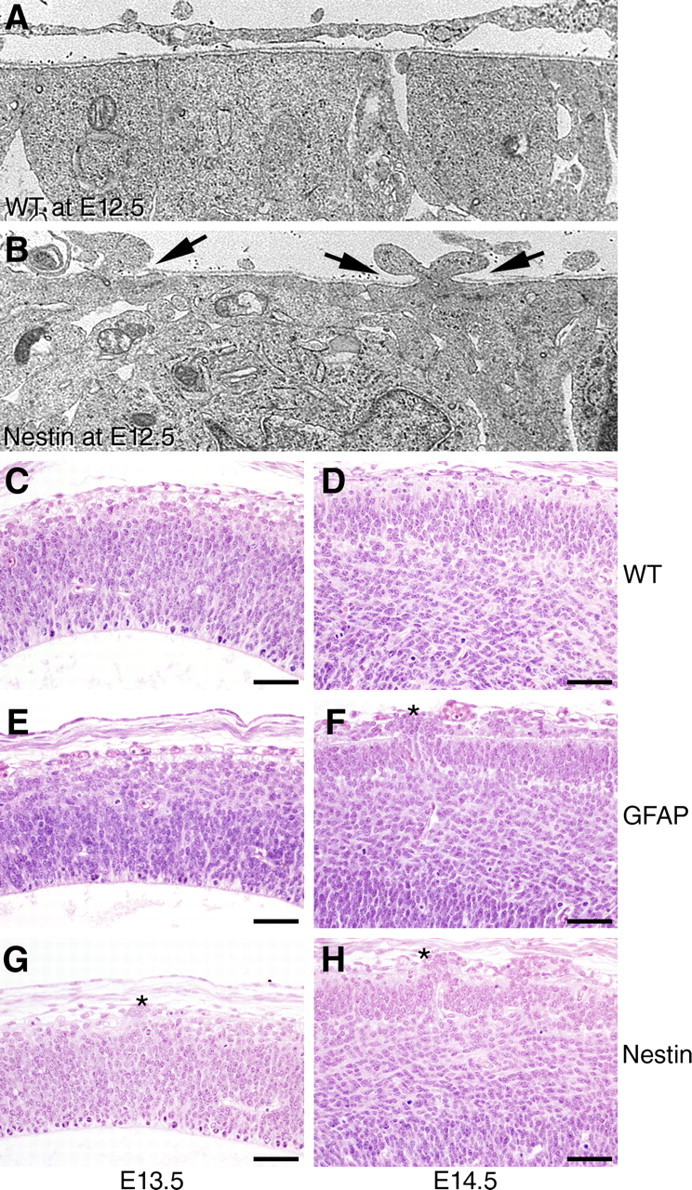
Temporal differences in cerebral cortex pathology in dystroglycan-null mice. A, B, Electron micrographs of E12.5 cerebral cortex (A, littermate control; B, Nestin-Cre/DG-null). Arrows in B mark the locations of basement membrane disruption where cell processes protrude into the subarachnoid space. C–H, Hematoxylin- and eosin-stained sections from E13.5 (C, E, G) and E14.5 (D, F, H) cerebral cortices from wild-type littermate (C, D), GFAP-Cre/DG-null (E, F), and Nestin-Cre/DG-null (G, H) mice, showing that pathology in Nestin-Cre/DG-null mice is first apparent at the light microscopy level at E13.5, and that pathology in GFAP-CRE/DG-null mice is first apparent at E14.5. Asterisks denote glial/neuron heterotopia. Scale bars, 50 μm.
Figure 5.
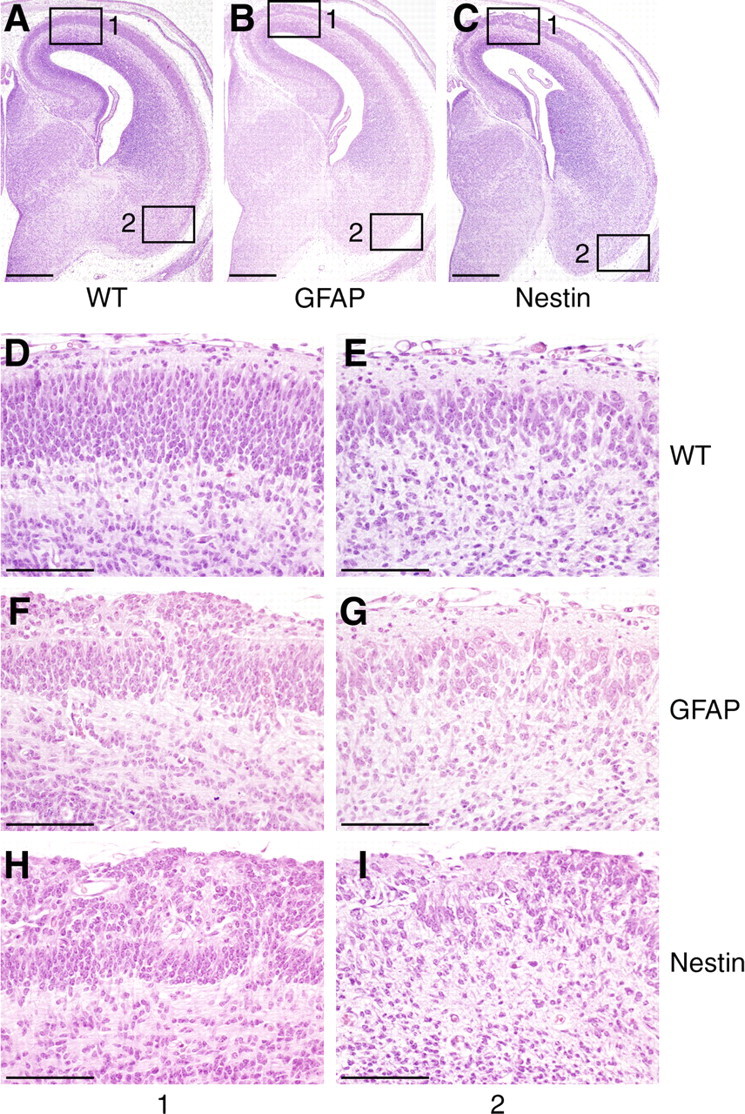
Spatial differences in cerebral cortex pathology in dystroglycan-null embryos. A–I, Sections of wild-type (A, D, E), GFAP-Cre/DG-null (B, F, G), and Nestin-Cre/DG-null (C, H, I) mice, showing a medial to lateral gradient of pathology that corresponds to the time of dystroglycan loss in the conditional DG-null mutants. Enlarged images of the medial (1) and lateral (2) cerebral cortex show diffuse glial/neuronal heterotopia in Nestin-Cre/DG-null mice, and pathology that is restricted to the medial cerebral cortex in GFAP-Cre/DG-null mice. Scale bars, A–C, 500 μm; D–I, 100 μm.
In the cerebral cortex, radial glia serve as neuronal precursors and also provide scaffolding for neuron migration (Noctor et al., 2001). The processes of radial glia extend from the ventricular zone and form foot processes abutting the basement membrane of the glia limitans at the pial surface. To assess the integrity of the glia limitans and the morphology of the processes of the radial glia, we labeled cryosections of embryonic brains with antibodies to nestin and laminin. In the brains of littermate control mice, the nestin antibody labeled radially oriented glial fibers with foot processes adjacent to the pial surface, and the laminin antibody stained the basement membrane at the glia limitans. In sections of Nestin-Cre/DG-null mouse brains, laminin expression was discontinuous at the glia limitans, and radially oriented glial processes were disorganized (Fig. 6 B,D). In some cases, remnants of basal lamina were present in the brain parenchyma, deep underneath the brain surface. Some radially oriented glia formed foot processes abutting these remnants, while others extended into the leptomeningeal heterotopia (Fig. 6 F). The lateral-most edge of heterotopia in both Nestin-Cre/DG-null and GFAP-Cre/DG-null mice (data not shown) transitioned abruptly from intact to disrupted glia limitans, with the heterotopia appearing as a wedge at the transition zone (supplemental Fig. 7, available at www.jneurosci.org as supplemental material).
Figure 6.

Disruption of the glia limitans, with abnormalities of the radial glia, in Nestin-Cre/DG-null mice. A–F, Sections of wild-type littermate (A, C, E) and Nestin-Cre/DG-null (B, D, F) cerebral cortex at E16.5, labeled with antibodies to laminin (green) and nestin (red), reveal disruption of the glia limitans and aberrant positioning of endfeet and processes of radial glia in Nestin-Cre/DG-null mice. Scale bars, 20 μm.
CNS development in mice lacking the cytoplasmic domain of dystroglycan (βcyt/βcyt)
Through the molecular interactions of its α- and β-subunits, dystroglycan forms a link between the extracellular matrix and the cytoskeleton. In skeletal muscle, disruption of this link causes muscular dystrophy (Ervasti and Campbell, 1993). The cytoplasmic domain of β-dystroglycan also binds rapsyn and the adapter protein Grb2, suggesting that dystroglycan may have additional roles in cell signaling (Moore and Winder, 2010). To test whether the β-dystroglycan cytoplasmic region is required for normal brain development, we examined the histology of the brain in transgenic mice lacking the entire cytoplasmic region of β-dystroglycan (βcyt/βcyt).
Generation of the βcyt/βcyt mice was previously described (Satz et al., 2009). To confirm that the βcyt dystroglycan mutant protein was expressed, brain sections were labeled with antibodies to α- and β-dystroglycan. Immunofluorescence analysis with the IIH6 antibody, which specifically recognizes α-dystroglycan (Michele et al., 2002), showed that dystroglycan was correctly localized in the βcyt/βcyt mice (Fig. 7 B). The AP83 antibody, which recognizes the C terminus of β-dystroglycan (Duclos et al., 1998), showed no signal in the βcyt/ βcyt mouse brain, confirming that the β-dystroglycan cytoplasmic domain was deleted (Fig. 7 D). In the βcyt/ βcyt mice, laminin was normally expressed at basement membrane interfaces formed by glial endfeet (Fig. 7 F), suggesting that the intracellular interactions of dystroglycan are dispensible for the organization of laminin at the glia limitans.
Figure 7.
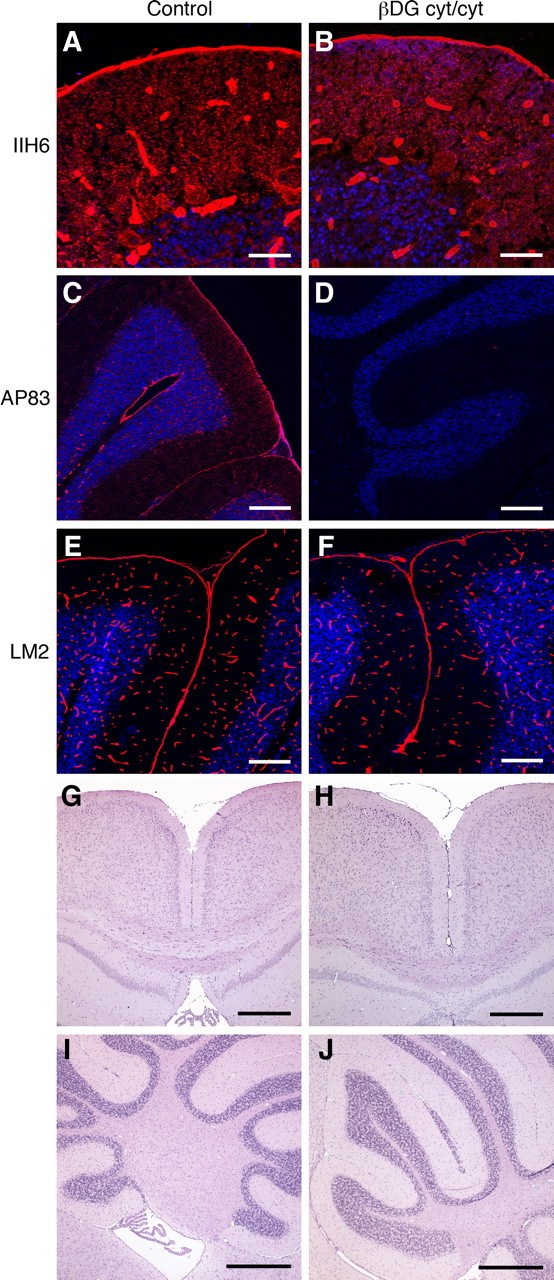
Mild migration defects limited to cerebellum in mutant mice that lack the cytoplasmic domain of β-dystroglycan. A–F, Sections of wild-type littermate control (A, C, E) and βcyt/cyt (B, D, F) cerebellum labeled with IIH6 (A, B), AP83 (C, D), and anti-laminin (E, F). G, H, Sections of cerebral cortices from wild-type littermate (G) and βcyt/cyt mice (H). I, J, Sections of cerebella from wild-type littermate (I) and βcyt/cyt mice (J). Small foci of granule cells fail to migrate in the βcyt/cyt cerebellum. Scale bars: A, B, 20 μm; C–F, 100 μm; G–J, 500 μm.
Although deletion of dystroglycan causes severe brain malformations, the deletion of the cytoplasmic domain of β-dystroglycan had a relatively small effect on neuron migration. The laminar organization of the cerebral cortex in βcyt/ βcyt mice was indistinguishable from that in littermate controls at the light microscopy level (Fig. 7 H). The cytoarchitecture of the cerebellum was also relatively preserved, but small foci of ectopic granule neurons were observed in the cerebella of βcyt/βcyt mice (Fig. 7 I).
Generation of NEX-Cre/DG-null mice
Previous studies indicate that dystroglycan is expressed in migrating neuroblasts as well as in glia (Henion et al., 2003; Ohtsuka-Tsurumi et al., 2004), and based on the above-described experiments we could not exclude the possibility that neuronal dystroglycan may be necessary for migration. To determine whether loss of dystroglycan from neurons would disrupt their migration, we generated mice with a NEX-Cre deletion of dystroglycan. NEX regulatory elements drive expression of the Cre recombinase beginning at embryonic day 10.5 (as in the case of Nestin-Cre), but NEX-Cre expression is restricted to neurons of the telencephalon, including cerebral cortical neurons and neurons of the hippocampus (Goebbels et al., 2006; Belvindrah et al., 2007).
To confirm the loss of dystroglycan in NEX-Cre/DG-null mice, brains were examined by immunoblotting with antibodies to α- and β-dystroglycan. To minimize contamination from brain regions that lack NEX-Cre expression, the cerebral cortices were dissected and run on SDS-PAGE. α- and β-dystroglycan expression was reduced in NEX-Cre/DG-null mice (Fig. 8 A) relative to wild-type mice. Macroscopic inspection of the NEX-Cre/DG-null brain revealed no structural abnormalities. The histology of the cerebral cortex also appeared normal in NEX-Cre/DG-null mice (Fig. 8 D), and immunofluorescence with antibodies to laminin demonstrated that the glia limitans was intact (Fig. 8 F).
Figure 8.
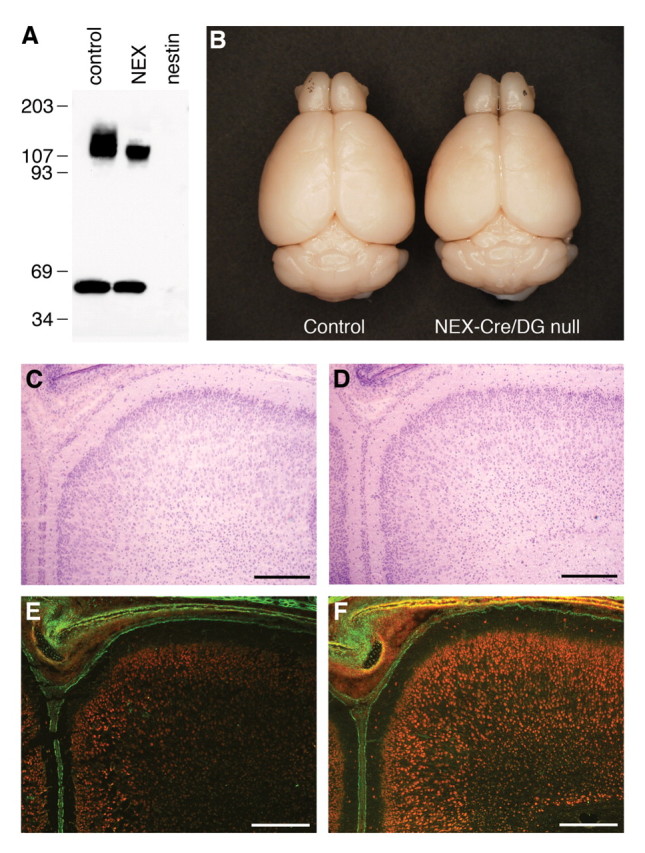
Neuron-specific loss of dystroglycan in the telencephalon fails to produce glial/neuronal heterotopia in NEX-Cre/DG-null mice. A, Immunoblot of control, NEX-Cre/DG-null, and Nestin-Cre/DG-null cerebral cortex. B, Gross photograph of control and NEX-Cre/DG-null brains. C, D, Sections of wild-type littermate (C) and NEX-Cre/DG-null (D) neonate cerebral cortex stained with hematoxylin and eosin. E, F, Sections of wild-type littermate (E) and NEX-Cre/DG-null (F) neonate cortex stained with anti-laminin (green) and propidium iodide (red). Scale bars: A–D, 100 μm.
Impaired long-term potentiation in NEX-Cre/DG-null mice and Largemyd
A light and electron microscopy study showed dystroglycan localization in the hippocampus at the postsynaptic apparatus of synapses established by the mossy fibers on cell bodies and dendrites of pyramidal neurons (Zaccaria et al., 2001). GFAP-Cre-driven deletion of dystroglycan was previously shown to blunt long-term potentiation, through an uncharacterized mechanism (Moore et al., 2002), suggesting that dystroglycan may modulate synaptic function. The effect on synaptic plasticity may be due to the loss of dystroglycan expression in either neurons or glia, which may integrate synaptic function (Haydon, 2001). Also, we cannot rule out the possibility that defects in brain development may have affected the electrophysiology of the GFAP-Cre/DG-null brain. To address these questions, we examined hippocampal long-term potentiation in NEX-Cre/DG-null mice.
To investigate whether synaptic transmission is affected in the NEX-Cre/DG-null mice, we measured fEPSPs in the isolated hippocampus slices. Data recorded in 17 slices from 8 mutant mice and 18 slices from 8 wild-type animals were collected and analyzed. Synaptic transmission in the NEX-Cre/DG-null mice did not differ from that in wild-type mice, as shown in Figure 9 A (black traces). However, the normal enhancement of synaptic transmission in response to a high-frequency stimulus was greatly blunted (Fig. 9 A,B), indicating that long-term potentiation at CA3–CA1 synapses in the hippocampus was impaired by the loss of neuronal dystroglycan.
Figure 9.
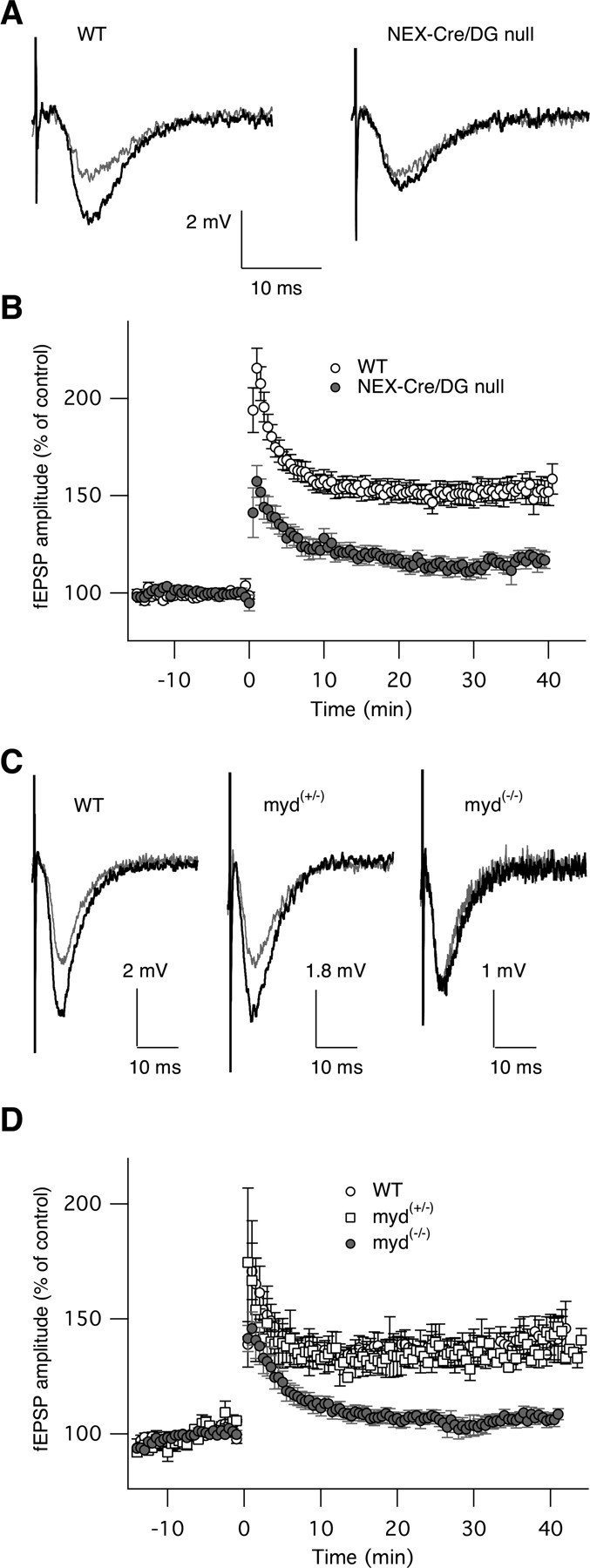
Neuron-specific deletion and hypoglycosylation of dystroglycan suppress long-term potentiation. A, Representative basic fEPSPs (black traces) and induced fEPSPs (gray traces) from wild-type (left) and NEX-Cre/DG-null (right) mice. B, Time course showing normalized fEPSPs before and after high-frequency stimulus in wild-type and Nex-Cre/DG-null mice. C, fEPSPs from wild-type, myd+/− and myd−/− mice (top). HFS induced LTP in wild-type (n = 8) and myd+/− (n = 3) mice but failed to induce LTP in myd-null myd−/− mice (n = 15). D, The amplitudes of fEPSPs normalized to the average fEPSPs before HFS are plotted as a function of time (each n represents the number of slices but not animals) (bottom).
In congenital muscular dystrophies, dystroglycan is present in a hypoglycosylated state (Michele et al., 2002; Satz et al., 2008), but long-term potentiation has not been previously examined in mice with defects in dystroglycan glycosylation. For this reason we examined long-term potentiation in Largemyd mice, a mouse model of cobblestone lissencephaly caused by mutation of Large (Grewal et al., 2001; Holzfeind et al., 2002; Michele et al., 2002), a glycosyltransferase necessary for the normal glycosylation of dystroglycan (Barresi et al., 2004; Barresi and Campbell, 2006). Similar to NEX-Cre/DG-null mice, the enhanced synaptic transmission induced by high-frequency stimulus was greatly inhibited (Fig. 9 C,D), indicating the long-term potentiation at CA3–CA1 synapses in the hippocampus is also impaired by defective dystroglycan glycosylation.
Cellular differences in dystroglycan glycosylation
The molecular weight of α-dystroglycan in brain has been reported as 120 kDa (Smalheiser and Schwartz, 1987). Immunoblotting of mouse brain proteins with the glycoepitope-specific IIH6 antibody revealed additional high molecular-weight bands in the cerebral cortex, hippocampus and cerebellum, suggesting that previously unrecognized differences in α-dystroglycan glycosylation are present in the brain. Cell type-specific differences in dystroglycan glycosylation have been previously observed in the retina (Moukhles et al., 2000) but have not been recognized in the brain. For these reasons, we compared the expression of dystroglycan in brain regions from conditional mutant mice that exhibited cell type-specific patterns of dystroglycan deletion.
In the control hippocampus and cerebral cortex, α-dystroglycan was detected as a broad band of higher molecular weight compared with the NEX-Cre/DG-null hippocampus and cerebral cortex, indicating that higher molecular weight glycoforms are present in the neurons that are targeted by the NEX-Cre transgene. Dramatic differences in dystroglycan glycosylation were observed in the cerebellum, where we detected a band of ∼180 kDa in addition to a band at 120 kDa using the IIH6 antibody. The higher molecular weight band is absent from immunoblots of Nestin-Cre/DG-null cerebella, which lack dystroglycan expression in both neurons and glia (Fig. 10 A). However, the 180 kDa band is present in GFAP-Cre/DG-null mice (Fig. 10 A), which show persistent expression of dystroglycan in Purkinje neurons (Moore et al., 2002), suggesting that the 180 kDa glycoform of α-dystroglycan is specific for Purkinje cells. We were able to demonstrate the specificity of the 180 kDa band by using Pcp2-Cre (Barski et al., 2000) to delete dystroglycan specifically in Purkinje cells. The 180 kDa band was severely reduced in PCP-Cre/DG-null mice. The high molecular-weight bands were capable of binding ligand (Fig. 10 B,C), demonstrating that they are functional receptors and confirming that the novel bands detected by the IIH6 antibody were dystroglycan.
Figure 10.
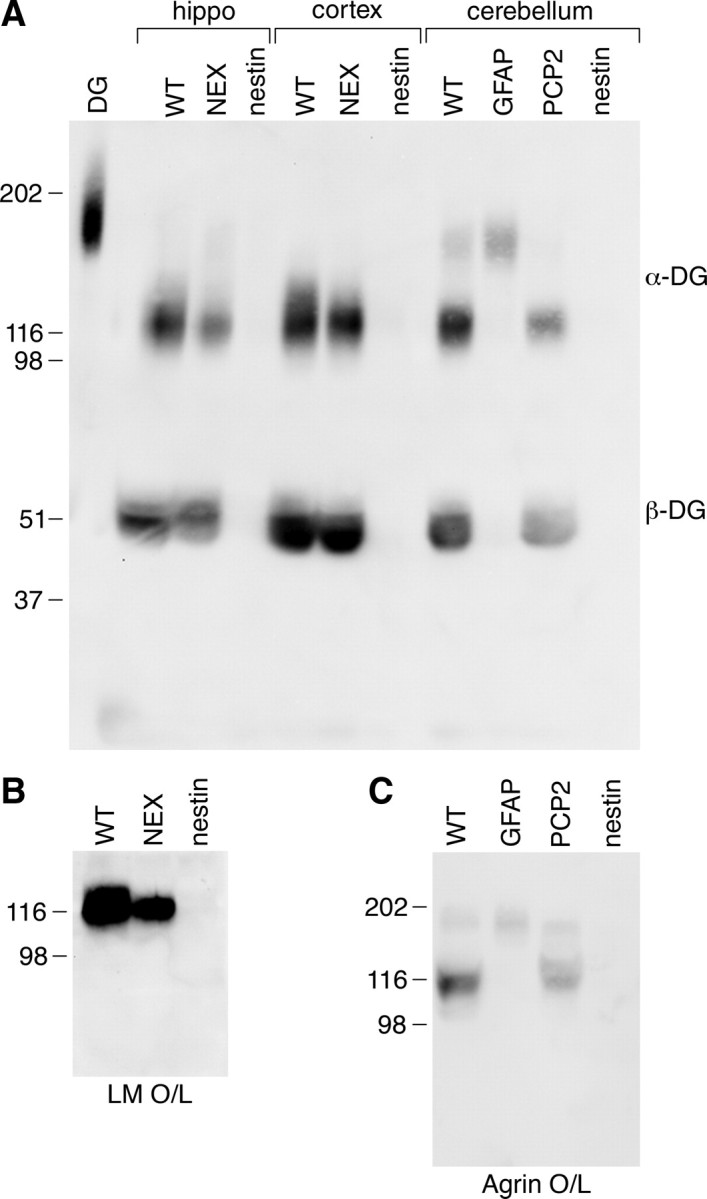
Differential glycosylation of dystroglycan in neurons and glia. A, WGA enriched homogenates of cortex and hippocampus from wild-type, NEX-Cre/DG-null and Nestin-Cre/DG-null mice and cerebellum from wild-type, GFAP-Cre/DG-null, PCP2-Cre/DG-null and Nestin-Cre/DG-null mice. B, Ligand overlay on wild-type, Nex-Cre/DG-null and Nestin-Cre/DG-null cerebral cortex. C, Ligand overlay on wild-type, GFAP-Cre/DG-null, PCP2-Cre/DG-null, and Nestin-Cre/DG-null cerebellum.
Discussion
Abnormal posttranslational processing of dystroglycan is associated with congenital muscular dystrophies with CNS involvement ranging from mental retardation to structural defects, including cobblestone (type II) lissencephaly and hydrocephalus (Muntoni and Voit, 2004; Barresi and Campbell, 2006). Defects in neuron migration that resemble cobblestone lissencephaly also occur in mice with tissue-specific deletion of dystroglycan (Moore et al., 2002; Satz et al., 2008). Prior studies indicate that dystroglycan is broadly expressed in neurons and glia of the developing and adult brain (Zaccaria et al., 2001; Henion et al., 2003; Ohtsuka-Tsurumi et al., 2004); however, little is known about its cellular functions. In this study we provide genetic evidence of distinct roles for glial dystroglycan in development, and for neuronal dystroglycan in synaptic plasticity.
We previously showed that MOX2-Cre mediated deletion of dystroglycan (MORE-DG-null) in all epiblast-derived tissues produces severe brain malformations (Satz et al., 2008) that are not characteristic of mice with GFAP-Cre mediated deletion of dystroglycan in the CNS (Moore et al., 2002), although migration defects are present in both mouse strains. Differences in the phenotypes of the two mouse strains might be attributable to later transgene expression driven by GFAP-Cre or residual dystroglycan expression in neurons. The GFAP-Cre transgene is expressed in radial glia and astrocytes, and subsequently affects the expression of targeted genes in a subset of radial glia-derived neurons (Zhuo et al., 2001). In addition, deletion of dystroglycan in tissues outside of the CNS could potentially have contributed to the severity of the brain malformation in the MORE-DG-null mice. To help address these questions, we used Nestin-Cre to delete CNS dystroglycan earlier and more globally than GFAP-Cre.
Nestin-Cre/DG-null mice exhibited earlier and more widespread disruptions of neuronal migration than GFAP-Cre/DG-null mice, and developed hydrocephalus. Hydrocephalus was observed in nearly 40% of the Nestin-Cre/DG-null mice, a prevalence similar to that seen in MORE-DG-null mice (Satz et al., 2008). In contrast, we observed severe hydrocephalus in just under 1% of GFAP-Cre/DG-null mice (data not shown). All three mouse colonies have a C57BL/6J background, in which spontaneous hydrocephalus is reported to occur at a frequency of only 0.03% (Jackson Laboratories), significantly lower than the frequency observed in both the MORE-DG-null and Nestin-Cre/DG-null mice. We previously suggested that glial/neuronal heterotopia filling the subarachnoid space might prevent the resorption of CSF (Satz et al., 2008). Although glial/neuronal heterotopia were present in both GFAP-Cre/DG-null and Nestin-Cre/DG-null mice, hydrocephalus was rarely observed in GFAP-Cre/DG-null mice. Spatial mapping of glial/neuronal heterotopia in Nestin-Cre/DG-null and GFAP-Cre/DG-null mice revealed earlier and much more widespread cerebral cortical dysplasia in the Nestin-Cre/DG-null mice, suggesting that more global obliteration of the subarachnoid space may underlie the development of severe hydrocephalus in the mice. Differences in residual dystroglycan expression between Nestin-Cre/DG-null and GFAP-Cre/DG-null mice also suggests that variations in residual dystroglycan function may contribute to the heterogeneity in disease severity observed in congenital muscular dystrophy patients.
Breaches of the glia limitans are associated with aberrant neuron migration in congenital muscular dystrophies (Muntoni and Voit, 2004) and mouse models (Holzfeind et al., 2002; Michele et al., 2002; Moore et al., 2002; Chiyonobu et al., 2005; Liu et al., 2006; Satz et al., 2008; Ackroyd et al., 2009), indicating that interactions of dystroglycan with the extracellular matrix are important for basement membrane integrity in the CNS. Dystroglycan forms a link between the extracellular matrix and the cytoskeleton (Ervasti and Campbell, 1993) and this structural function is important for muscle cell integrity but the importance in brain is not understood. A large number of studies suggest that the cytoplasmic tail of dystroglycan may also have important signaling functions in addition to a structural role (Langenbach and Rando, 2002; Oak et al., 2003; Zhou et al., 2005; Lai et al., 2009; Xiong et al., 2009; Moore and Winder, 2010). In the current study, deletion of the cytoplasmic domain of β-dystroglycan did not cause disruptions of the glia limitans or of neuron migration in the cerebral cortex, indicating that the feature of dystroglycan that is relevant to cerebral cortical development is primarily its ability to interact with extracellular proteins. Interestingly, small foci of ectopic cerebellar granule neurons were observed in the βcyt/βcyt mice, suggesting that an additional mechanism may play a role in development of the cerebellum. However, the cerebellar cytoarchitecture was well preserved in βcyt/βcyt mice relative to the Largemyd, GFAP-Cre/DG-null or Nestin-Cre/DG-null mice, indicating that dystroglycan's extracellular interactions have a greater significance to cerebellar granule neuron migration.
Previous immunohistochemical and in situ hybridization studies indicate that dystroglycan is expressed in migrating neurons as well as in radially oriented glia during CNS development (Ohtsuka-Tsurumi et al., 2004), and we could not exclude the possibility that neuronal dystroglycan may be necessary for migration. To better understand the cell type underlying the migration defects in dystroglycan-deficient mice, we used NEX-Cre to disrupt dystroglycan expression solely in neurons. In contrast to the cytoarchitecture of Nestin-Cre/DG-null mice, that of the NEX-Cre/DG-null cerebral cortex appeared normal, suggesting that loss of dystroglycan expression in glial cells is the primary mechanism responsible for the abnormal cerebral cortical development observed in Nestin-Cre/DG-null and GFAP-Cre/DG-null mice. Dystroglycan stabilizes the basement membrane at the glia limitans and thus supports the cellular infrastructure necessary for normal laminar development of the cerebral cortex.
Similar to dystroglycan, integrin family members act as ECM receptors and contribute to cell adhesion as well as various cellular processes. In glial cells, integrin β1 expression (Itgb1) is important for assembly of the basal lamina, the proliferation and migration of neurons, and the differentiation and survival of radial glia (Graus-Porta et al., 2001; Blaess et al., 2004; Belvindrah et al., 2006, 2007; Radakovits et al., 2009). In neurons, in contrast, Itgb1 is dispensable for normal cortical organization, yet is required for maintenance of the normal response to high-frequency simulation and for the induction of long-term potentiation within the hippocampus (Huang et al., 2006). Despite the resemblance of the cortical and cerebellar malformations in Itgb1-null and DG-null mice, it appears that the extracellular ligand interactions are most relevant with respect to α-dystroglycan function whereas intracellular signaling is critical with respect to Itgb1 function. In fact, deletion of integrin-linked kinase (Ilk), an intracellular effector of Itgb1, in the CNS results in abnormal cerebellar development associated with defects in the outgrowth of glial processes, assembly of the meningeal basement membrane assembly, and proliferation of granule cells (Belvindrah et al., 2006; Mills et al., 2006).
Analysis of the Nex-Cre/DG-null mice revealed that even though neuronal dystroglycan does not play a major role in forebrain development, dystroglycan expression in neurons is required for normal hippocampal long-term potentiation. Although dystroglycan is present in cobblestone lissencephalies, α-dystroglycan is hypoglycosylated in this context (Michele et al., 2002; Barresi and Campbell, 2006; Satz et al., 2008). Since long-term potentiation had not been examined in mice with defects in α-dystroglycan glycosylation, we studied Largemyd mice and showed that the disruption of α-dystroglycan glycosylation is sufficient to impair long-term potentiation at CA3–CA1 synapses. Dystroglycan is expressed at the postsynaptic apparatus of synapses established by the mossy fibers on cell bodies and dendrites of CA3 and CA1 pyramidal neurons (Zaccaria et al., 2001). In cultures of hippocampal neurons, dystroglycan colocalizes with a subset of inhibitory synapses containing GABAAR subunits a1, a2, and y2, but the clustering of GABAergic synapses was normal in the absence of dystroglycan (Lévi et al., 2002). The precise mechanism mediating the impairment of synaptic plasticity in dystroglycan deficient mice is unknown and a subject for future inquiry.
The binding of extracellular ligands by α-dystroglycan is dependent on posttranslational processing (Michele et al., 2002; Barresi et al., 2004; Satz et al., 2008). Although the primary sequence of α-dystroglycan predicts a molecular weight of 67 kDa, the observed molecular weight is 156 kDa in skeletal muscle due to the posttranslational addition of glycans (Smalheiser and Schwartz, 1987; Ibraghimov-Beskrovnaya et al., 1992). Both N-linked and O-linked sugar moieties are present on α-dystroglycan, but chemical removal of N-linked sugars reduces the molecular weight of the protein by only 4 kDa (Ervasti and Campbell, 1993; Barresi and Campbell, 2006). Thus, the greater-than-predicted molecular weight is largely due to the addition of O-linked sugars. Disruption of N-linked glycosylation has no effect on the ability of dystroglycan to bind laminin, while disruption of O-linked glycosylation reduces its binding to ligand (Ervasti and Campbell, 1993). Antibody IIH6, which recognizes a unique glycoepitope on α-dystroglycan, competes with dystroglycan ligand binding (Brown et al., 1999). Immunoblotting of lysates generated from skeletal muscle, brain, or patient (FCMD, WWS, or MEB)-derived cultured cells with IIH6 failed to detect expression of α-dystroglycan, whereas immunoblotting with antibodies to the core peptide detected α-dystroglycan at a lower molecular weight (Michele et al., 2002; Barresi et al., 2004; Satz et al., 2008). We identified previously unrecognized differences in dystroglycan molecular weight in neurons and glial cells, indicating that dystroglycan is differentially glycosylated in the brain. The ligand binding affinity of dystroglycan is dependent on the posttranslational addition of glycans to α-dystroglycan (Oak et al., 2003), suggesting that the diversity of dystroglycan function may be attributable to cellular differences in glycosylation and the diversity of dystroglycan ligands.
In summary, the findings demonstrate that the extracellular interactions of dystroglycan expressed on glial cells stabilize the glia limitans and the scaffolding for neuron migration (Fig. 11), whereas dystroglycan expressed on neurons facilitates hippocampal long-term potentiation. The former functions involve extracellular dystroglycan interactions, since the cerebral cortex develops normally in mice that lack only the dystroglycan intracellular domain. Also notable is the fact that we found cell type-specific differences in dystroglycan glycosylation, which suggest that the diversity of dystroglycan function in the CNS, and the broad clinical spectrum of type II lissencephalies, may be consequences of cellular differences in glycosylation and ligand binding affinity, as well as of the diversity of dystroglycan ligands.
Figure 11.
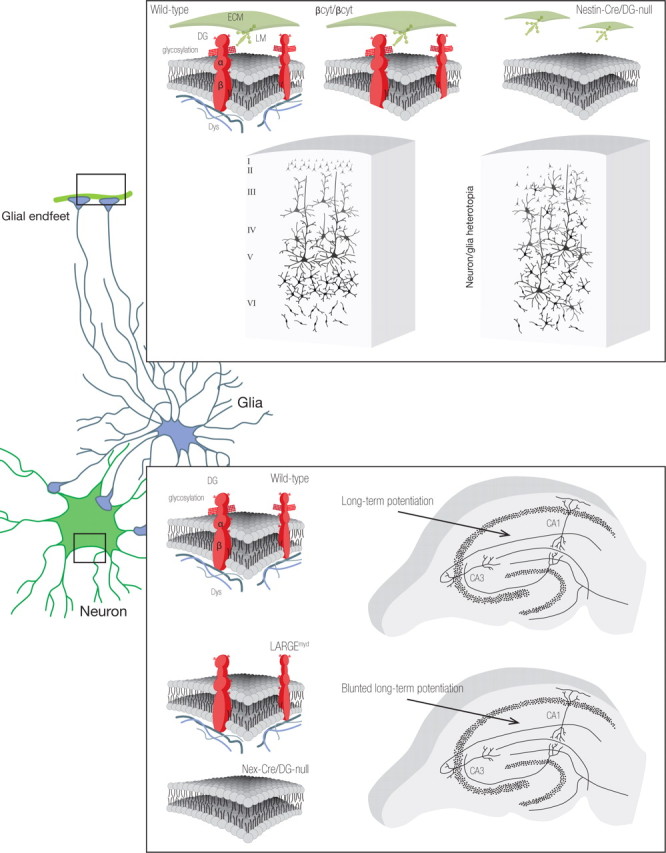
The diversity of dystroglycan function in the CNS.
Footnotes
Grant support was provided by National Institutes of Health Grants R21 NS041407 (S.A.M., K.P.C.) and U54 NS053672 (S.A.M., K.P.C., A.P.O). K.P.C. is an investigator of the Howard Hughes Medical Institute. Joel Carl, Department of Pathology, assisted with the preparation of many of the figures.
References
- Ackroyd MR, Skordis L, Kaluarachchi M, Godwin J, Prior S, Fidanboylu M, Piercy RJ, Muntoni F, Brown SC. Reduced expression of fukutin related protein in mice results in a model for fukutin related protein associated muscular dystrophies. Brain. 2009;132:439–451. doi: 10.1093/brain/awn335. [DOI] [PubMed] [Google Scholar]
- Barresi R, Campbell KP. Dystroglycan: from biosynthesis to pathogenesis of human disease. J Cell Sci. 2006;119:199–207. doi: 10.1242/jcs.02814. [DOI] [PubMed] [Google Scholar]
- Barresi R, Michele DE, Kanagawa M, Harper HA, Dovico SA, Satz JS, Moore SA, Zhang W, Schachter H, Dumanski JP, Cohn RD, Nishino I, Campbell KP. LARGE can functionally bypass alpha-dystroglycan glycosylation defects in distinct congenital muscular dystrophies. Nat Med. 2004;10:696–703. doi: 10.1038/nm1059. [DOI] [PubMed] [Google Scholar]
- Barski JJ, Dethleffsen K, Meyer M. Cre recombinase expression in cerebellar Purkinje cells. Genesis. 2000;28:93–98. [PubMed] [Google Scholar]
- Beltrán-Valero de Bernabé D, Currier S, Steinbrecher A, Celli J, van Beusekom E, van der Zwaag B, Kayserili H, Merlini L, Chitayat D, Dobyns WB, Cormand B, Lehesjoki AE, Cruces J, Voit T, Walsh CA, van Bokhoven H, Brunner HG. Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg syndrome. Am J Hum Genet. 2002;71:1033–1043. doi: 10.1086/342975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrán-Valero de Bernabé D, Voit T, Longman C, Steinbrecher A, Straub V, Yuva Y, Herrmann R, Sperner J, Korenke C, Diesen C, Dobyns WB, Brunner HG, van Bokhoven H, Brockington M, Muntoni F. Mutations in the FKRP gene can cause muscle-eye-brain disease and Walker-Warburg syndrome. J Med Genet. 2004;41:e61. doi: 10.1136/jmg.2003.013870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belvindrah R, Nalbant P, Ding S, Wu C, Bokoch GM, Müller U. Integrin-linked kinase regulates Bergmann glial differentiation during cerebellar development. Mol Cell Neurosci. 2006;33:109–125. doi: 10.1016/j.mcn.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Belvindrah R, Graus-Porta D, Goebbels S, Nave KA, Müller U. Beta1 integrins in radial glia but not in migrating neurons are essential for the formation of cell layers in the cerebral cortex. J Neurosci. 2007;27:13854–13865. doi: 10.1523/JNEUROSCI.4494-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaess S, Graus-Porta D, Belvindrah R, Radakovits R, Pons S, Littlewood-Evans A, Senften M, Guo H, Li Y, Miner JH, Reichardt LF, Müller U. Beta1-integrins are critical for cerebellar granule cell precursor proliferation. J Neurosci. 2004;24:3402–3412. doi: 10.1523/JNEUROSCI.5241-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowe MA, Deyst KA, Leszyk JD, Fallon JR. Identification and purification of an agrin receptor from Torpedo postsynaptic membranes: a heteromeric complex related to the dystroglycans. Neuron. 1994;12:1173–1180. doi: 10.1016/0896-6273(94)90324-7. [DOI] [PubMed] [Google Scholar]
- Brown SC, Fassati A, Popplewell L, Page AM, Henry MD, Campbell KP, Dickson G. Dystrophic phenotype induced in vitro by antibody blockade of muscle alpha-dystroglycan-laminin interaction. J Cell Sci. 1999;112:209–216. doi: 10.1242/jcs.112.2.209. [DOI] [PubMed] [Google Scholar]
- Chen ZL, Haegeli V, Yu H, Strickland S. Cortical deficiency of laminin gamma1 impairs the AKT/GSK-3beta signaling pathway and leads to defects in neurite outgrowth and neuronal migration. Dev Biol. 2009;327:158–168. doi: 10.1016/j.ydbio.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiyonobu T, Sasaki J, Nagai Y, Takeda S, Funakoshi H, Nakamura T, Sugimoto T, Toda T. Effects of fukutin deficiency in the developing mouse brain. Neuromuscul Disord. 2005;15:416–426. doi: 10.1016/j.nmd.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Clement E, Mercuri E, Godfrey C, Smith J, Robb S, Kinali M, Straub V, Bushby K, Manzur A, Talim B, Cowan F, Quinlivan R, Klein A, Longman C, McWilliam R, Topaloglu H, Mein R, Abbs S, North K, Barkovich AJ, et al. Brain involvement in muscular dystrophies with defective dystroglycan glycosylation. Ann Neurol. 2008;64:573–582. doi: 10.1002/ana.21482. [DOI] [PubMed] [Google Scholar]
- Cohn RD, Henry MD, Michele DE, Barresi R, Saito F, Moore SA, Flanagan JD, Skwarchuk MW, Robbins ME, Mendell JR, Williamson RA, Campbell KP. Disruption of DAG1 in differentiated skeletal muscle reveals a role for dystroglycan in muscle regeneration. Cell. 2002;110:639–648. doi: 10.1016/s0092-8674(02)00907-8. [DOI] [PubMed] [Google Scholar]
- Costell M, Gustafsson E, Aszódi A, Mörgelin M, Bloch W, Hunziker E, Addicks K, Timpl R, Fässler R. Perlecan maintains the integrity of cartilage and some basement membranes. J Cell Biol. 1999;147:1109–1122. doi: 10.1083/jcb.147.5.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz AL, Gleeson JG. The molecular and genetic mechanisms of neocortex development. Clin Perinatol. 2009;36:503–512. doi: 10.1016/j.clp.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duclos F, Straub V, Moore SA, Venzke DP, Hrstka RF, Crosbie RH, Durbeej M, Lebakken CS, Ettinger AJ, van der Meulen J, Holt KH, Lim LE, Sanes JR, Davidson BL, Faulkner JA, Williamson R, Campbell KP. Progressive muscular dystrophy in alpha-sarcoglycan-deficient mice. J Cell Biol. 1998;142:1461–1471. doi: 10.1083/jcb.142.6.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ervasti JM, Campbell KP. Membrane organization of the dystrophin-glucoprotein complex. Cell. 1991;66:1121–1131. doi: 10.1016/0092-8674(91)90035-w. [DOI] [PubMed] [Google Scholar]
- Ervasti JM, Campbell KP. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol. 1993;122:809–823. doi: 10.1083/jcb.122.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee SH, Montanaro F, Lindenbaum MH, Carbonetto S. Dystroglycan-alpha, a dystrophin-associated glycoprotein, is a functional agrin receptor. Cell. 1994;77:675–686. doi: 10.1016/0092-8674(94)90052-3. [DOI] [PubMed] [Google Scholar]
- Godfrey C, Clement E, Mein R, Brockington M, Smith J, Talim B, Straub V, Robb S, Quinlivan R, Feng L, Jimenez-Mallebrera C, Mercuri E, Manzur AY, Kinali M, Torelli S, Brown SC, Sewry CA, Bushby K, Topaloglu H, North K, et al. Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain. 2007;130:2725–2735. doi: 10.1093/brain/awm212. [DOI] [PubMed] [Google Scholar]
- Goebbels S, Bormuth I, Bode U, Hermanson O, Schwab MH, Nave KA. Genetic targeting of principal neurons in neocortex and hippocampus of NEX-Cre mice. Genesis. 2006;44:611–621. doi: 10.1002/dvg.20256. [DOI] [PubMed] [Google Scholar]
- Graus-Porta D, Blaess S, Senften M, Littlewood-Evans A, Damsky C, Huang Z, Orban P, Klein R, Schittny JC, Müller U. Beta1-class integrins regulate the development of laminae and folia in the cerebral and cerebellar cortex. Neuron. 2001;31:367–379. doi: 10.1016/s0896-6273(01)00374-9. [DOI] [PubMed] [Google Scholar]
- Grewal PK, Holzfeind PJ, Bittner RE, Hewitt JE. Mutant glycosyltransferase and altered glycosylation of alpha-dystroglycan in the myodystrophy mouse. Nat Genet. 2001;28:151–154. doi: 10.1038/88865. [DOI] [PubMed] [Google Scholar]
- Guerrini R, Carrozzo R. Epilepsy and genetic malformations of the cerebral cortex. Am J Med Genet. 2001;106:160–173. doi: 10.1002/ajmg.1569. [DOI] [PubMed] [Google Scholar]
- Guerrini R, Dobyns WB, Barkovich AJ. Abnormal development of the human cerebral cortex: genetics, functional consequences and treatment options. Trends Neurosci. 2008;31:154–162. doi: 10.1016/j.tins.2007.12.004. [DOI] [PubMed] [Google Scholar]
- Haydon PG. GLIA: listening and talking to the synapse. Nat Rev Neurosci. 2001;2:185–193. doi: 10.1038/35058528. [DOI] [PubMed] [Google Scholar]
- Henion TR, Qu Q, Smith FI. Expression of dystroglycan, fukutin and POMGnT1 during mouse cerebellar development. Brain Res Mol Brain Res. 2003;112:177–181. doi: 10.1016/s0169-328x(03)00055-x. [DOI] [PubMed] [Google Scholar]
- Holzfeind PJ, Grewal PK, Reitsamer HA, Kechvar J, Lassmann H, Hoeger H, Hewitt JE, Bittner RE. Skeletal, cardiac and tongue muscle pathology, defective retinal transmission, and neuronal migration defects in the Large(myd) mouse defines a natural model for glycosylation-deficient muscle-eye-brain disorders. Hum Mol Genet. 2002;11:2673–2687. doi: 10.1093/hmg/11.21.2673. [DOI] [PubMed] [Google Scholar]
- Huang Z, Shimazu K, Woo NH, Zang K, Müller U, Lu B, Reichardt LF. Distinct roles of the beta 1-class integrins at the developing and the mature hippocampal excitatory synapse. J Neurosci. 2006;26:11208–11219. doi: 10.1523/JNEUROSCI.3526-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibraghimov-Beskrovnaya O, Ervasti JM, Leveille CJ, Slaughter CA, Sernett SW, Campbell KP. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature. 1992;355:696–702. doi: 10.1038/355696a0. [DOI] [PubMed] [Google Scholar]
- Ibraghimov-Beskrovnaya O, Milatovich A, Ozcelik T, Yang B, Koepnick K, Francke U, Campbell KP. Human dystroglycan: skeletal muscle cDNA, genomic structure, origin of tissue specific isoforms and chromosomal localization. Hum Mol Genet. 1993;2:1651–1657. doi: 10.1093/hmg/2.10.1651. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Nakahori Y, Miyake M, Matsumura K, Kondo-Iida E, Nomura Y, Segawa M, Yoshioka M, Saito K, Osawa M, Hamano K, Sakakihara Y, Nonaka I, Nakagome Y, Kanazawa I, Nakamura Y, Tokunaga K, Toda T. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature. 1998;394:388–392. doi: 10.1038/28653. [DOI] [PubMed] [Google Scholar]
- Lai Y, Thomas GD, Yue Y, Yang HT, Li D, Long C, Judge L, Bostick B, Chamberlain JS, Terjung RL, Duan D. Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J Clin Invest. 2009;119:624–635. doi: 10.1172/JCI36612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langenbach KJ, Rando TA. Inhibition of dystroglycan binding to laminin disrupts the PI3K/AKT pathway and survival signaling in muscle cells. Muscle Nerve. 2002;26:644–653. doi: 10.1002/mus.10258. [DOI] [PubMed] [Google Scholar]
- Lévi S, Grady RM, Henry MD, Campbell KP, Sanes JR, Craig AM. Dystroglycan is selectively associated with inhibitory GABAergic synapses but is dispensable for their differentiation. J Neurosci. 2002;22:4274–4285. doi: 10.1523/JNEUROSCI.22-11-04274.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Ball SL, Yang Y, Mei P, Zhang L, Shi H, Kaminski HJ, Lemmon VP, Hu H. A genetic model for muscle-eye-brain disease in mice lacking protein O-mannose 1,2-N-acetylglucosaminyltransferase (POMGnT1) Mech Dev. 2006;123:228–240. doi: 10.1016/j.mod.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Michele DE, Barresi R, Kanagawa M, Saito F, Cohn RD, Satz JS, Dollar J, Nishino I, Kelley RI, Somer H, Straub V, Mathews KD, Moore SA, Campbell KP. Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature. 2002;418:417–422. doi: 10.1038/nature00837. [DOI] [PubMed] [Google Scholar]
- Mills J, Niewmierzycka A, Oloumi A, Rico B, St-Arnaud R, Mackenzie IR, Mawji NM, Wilson J, Reichardt LF, Dedhar S. Critical role of integrin-linked kinase in granule cell precursor proliferation and cerebellar development. J Neurosci. 2006;26:830–840. doi: 10.1523/JNEUROSCI.1852-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miner JH, Cunningham J, Sanes JR. Roles for laminin in embryogenesis: exencephaly, syndactyly, and placentopathy in mice lacking the laminin alpha5 chain. J Cell Biol. 1998;143:1713–1723. doi: 10.1083/jcb.143.6.1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochida GH, Walsh CA. Genetic basis of developmental malformations of the cerebral cortex. Arch Neurol. 2004;61:637–640. doi: 10.1001/archneur.61.5.637. [DOI] [PubMed] [Google Scholar]
- Moore CJ, Winder SJ. Dystroglycan versatility in cell adhesion: a tale of multiple motifs. Cell Commun Signal. 2010;8:3. doi: 10.1186/1478-811X-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore SA, Saito F, Chen J, Michele DE, Henry MD, Messing A, Cohn RD, Ross-Barta SE, Westra S, Williamson RA, Hoshi T, Campbell KP. Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature. 2002;418:422–425. doi: 10.1038/nature00838. [DOI] [PubMed] [Google Scholar]
- Moukhles H, Roque R, Carbonetto S. alpha-dystroglycan isoforms are differentially distributed in adult rat retina. J Comp Neurol. 2000;420:182–194. doi: 10.1002/(sici)1096-9861(20000501)420:2<182::aid-cne3>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Muntoni F, Voit T. The congenital muscular dystrophies in 2004: a century of exciting progress. Neuromuscul Disord. 2004;14:635–649. doi: 10.1016/j.nmd.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Flint AC, Weissman TA, Dammerman RS, Kriegstein AR. Neurons derived from radial glial cells establish radial units in neocortex. Nature. 2001;409:714–720. doi: 10.1038/35055553. [DOI] [PubMed] [Google Scholar]
- Oak SA, Zhou YW, Jarrett HW. Skeletal muscle signaling pathway through the dystrophin glycoprotein complex and Rac1. J Biol Chem. 2003;278:39287–39295. doi: 10.1074/jbc.M305551200. [DOI] [PubMed] [Google Scholar]
- Occhi S, Zambroni D, Del Carro U, Amadio S, Sirkowski EE, Scherer SS, Campbell KP, Moore SA, Chen ZL, Strickland S, Di Muzio A, Uncini A, Wrabetz L, Feltri ML. Both laminin and Schwann cell dystroglycan are necessary for proper clustering of sodium channels at nodes of Ranvier. J Neurosci. 2005;25:9418–9427. doi: 10.1523/JNEUROSCI.2068-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsuka-Tsurumi E, Saito Y, Yamamoto T, Voit T, Kobayashi M, Osawa M. Co-localization of fukutin and alpha-dystroglycan in the mouse central nervous system. Brain Res Dev Brain Res. 2004;152:121–127. doi: 10.1016/j.devbrainres.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Olson EC, Walsh CA. Smooth, rough and upside-down neocortical development. Curr Opin Genet Dev. 2002;12:320–327. doi: 10.1016/s0959-437x(02)00305-2. [DOI] [PubMed] [Google Scholar]
- Radakovits R, Barros CS, Belvindrah R, Patton B, Müller U. Regulation of radial glial survival by signals from the meninges. J Neurosci. 2009;29:7694–7705. doi: 10.1523/JNEUROSCI.5537-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross ME, Walsh CA. Human brain malformations and their lessons for neuronal migration. Annu Rev Neurosci. 2001;24:1041–1070. doi: 10.1146/annurev.neuro.24.1.1041. [DOI] [PubMed] [Google Scholar]
- Ruegg MA, Bixby JL. Agrin orchestrates synaptic differentiation at the vertebrate neuromuscular junction. Trends Neurosci. 1998;21:22–27. doi: 10.1016/s0166-2236(97)01154-5. [DOI] [PubMed] [Google Scholar]
- Sato S, Omori Y, Katoh K, Kondo M, Kanagawa M, Miyata K, Funabiki K, Koyasu T, Kajimura N, Miyoshi T, Sawai H, Kobayashi K, Tani A, Toda T, Usukura J, Tano Y, Fujikado T, Furukawa T. Pikachurin, a dystroglycan ligand, is essential for photoreceptor ribbon synapse formation. Nat Neurosci. 2008;11:923–931. doi: 10.1038/nn.2160. [DOI] [PubMed] [Google Scholar]
- Satz JS, Barresi R, Durbeej M, Willer T, Turner A, Moore SA, Campbell KP. Brain and eye malformations resembling Walker-Warburg syndrome are recapitulated in mice by dystroglycan deletion in the epiblast. J Neurosci. 2008;28:10567–10575. doi: 10.1523/JNEUROSCI.2457-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satz JS, Philp AR, Nguyen H, Kusano H, Lee J, Turk R, Riker MJ, Hernández J, Weiss RM, Anderson MG, Mullins RF, Moore SA, Stone EM, Campbell KP. Visual impairment in the absence of dystroglycan. J Neurosci. 2009;29:13136–13146. doi: 10.1523/JNEUROSCI.0474-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smalheiser NR, Schwartz NB. Cranin: a laminin-binding protein of cell membranes. Proc Natl Acad Sci U S A. 1987;84:6457–6461. doi: 10.1073/pnas.84.18.6457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugita S, Saito F, Tang J, Satz J, Campbell K, Südhof TC. A stoichiometric complex of neurexins and dystroglycan in brain. J Cell Biol. 2001;154:435–445. doi: 10.1083/jcb.200105003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talts JF, Andac Z, Göhring W, Brancaccio A, Timpl R. Binding of the G domains of laminin alpha1 and alpha2 chains and perlecan to heparin, sulfatides, alpha-dystroglycan and several extracellular matrix proteins. EMBO J. 1999;18:863–870. doi: 10.1093/emboj/18.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schütz G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- van Reeuwijk J, Brunner HG, van Bokhoven H. Glyc-O-genetics of Walker-Warburg syndrome. Clin Genet. 2005;67:281–289. doi: 10.1111/j.1399-0004.2004.00368.x. [DOI] [PubMed] [Google Scholar]
- van Reeuwijk J, Grewal PK, Salih MA, Beltrán-Valero de Bernabé D, McLaughlan JM, Michielse CB, Herrmann R, Hewitt JE, Steinbrecher A, Seidahmed MZ, Shaheed MM, Abomelha A, Brunner HG, van Bokhoven H, Voit T. Intragenic deletion in the LARGE gene causes Walker-Warburg syndrome. Hum Genet. 2007;121:685–690. doi: 10.1007/s00439-007-0362-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson RA, Henry MD, Daniels KJ, Hrstka RF, Lee JC, Sunada Y, Ibraghimov-Beskrovnaya O, Campbell KP. Dystroglycan is essential for early embryonic development: disruption of Reichert's membrane in Dag1-null mice. Hum Mol Genet. 1997;6:831–841. doi: 10.1093/hmg/6.6.831. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Zhou Y, Jarrett HW. Dystrophin glycoprotein complex-associated Gbetagamma subunits activate phosphatidylinositol-3-kinase/Akt signaling in skeletal muscle in a laminin-dependent manner. J Cell Physiol. 2009;219:402–414. doi: 10.1002/jcp.21684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Jung D, Motto D, Meyer J, Koretzky G, Campbell KP. SH3 domain-mediated interaction of dystroglycan and Grb2. J Biol Chem. 1995;270:11711–11714. doi: 10.1074/jbc.270.20.11711. [DOI] [PubMed] [Google Scholar]
- Yoshida A, Kobayashi K, Manya H, Taniguchi K, Kano H, Mizuno M, Inazu T, Mitsuhashi H, Takahashi S, Takeuchi M, Herrmann R, Straub V, Talim B, Voit T, Topaloglu H, Toda T, Endo T. Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev Cell. 2001;1:717–724. doi: 10.1016/s1534-5807(01)00070-3. [DOI] [PubMed] [Google Scholar]
- Zaccaria ML, Di Tommaso F, Brancaccio A, Paggi P, Petrucci TC. Dystroglycan distribution in adult mouse brain: a light and electron microscopy study. Neuroscience. 2001;104:311–324. doi: 10.1016/s0306-4522(01)00092-6. [DOI] [PubMed] [Google Scholar]
- Zhou YW, Oak SA, Senogles SE, Jarrett HW. Laminin-alpha1 globular domains 3 and 4 induce heterotrimeric G protein binding to alpha-syntrophin's PDZ domain and alter intracellular Ca2+ in muscle. Am J Physiol Cell Physiol. 2005;288:C377–388. doi: 10.1152/ajpcell.00279.2004. [DOI] [PubMed] [Google Scholar]
- Zhuo L, Theis M, Alvarez-Maya I, Brenner M, Willecke K, Messing A. hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis. 2001;31:85–94. doi: 10.1002/gene.10008. [DOI] [PubMed] [Google Scholar]