

Abstract
In the title N-substituted benzimidazol-2-one, C10H10N2O, the fused ring system is almost planar (r.m.s. deviation = 0.01 Å) and aligned at 57.9 (1)° with respect to the propenyl fragment. In the crystal, adjacent molecules are linked by pairs of N—H⋯O hydrogen bonds into inversion dimers.
Related literature
For the transformation of 1-isopropenyl-1,3-benzimidazol-2-one to other biologically-active compounds, see: Lakhrissi et al. (2010 ▶); Li et al. (2010 ▶). A shorter heating time in the synthesis leads to the formation of 4-methyl-2,3-dihydro-1H-1,5-benzodiazepin-2-one; see: Saber et al. (2010 ▶).
Experimental
Crystal data
C10H10N2O
M r = 174.20
Monoclinic,
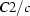
a = 15.8724 (2) Å
b = 6.0971 (1) Å
c = 17.9313 (3) Å
β = 90.961 (2)°
V = 1735.07 (5) Å3
Z = 8
Mo Kα radiation
μ = 0.09 mm−1
T = 100 K
0.35 × 0.30 × 0.18 mm
Data collection
Bruker X8 APEXII diffractometer
13930 measured reflections
2506 independent reflections
2231 reflections with I > 2σ(I)
R int = 0.023
Refinement
R[F 2 > 2σ(F 2)] = 0.039
wR(F 2) = 0.114
S = 0.98
2506 reflections
123 parameters
1 restraint
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.39 e Å−3
Δρmin = −0.21 e Å−3
Data collection: APEX2 (Bruker, 2008 ▶); cell refinement: SAINT (Bruker, 2008 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: X-SEED (Barbour, 2001 ▶); software used to prepare material for publication: publCIF (Westrip, 2010 ▶).
Supplementary Material
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536810017897/nc2184sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810017897/nc2184Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1⋯O1i | 0.87 (1) | 1.95 (1) | 2.811 (1) | 172 (2) |
Symmetry code: (i) 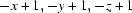 .
.
Acknowledgments
We thank Université Mohammed V-Agdal and the University of Malaya for supporting this study.
supplementary crystallographic information
Comment
Benzimidazol-2-one derivatives possess a range of biological and pharmacological activities. Among the many compounds is N-isopropenyl benzimidazol-2-one, which can be further converted to 1-acyl-3-isopropenyl benzimidazol-2-ones that are active against Botrytis cinerea fungi that affect vegetables and fruits (Li et al. 2010). The reagent is also commercially available. We have recently reported the use of this reagent in the synthesis of some glucose-substituted benzimidazol-2-ones (Lakhrissi et al., 2010). For the purpose of understanding the chemistry of these compounds, the crystal structure of the reagent is determined in the present study.
In the molecule of C10H10N2O (Scheme I, Fig. 1), the fused-ring is planar (r.m.s. deviation 0.01 Å); the propenyl fragment is aligned at 57.9 (1) ° with respect to the fused-ring. Adjacent molecules are linked about a center-of-inversion by an N–H···O hydrogen bond.
Experimental
o-Phenylenediamine (1.0 g, 9 mmol) and ethyl acetoacetate (1.2 ml, 9 mmol) were heated in xylene (10 ml) for 6 hours. The mixture was set aside for the growth of colorless crystals of N-isopropenyl benzimidazol-2-one; yield 90%. When the heating time is shortened to 1 hour, the product is 4-methyl-2,3-dihydro-1H-1,5-benzodiazepin-2-one; details are given in another report (Saber et al., 2010).
Refinement
Carbon-bound H-atoms were placed in calculated positions (C—H 0.95–0.98 Å) and were included in the refinement in the riding model approximation, with U(H) set to 1.2–1.5Ueq(C).
The amino H-atom was located in a difference Fourier map; the N–H distance was restrained to 0.86±0.01 Å. T; the temperature factor of the amino hydrogen atom was freely refined.
Figures
Fig. 1.

Thermal ellipsoid plot (Barbour, 2001) of the molecule of C10H10N2O at the 70% probability level shown as a hydrogen-bonded dimer; hydrogen atoms are drawn as spheres of arbitrary radius. Symmetry code: i = 1 - x, 1 - y, 1 - z.
Crystal data
| C10H10N2O | F(000) = 736 |
| Mr = 174.20 | Dx = 1.334 Mg m−3 |
| Monoclinic, C2/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -C 2yc | Cell parameters from 8296 reflections |
| a = 15.8724 (2) Å | θ = 2.3–35.0° |
| b = 6.0971 (1) Å | µ = 0.09 mm−1 |
| c = 17.9313 (3) Å | T = 100 K |
| β = 90.961 (2)° | Block, colorless |
| V = 1735.07 (5) Å3 | 0.35 × 0.30 × 0.18 mm |
| Z = 8 |
Data collection
| Bruker X8 APEXII diffractometer | 2231 reflections with I > 2σ(I) |
| Radiation source: fine-focus sealed tube | Rint = 0.023 |
| graphite | θmax = 30.0°, θmin = 3.4° |
| φ and ω scans | h = −21→21 |
| 13930 measured reflections | k = −8→8 |
| 2506 independent reflections | l = −25→25 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.039 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.114 | H atoms treated by a mixture of independent and constrained refinement |
| S = 0.98 | w = 1/[σ2(Fo2) + (0.0716P)2 + 0.9009P] where P = (Fo2 + 2Fc2)/3 |
| 2506 reflections | (Δ/σ)max = 0.001 |
| 123 parameters | Δρmax = 0.39 e Å−3 |
| 1 restraint | Δρmin = −0.21 e Å−3 |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| O1 | 0.58630 (4) | 0.41270 (11) | 0.43646 (4) | 0.02815 (17) | |
| N1 | 0.45280 (5) | 0.26700 (13) | 0.45678 (4) | 0.02451 (18) | |
| H1 | 0.4355 (10) | 0.366 (2) | 0.4880 (7) | 0.048 (4)* | |
| N2 | 0.53743 (5) | 0.08911 (12) | 0.38109 (4) | 0.02211 (17) | |
| C1 | 0.40921 (5) | 0.08279 (14) | 0.43226 (5) | 0.02174 (18) | |
| C2 | 0.32851 (6) | 0.00950 (17) | 0.44634 (5) | 0.0275 (2) | |
| H2 | 0.2922 | 0.0883 | 0.4783 | 0.033* | |
| C3 | 0.30248 (6) | −0.18432 (17) | 0.41176 (6) | 0.0296 (2) | |
| H3 | 0.2473 | −0.2386 | 0.4201 | 0.036* | |
| C4 | 0.35602 (6) | −0.29936 (17) | 0.36524 (6) | 0.0296 (2) | |
| H4 | 0.3367 | −0.4314 | 0.3426 | 0.035* | |
| C5 | 0.43747 (6) | −0.22577 (15) | 0.35092 (5) | 0.0259 (2) | |
| H5 | 0.4740 | −0.3051 | 0.3193 | 0.031* | |
| C6 | 0.46266 (5) | −0.03240 (14) | 0.38485 (4) | 0.02048 (18) | |
| C7 | 0.53105 (6) | 0.27304 (14) | 0.42624 (5) | 0.02226 (18) | |
| C8 | 0.60775 (5) | 0.04385 (15) | 0.33453 (5) | 0.02371 (19) | |
| C9 | 0.64627 (6) | −0.14799 (17) | 0.33983 (6) | 0.0338 (2) | |
| H9A | 0.6272 | −0.2545 | 0.3743 | 0.041* | |
| H9B | 0.6930 | −0.1793 | 0.3091 | 0.041* | |
| C10 | 0.62872 (7) | 0.21916 (17) | 0.27950 (6) | 0.0327 (2) | |
| H10A | 0.6743 | 0.1682 | 0.2476 | 0.049* | |
| H10B | 0.6469 | 0.3517 | 0.3062 | 0.049* | |
| H10C | 0.5788 | 0.2524 | 0.2487 | 0.049* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O1 | 0.0268 (3) | 0.0240 (3) | 0.0338 (4) | −0.0062 (2) | 0.0053 (3) | −0.0065 (3) |
| N1 | 0.0231 (4) | 0.0223 (4) | 0.0284 (4) | −0.0013 (3) | 0.0057 (3) | −0.0049 (3) |
| N2 | 0.0214 (4) | 0.0209 (3) | 0.0242 (3) | −0.0038 (3) | 0.0054 (3) | −0.0036 (3) |
| C1 | 0.0218 (4) | 0.0219 (4) | 0.0216 (4) | −0.0003 (3) | 0.0012 (3) | 0.0005 (3) |
| C2 | 0.0215 (4) | 0.0315 (5) | 0.0298 (4) | −0.0009 (3) | 0.0044 (3) | −0.0010 (3) |
| C3 | 0.0222 (4) | 0.0350 (5) | 0.0316 (5) | −0.0075 (3) | 0.0017 (3) | 0.0001 (4) |
| C4 | 0.0282 (5) | 0.0304 (5) | 0.0302 (4) | −0.0090 (4) | 0.0004 (3) | −0.0036 (4) |
| C5 | 0.0265 (4) | 0.0258 (4) | 0.0255 (4) | −0.0044 (3) | 0.0034 (3) | −0.0044 (3) |
| C6 | 0.0199 (4) | 0.0216 (4) | 0.0200 (4) | −0.0022 (3) | 0.0016 (3) | 0.0008 (3) |
| C7 | 0.0234 (4) | 0.0204 (4) | 0.0230 (4) | −0.0008 (3) | 0.0027 (3) | −0.0013 (3) |
| C8 | 0.0215 (4) | 0.0263 (4) | 0.0235 (4) | −0.0056 (3) | 0.0057 (3) | −0.0045 (3) |
| C9 | 0.0292 (5) | 0.0292 (5) | 0.0433 (6) | −0.0002 (4) | 0.0114 (4) | −0.0055 (4) |
| C10 | 0.0352 (5) | 0.0346 (5) | 0.0287 (5) | −0.0088 (4) | 0.0090 (4) | 0.0014 (4) |
Geometric parameters (Å, °)
| O1—C7 | 1.2338 (11) | C3—H3 | 0.9500 |
| N1—C7 | 1.3663 (11) | C4—C5 | 1.3963 (13) |
| N1—C1 | 1.3868 (11) | C4—H4 | 0.9500 |
| N1—H1 | 0.871 (9) | C5—C6 | 1.3827 (12) |
| N2—C7 | 1.3878 (11) | C5—H5 | 0.9500 |
| N2—C6 | 1.4016 (10) | C8—C9 | 1.3225 (14) |
| N2—C8 | 1.4319 (11) | C8—C10 | 1.4960 (13) |
| C1—C2 | 1.3839 (12) | C9—H9A | 0.9500 |
| C1—C6 | 1.3999 (11) | C9—H9B | 0.9500 |
| C2—C3 | 1.3939 (14) | C10—H10A | 0.9800 |
| C2—H2 | 0.9500 | C10—H10B | 0.9800 |
| C3—C4 | 1.3907 (14) | C10—H10C | 0.9800 |
| C7—N1—C1 | 110.29 (7) | C4—C5—H5 | 121.5 |
| C7—N1—H1 | 122.5 (11) | C5—C6—C1 | 121.42 (8) |
| C1—N1—H1 | 127.2 (11) | C5—C6—N2 | 131.94 (8) |
| C7—N2—C6 | 109.21 (7) | C1—C6—N2 | 106.63 (7) |
| C7—N2—C8 | 124.13 (7) | O1—C7—N1 | 127.42 (8) |
| C6—N2—C8 | 126.52 (7) | O1—C7—N2 | 125.86 (8) |
| C2—C1—N1 | 131.46 (8) | N1—C7—N2 | 106.72 (7) |
| C2—C1—C6 | 121.39 (8) | C9—C8—N2 | 119.50 (8) |
| N1—C1—C6 | 107.14 (7) | C9—C8—C10 | 124.87 (9) |
| C1—C2—C3 | 117.45 (9) | N2—C8—C10 | 115.54 (8) |
| C1—C2—H2 | 121.3 | C8—C9—H9A | 120.0 |
| C3—C2—H2 | 121.3 | C8—C9—H9B | 120.0 |
| C4—C3—C2 | 120.99 (9) | H9A—C9—H9B | 120.0 |
| C4—C3—H3 | 119.5 | C8—C10—H10A | 109.5 |
| C2—C3—H3 | 119.5 | C8—C10—H10B | 109.5 |
| C3—C4—C5 | 121.69 (9) | H10A—C10—H10B | 109.5 |
| C3—C4—H4 | 119.2 | C8—C10—H10C | 109.5 |
| C5—C4—H4 | 119.2 | H10A—C10—H10C | 109.5 |
| C6—C5—C4 | 117.05 (9) | H10B—C10—H10C | 109.5 |
| C6—C5—H5 | 121.5 | ||
| C7—N1—C1—C2 | −178.46 (10) | C8—N2—C6—C5 | 4.24 (15) |
| C7—N1—C1—C6 | 0.49 (10) | C7—N2—C6—C1 | 1.11 (10) |
| N1—C1—C2—C3 | 179.14 (9) | C8—N2—C6—C1 | −174.64 (8) |
| C6—C1—C2—C3 | 0.31 (14) | C1—N1—C7—O1 | −179.45 (9) |
| C1—C2—C3—C4 | 0.29 (15) | C1—N1—C7—N2 | 0.19 (10) |
| C2—C3—C4—C5 | −0.32 (16) | C6—N2—C7—O1 | 178.83 (9) |
| C3—C4—C5—C6 | −0.25 (15) | C8—N2—C7—O1 | −5.29 (15) |
| C4—C5—C6—C1 | 0.85 (13) | C6—N2—C7—N1 | −0.81 (10) |
| C4—C5—C6—N2 | −177.89 (9) | C8—N2—C7—N1 | 175.06 (8) |
| C2—C1—C6—C5 | −0.91 (13) | C7—N2—C8—C9 | 127.09 (10) |
| N1—C1—C6—C5 | −179.99 (8) | C6—N2—C8—C9 | −57.76 (13) |
| C2—C1—C6—N2 | 178.11 (8) | C7—N2—C8—C10 | −56.22 (12) |
| N1—C1—C6—N2 | −0.97 (9) | C6—N2—C8—C10 | 118.93 (9) |
| C7—N2—C6—C5 | 179.99 (9) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1···O1i | 0.87 (1) | 1.95 (1) | 2.811 (1) | 172 (2) |
Symmetry codes: (i) −x+1, −y+1, −z+1.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: NC2184).
References
- Barbour, L. J. (2001). J. Supramol. Chem.1, 189–191.
- Bruker (2008). APEX2 and SAINT Bruker AXS Inc., Madison, Wisconsin, USA.
- Lakhrissi, B., Benksim, A., Massoui, M., Essassi, E. M., Lequart, V., Joly, N., Beaupeŕe, D., Wadouachi, A. & Martin, P. (2010). Carbohydr. Res.343, 421–433. [DOI] [PubMed]
- Li, S.-K., Ji, Z.-Q., Zhang, J.-W., Guo, Z.-Y. & Wu, W.-J. (2010). J. Agric. Food Chem.58, 2668–2672. [DOI] [PubMed]
- Saber, A., Zouihri, H., Essassi, E. M. & Ng, S. W. (2010). Acta Cryst. E66, o1408. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Westrip, S. P. (2010). J. Appl. Cryst.43 Submitted.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536810017897/nc2184sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810017897/nc2184Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report