

Abstract
The title crystal structure, C11H18N4O3, is the first diazoacetamide in which the diazoacetyl group is attached to an N atom. The piperazine ring is in a chair form and hence the molecule has an extended conformation. Both ring N atoms are bonded in an essentially planar configuration with the sum of the C—N—C angles being 359.8 (2) and 357.7 (2)°. In the crystal structure, the O atom of the diazoacetyl group accepts two H atoms from C—H donors, thus generating chains of weak hydrogen-bonded R 2 1(7) rings.
Related literature
For the only other reported synthesis of a diazoacetamide in the Chemical Abstracts Service (CAS, American Chemical Society, 2008 ▶) with a 1,4-diaza six-membered ring, see: Mickelson et al. (1996 ▶). For other diazoacetamides, see: Ouihia et al. (1993 ▶). For related structures, see: Fenlon et al. (2007 ▶); Wang et al. (2006 ▶); Miller et al. (1991 ▶). For synthetic details, see: Kaupang (2010 ▶); Toma et al. (2007 ▶). For hydrogen-bond graph-set notation, see: Bernstein et al. (1995 ▶). For a description of the Cambridge Structural Database, see: Allen (2002 ▶).
Experimental
Crystal data
C11H18N4O3
M r = 254.29
Monoclinic,
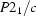
a = 14.654 (10) Å
b = 10.548 (7) Å
c = 8.553 (6) Å
β = 91.122 (6)°
V = 1321.8 (15) Å3
Z = 4
Mo Kα radiation
μ = 0.10 mm−1
T = 105 K
0.55 × 0.42 × 0.08 mm
Data collection
Bruker APEXII CCD diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2007 ▶) T min = 0.870, T max = 0.992
7308 measured reflections
2692 independent reflections
2111 reflections with I > 2σ(I)
R int = 0.041
Refinement
R[F 2 > 2σ(F 2)] = 0.038
wR(F 2) = 0.103
S = 1.03
2692 reflections
169 parameters
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.17 e Å−3
Δρmin = −0.23 e Å−3
Data collection: APEX2 (Bruker, 2007 ▶); cell refinement: SAINT-Plus (Bruker, 2007 ▶); data reduction: SAINT-Plus; program(s) used to solve structure: SHELXTL (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXTL; molecular graphics: SHELXTL; software used to prepare material for publication: SHELXTL.
Supplementary Material
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536810016211/lh5033sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810016211/lh5033Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C1—H1⋯O1i | 0.946 (17) | 2.313 (18) | 3.250 (3) | 170.6 (14) |
| C3—H32⋯O1i | 0.99 | 2.36 | 3.327 (3) | 164 |
Symmetry code: (i) 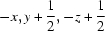 .
.
supplementary crystallographic information
Comment
The tert-butyl 4-(2-diazoacetyl)piperazine-1-carboxylate (I) was prepared as part of a series of diazoacetamides, to be used in intramolecular C—H insertion reactions (Kaupang, 2010). It was synthesized from tert-butyl 4-(2-bromoacetyl)piperazine-1-carboxylate by a slight modification of the procedure reported by Toma et al. (2007), employing 1,1,3,3-tetramethylguanidine as the base instead of 1,8-diazabicyclo[5.4.0]undec-7-ene.
Diffraction data were first collected at ambient temperature, yielding a structure with a massively disordered six-membered ring. At 105 K the ring is completely ordered in a well defined chair conformation, Fig. 1.
The diazoacetyl moiety is not an uncommon functional group in organic molecules, however only 15 occurrences were found in the Cambridge Structural Database (Version 5.31 of November 2009; Allen, 2002), and none where, as here, the group is attached to a N atom. In only two structures the group sits on a non-aromatic ring (Miller et al., 1991; Fenlon et al., 2007).
In a model molecule like trimethylamine the N atom is located about 0.45 Å above the plane defined by the three C atoms. In the structure of (I) N3 is 0.125 (2) Å above the plane defined by C2, C3 and C4, while N4 is only 0.039 (2) Å above the plane defined by C5, C6 and C7, which essentially shows a planar configuration (sum of C—N—C angles 359.8 (2) °). This is due to the double bond character of the amidic N3—C2 and N4—C7 bonds measuring 1.3515 (18) and 1.3483 (18) Å, respectively. An example of a related structure is 1,4-di(chloroacetyl)piperazine (Wang et al., 2006).
In the crystal structure, the O atom of the diazoacetyl group accepts two H atoms from C—H donors, thus generating chains of hydrogen-bonded R12(7) rings (Bernstein et al., 1995).
Experimental
A 2.5 ml vial containing tert-butyl 4-(2-diazoacetyl)piperazine-1-carboxylate (10.8 mg) and dichloromethane (1000 ml) was capped and a pinhole (0.5 mm) was made in the cap to allow for vapour diffusion of solvents. This vial was placed inside a 25 ml vial containing n-pentane (8 ml) that was subsequently capped and stored in the dark at ambient temperature for approximately 48 hours, affording bright yellow plate-shaped crystals.
Refinement
Coordinates were refined for H1 (bonded to C1), which is involved in the shortest intermolcular interaction. Other H atoms were positioned with idealized geometry and fixed C–H distances set to 0.98 Å (methyl) or 0.99 Å (methylene). Free rotation was permitted for the methyl groups. Uiso values were 1.2Ueq of the carrier atom or 1.5Ueq for methyl groups.
Figures
Fig. 1.

The asymmetric unit of (I), with atomic numbering indicated, together with selected atoms of neighbouring molecules connected by weak hydrogen bonds shown as dotted lines (see Table 1, symmetry operations are -x,1/2+y,1/2-z for O', -x,-1/2+y,1/2-z for H1" and H32"). Displacement ellipsoids are shown at the 50% probability level with H atoms as spheres of arbitrary size.
Crystal data
| C11H18N4O3 | F(000) = 544 |
| Mr = 254.29 | Dx = 1.278 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| a = 14.654 (10) Å | Cell parameters from 2404 reflections |
| b = 10.548 (7) Å | θ = 2.4–26.4° |
| c = 8.553 (6) Å | µ = 0.10 mm−1 |
| β = 91.122 (6)° | T = 105 K |
| V = 1321.8 (15) Å3 | Plate, yellow |
| Z = 4 | 0.55 × 0.42 × 0.08 mm |
Data collection
| Bruker APEXII CCD diffractometer | 2692 independent reflections |
| Radiation source: fine-focus sealed tube | 2111 reflections with I > 2σ(I) |
| graphite | Rint = 0.041 |
| Detector resolution: 8.3 pixels mm-1 | θmax = 26.4°, θmin = 2.4° |
| Three sets of frames each taken over 0.3° ω rotation with 20 s exposure time. Detector set at 2θ = 26°, crystal–to–detector distance 6.00 cm. scans | h = −17→18 |
| Absorption correction: multi-scan (SADABS; Bruker, 2007) | k = −10→13 |
| Tmin = 0.870, Tmax = 0.992 | l = −10→10 |
| 7308 measured reflections |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.038 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.103 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.03 | w = 1/[σ2(Fo2) + (0.0463P)2 + 0.1671P] where P = (Fo2 + 2Fc2)/3 |
| 2692 reflections | (Δ/σ)max = 0.001 |
| 169 parameters | Δρmax = 0.17 e Å−3 |
| 0 restraints | Δρmin = −0.22 e Å−3 |
Special details
| Experimental. Crystallized from dichloromethane and n-pentane. |
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| O1 | 0.02567 (7) | 0.20344 (9) | 0.19361 (12) | 0.0353 (3) | |
| O2 | 0.38950 (6) | 0.35633 (9) | 0.62995 (11) | 0.0296 (2) | |
| O3 | 0.33388 (6) | 0.55670 (8) | 0.64091 (11) | 0.0294 (2) | |
| N1 | −0.14125 (10) | 0.34028 (15) | −0.00383 (19) | 0.0562 (4) | |
| N2 | −0.08817 (8) | 0.37402 (12) | 0.08229 (16) | 0.0377 (3) | |
| N3 | 0.10933 (7) | 0.34931 (10) | 0.32642 (14) | 0.0293 (3) | |
| N4 | 0.27460 (8) | 0.42179 (11) | 0.46913 (15) | 0.0350 (3) | |
| C1 | −0.02502 (10) | 0.41229 (14) | 0.18201 (17) | 0.0324 (3) | |
| H1 | −0.0254 (10) | 0.4999 (17) | 0.2058 (19) | 0.039* | |
| C2 | 0.03721 (9) | 0.31392 (13) | 0.23569 (16) | 0.0276 (3) | |
| C3 | 0.11676 (9) | 0.47080 (12) | 0.40835 (18) | 0.0301 (3) | |
| H31 | 0.0971 | 0.4604 | 0.5177 | 0.036* | |
| H32 | 0.0760 | 0.5337 | 0.3568 | 0.036* | |
| C4 | 0.17261 (10) | 0.25284 (13) | 0.38538 (18) | 0.0317 (3) | |
| H41 | 0.1681 | 0.1763 | 0.3188 | 0.038* | |
| H42 | 0.1561 | 0.2287 | 0.4931 | 0.038* | |
| C5 | 0.21342 (9) | 0.51824 (13) | 0.40729 (18) | 0.0322 (3) | |
| H51 | 0.2304 | 0.5397 | 0.2990 | 0.039* | |
| H52 | 0.2188 | 0.5960 | 0.4717 | 0.039* | |
| C6 | 0.26896 (10) | 0.30251 (13) | 0.38538 (19) | 0.0349 (4) | |
| H61 | 0.3104 | 0.2399 | 0.4357 | 0.042* | |
| H62 | 0.2886 | 0.3150 | 0.2763 | 0.042* | |
| C7 | 0.33775 (9) | 0.43825 (12) | 0.58409 (16) | 0.0259 (3) | |
| C8 | 0.39795 (9) | 0.59837 (13) | 0.76528 (15) | 0.0275 (3) | |
| C9 | 0.36780 (11) | 0.73381 (14) | 0.79087 (18) | 0.0387 (4) | |
| H91 | 0.3721 | 0.7811 | 0.6927 | 0.058* | |
| H92 | 0.4073 | 0.7734 | 0.8707 | 0.058* | |
| H93 | 0.3045 | 0.7346 | 0.8258 | 0.058* | |
| C10 | 0.49473 (10) | 0.59327 (15) | 0.70952 (17) | 0.0354 (4) | |
| H101 | 0.4989 | 0.6386 | 0.6099 | 0.053* | |
| H102 | 0.5129 | 0.5047 | 0.6951 | 0.053* | |
| H103 | 0.5354 | 0.6333 | 0.7873 | 0.053* | |
| C11 | 0.38403 (10) | 0.52052 (14) | 0.91124 (17) | 0.0351 (4) | |
| H111 | 0.3190 | 0.5190 | 0.9358 | 0.053* | |
| H112 | 0.4186 | 0.5583 | 0.9987 | 0.053* | |
| H113 | 0.4055 | 0.4337 | 0.8939 | 0.053* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O1 | 0.0331 (6) | 0.0239 (5) | 0.0487 (6) | −0.0044 (4) | −0.0025 (5) | −0.0072 (5) |
| O2 | 0.0298 (5) | 0.0242 (5) | 0.0348 (5) | 0.0047 (4) | −0.0017 (4) | 0.0004 (4) |
| O3 | 0.0326 (5) | 0.0217 (5) | 0.0336 (5) | 0.0019 (4) | −0.0074 (4) | −0.0042 (4) |
| N1 | 0.0446 (8) | 0.0565 (10) | 0.0665 (10) | −0.0058 (7) | −0.0207 (8) | −0.0054 (8) |
| N2 | 0.0320 (7) | 0.0341 (7) | 0.0469 (8) | −0.0010 (5) | −0.0060 (6) | −0.0007 (6) |
| N3 | 0.0279 (6) | 0.0200 (6) | 0.0398 (7) | 0.0018 (5) | −0.0042 (5) | −0.0041 (5) |
| N4 | 0.0304 (6) | 0.0239 (6) | 0.0503 (8) | 0.0057 (5) | −0.0125 (6) | −0.0096 (5) |
| C1 | 0.0297 (8) | 0.0281 (8) | 0.0390 (8) | −0.0023 (6) | −0.0063 (6) | −0.0020 (6) |
| C2 | 0.0257 (7) | 0.0241 (7) | 0.0332 (7) | −0.0038 (5) | 0.0042 (6) | −0.0018 (6) |
| C3 | 0.0324 (8) | 0.0204 (7) | 0.0373 (8) | 0.0039 (6) | −0.0050 (6) | −0.0041 (6) |
| C4 | 0.0342 (8) | 0.0202 (7) | 0.0406 (8) | 0.0031 (6) | −0.0035 (6) | −0.0040 (6) |
| C5 | 0.0341 (8) | 0.0224 (7) | 0.0396 (8) | −0.0002 (6) | −0.0089 (6) | −0.0014 (6) |
| C6 | 0.0325 (8) | 0.0264 (7) | 0.0455 (9) | 0.0053 (6) | −0.0054 (7) | −0.0121 (6) |
| C7 | 0.0241 (7) | 0.0237 (7) | 0.0300 (7) | −0.0013 (5) | 0.0030 (5) | −0.0008 (6) |
| C8 | 0.0314 (7) | 0.0259 (7) | 0.0249 (7) | −0.0041 (6) | −0.0037 (6) | −0.0013 (5) |
| C9 | 0.0523 (10) | 0.0268 (8) | 0.0367 (8) | −0.0006 (7) | −0.0048 (7) | −0.0049 (6) |
| C10 | 0.0346 (8) | 0.0390 (8) | 0.0325 (8) | −0.0078 (7) | −0.0006 (6) | 0.0005 (6) |
| C11 | 0.0398 (8) | 0.0354 (8) | 0.0302 (8) | −0.0032 (7) | 0.0025 (6) | 0.0019 (6) |
Geometric parameters (Å, °)
| O1—C2 | 1.2303 (17) | C4—H41 | 0.9900 |
| O2—C7 | 1.2099 (16) | C4—H42 | 0.9900 |
| O3—C7 | 1.3422 (17) | C5—H51 | 0.9900 |
| O3—C8 | 1.4722 (16) | C5—H52 | 0.9900 |
| N1—N2 | 1.1189 (18) | C6—H61 | 0.9900 |
| N2—C1 | 1.3099 (19) | C6—H62 | 0.9900 |
| N3—C2 | 1.3515 (18) | C8—C10 | 1.506 (2) |
| N3—C4 | 1.4600 (18) | C8—C11 | 1.511 (2) |
| N3—C3 | 1.4635 (18) | C8—C9 | 1.513 (2) |
| N4—C7 | 1.3483 (18) | C9—H91 | 0.9800 |
| N4—C5 | 1.4490 (18) | C9—H92 | 0.9800 |
| N4—C6 | 1.4494 (19) | C9—H93 | 0.9800 |
| C1—C2 | 1.450 (2) | C10—H101 | 0.9800 |
| C1—H1 | 0.946 (17) | C10—H102 | 0.9800 |
| C3—C5 | 1.503 (2) | C10—H103 | 0.9800 |
| C3—H31 | 0.9900 | C11—H111 | 0.9800 |
| C3—H32 | 0.9900 | C11—H112 | 0.9800 |
| C4—C6 | 1.506 (2) | C11—H113 | 0.9800 |
| C7—O3—C8 | 120.57 (10) | N4—C6—H61 | 109.6 |
| N1—N2—C1 | 179.04 (18) | C4—C6—H61 | 109.6 |
| C2—N3—C4 | 119.35 (12) | N4—C6—H62 | 109.6 |
| C2—N3—C3 | 124.53 (11) | C4—C6—H62 | 109.6 |
| C4—N3—C3 | 113.83 (11) | H61—C6—H62 | 108.1 |
| C7—N4—C5 | 125.93 (12) | O2—C7—O3 | 125.31 (12) |
| C7—N4—C6 | 120.26 (11) | O2—C7—N4 | 124.15 (13) |
| C5—N4—C6 | 113.59 (12) | O3—C7—N4 | 110.52 (11) |
| N2—C1—C2 | 114.68 (13) | O3—C8—C10 | 110.59 (12) |
| N2—C1—H1 | 115.7 (9) | O3—C8—C11 | 109.89 (11) |
| C2—C1—H1 | 129.6 (9) | C10—C8—C11 | 112.67 (12) |
| O1—C2—N3 | 122.08 (12) | O3—C8—C9 | 101.68 (11) |
| O1—C2—C1 | 120.21 (13) | C10—C8—C9 | 111.04 (12) |
| N3—C2—C1 | 117.64 (12) | C11—C8—C9 | 110.43 (13) |
| N3—C3—C5 | 110.49 (12) | C8—C9—H91 | 109.5 |
| N3—C3—H31 | 109.6 | C8—C9—H92 | 109.5 |
| C5—C3—H31 | 109.6 | H91—C9—H92 | 109.5 |
| N3—C3—H32 | 109.6 | C8—C9—H93 | 109.5 |
| C5—C3—H32 | 109.6 | H91—C9—H93 | 109.5 |
| H31—C3—H32 | 108.1 | H92—C9—H93 | 109.5 |
| N3—C4—C6 | 110.29 (12) | C8—C10—H101 | 109.5 |
| N3—C4—H41 | 109.6 | C8—C10—H102 | 109.5 |
| C6—C4—H41 | 109.6 | H101—C10—H102 | 109.5 |
| N3—C4—H42 | 109.6 | C8—C10—H103 | 109.5 |
| C6—C4—H42 | 109.6 | H101—C10—H103 | 109.5 |
| H41—C4—H42 | 108.1 | H102—C10—H103 | 109.5 |
| N4—C5—C3 | 109.92 (12) | C8—C11—H111 | 109.5 |
| N4—C5—H51 | 109.7 | C8—C11—H112 | 109.5 |
| C3—C5—H51 | 109.7 | H111—C11—H112 | 109.5 |
| N4—C5—H52 | 109.7 | C8—C11—H113 | 109.5 |
| C3—C5—H52 | 109.7 | H111—C11—H113 | 109.5 |
| H51—C5—H52 | 108.2 | H112—C11—H113 | 109.5 |
| N4—C6—C4 | 110.27 (12) | ||
| C4—N3—C2—O1 | −4.1 (2) | C7—N4—C6—C4 | 128.14 (15) |
| C3—N3—C2—O1 | −165.84 (14) | C5—N4—C6—C4 | −57.04 (17) |
| C4—N3—C2—C1 | 178.83 (13) | N3—C4—C6—N4 | 53.39 (17) |
| C3—N3—C2—C1 | 17.1 (2) | C8—O3—C7—O2 | 2.4 (2) |
| N2—C1—C2—O1 | −3.6 (2) | C8—O3—C7—N4 | −178.51 (11) |
| N2—C1—C2—N3 | 173.55 (13) | C5—N4—C7—O2 | −177.86 (14) |
| C2—N3—C3—C5 | −143.07 (14) | C6—N4—C7—O2 | −3.7 (2) |
| C4—N3—C3—C5 | 54.32 (16) | C5—N4—C7—O3 | 3.0 (2) |
| C2—N3—C4—C6 | 142.52 (13) | C6—N4—C7—O3 | 177.17 (13) |
| C3—N3—C4—C6 | −53.89 (17) | C7—O3—C8—C10 | 62.39 (16) |
| C7—N4—C5—C3 | −128.35 (15) | C7—O3—C8—C11 | −62.62 (16) |
| C6—N4—C5—C3 | 57.18 (17) | C7—O3—C8—C9 | −179.61 (12) |
| N3—C3—C5—N4 | −53.86 (17) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C1—H1···O1i | 0.946 (17) | 2.313 (18) | 3.250 (3) | 170.6 (14) |
| C3—H32···O1i | 0.99 | 2.36 | 3.327 (3) | 164 |
Symmetry codes: (i) −x, y+1/2, −z+1/2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: LH5033).
References
- Allen, F. H. (2002). Acta Cryst. B58, 380–388. [DOI] [PubMed]
- American Chemical Society (2008). Chemical Abstracts Service, American Chemical Society, Columbus, OH, USA; accessed Apr 27, 2010.
- Bernstein, J., Davis, R. E., Shimoni, L. & Chang, N.-L. (1995). Angew. Chem. Int. Ed. Engl.34, 1555–1573.
- Bruker (2007). APEX2, SAINT-Plus and SADABS Bruker AXS Inc., Madison, Wisconsin, USA.
- Fenlon, T. W., Schwaebisch, D., Mayweg, A. V. W., Lee, V., Adlington, R. M. & Baldwin, J. E. (2007). Synlett, pp. 2679–2682.
- Kaupang, Å. (2010). Masters thesis, University of Oslo, Norway. Available at http://www.duo.uio.no/.
- Mickelson, J. W., Jacobsen, E. J., Carter, D. B., Im, H. K., Im, W. B., Schreur, P. J. K. D., Sethy, V. H., Tang, A. H., McGee, J. E. & Petke, J. D. (1996). J. Med. Chem.39, 4654—4666. [DOI] [PubMed]
- Miller, R. D., Theis, W., Heilig, G. & Kirchmeyer, S. (1991). J. Org. Chem.56, 1453–1463.
- Ouihia, A., Rene, L., Guilhem, J., Pascard, C. & Badet, B. (1993). J. Org. Chem.58, 1641–1642.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Toma, T., Shimokawa, J. & Fukuyama, T. (2007). Org. Lett.9, 3195–3197. [DOI] [PubMed]
- Wang, J., Zeng, T., Li, M.-L., Duan, E.-H. & Li, J.-S. (2006). Acta Cryst. E62, o2912–o2913.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536810016211/lh5033sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810016211/lh5033Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report