

Abstract
In the title compound, C21H20N2O3, the naphthalimide unit is almost planar (r.m.s. deviation for the 15 non-H atoms = 0.059 Å). The carboximide N atom and the five C atoms of the 2-methylprop-2-enoyl substituent also lie in a plane (r.m.s. deviation = 0.009 Å), which subtends an angle of 84.34 (7)° to the naphthalamide plane. This orients the =CH2 group of the vinyl fragment towards the naphthalimide rings, giving the molecule an extended configuration. The piperidine ring adopts a chair conformation and there is evidence for some delocalization between the naphthalene and piperidine units, the C—Npip bond length being 1.404 (4) Å. In the crystal structure, π–π contacts with centroid–centroid distances of 3.5351 (18) and 3.7794 (18) Å supported by C—H⋯O hydrogen bonds link adjacent molecules in a head-to-tail fashion, forming dimers. These are further stabilized by other C—H⋯O contacts of varying strength, which stack the molecules down the b axis.
Related literature
For background to the applications of 1,8-naphthalamides, see: McAdam et al. (2003 ▶, 2010 ▶); Flood et al. (2007 ▶). For their incorporation into polymer systems, see: Dana et al. (2007 ▶); Munro et al. (2008 ▶). For related structures, see: McAdam et al. (2003 ▶); Easton et al. (1992 ▶); Batchelor et al. (1997 ▶); Tagg et al. (2008 ▶). For comparative bond-length data, see: Allen et al. (1987 ▶) and for ring conformations, see: Cremer & Pople (1975 ▶).
Experimental
Crystal data
C21H20N2O3
M r = 348.39
Monoclinic,
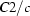
a = 29.049 (3) Å
b = 6.9852 (7) Å
c = 17.1503 (17) Å
β = 102.013 (6)°
V = 3403.7 (6) Å3
Z = 8
Mo Kα radiation
μ = 0.09 mm−1
T = 90 K
0.65 × 0.11 × 0.04 mm
Data collection
Bruker APEXII CCD diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2006 ▶) T min = 0.838, T max = 1.000
12211 measured reflections
1843 independent reflections
1440 reflections with I > 2σ(I)
R int = 0.075
θmax = 21.2°
Refinement
R[F 2 > 2σ(F 2)] = 0.054
wR(F 2) = 0.152
S = 1.12
1843 reflections
236 parameters
H-atom parameters constrained
Δρmax = 0.20 e Å−3
Δρmin = −0.21 e Å−3
Data collection: APEX2 (Bruker, 2006 ▶); cell refinement: APEX2 and SAINT (Bruker, 2006 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶) and TITAN2000 (Hunter & Simpson, 1999 ▶); molecular graphics: SHELXTL (Sheldrick, 2008 ▶) and Mercury (Macrae et al., 2008 ▶); software used to prepare material for publication: SHELXL97, enCIFer (Allen et al., 2004 ▶), PLATON (Spek, 2009 ▶) and publCIF (Westrip, 2010 ▶).
Supplementary Material
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536810018994/hb5457sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810018994/hb5457Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C21—H21B⋯O12i | 0.99 | 2.54 | 3.485 (4) | 160 |
| C24—H24A⋯O12ii | 0.99 | 2.67 | 3.365 (4) | 127 |
| C21—H21A⋯O11iii | 0.99 | 2.36 | 3.258 (4) | 150 |
| C25—H25B⋯O11iv | 0.99 | 2.72 | 3.278 (4) | 117 |
Symmetry codes: (i)  ; (ii)
; (ii)  ; (iii)
; (iii) 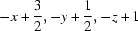 ; (iv)
; (iv) 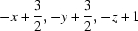 .
.
Acknowledgments
We thank the New Economy Research Fund; grant No. UOO-X0808 for support of this work and the University of Otago for purchase of the diffractometer.
supplementary crystallographic information
Comment
We have recently been interested in naphthalimide derivatives as components of donor-acceptor arrays because, as good acceptors, they often exhibit strong fluorescence together with redox triggered LMCT transitions in the near-IR (McAdam et al. 2003, 2010; Flood et al. 2007). We have also incorporated fluorescent naphthalimides into polymer systems (Dana et al., 2007; Munro et al., 2008). Methacrylate derivatives are polymer precursors and the title compound, I, Fig 1, was synthesised to further scope the possibilities of incorporating fluorescent naphthalimide derivatives into polymers.
The title compound comprises a 1,8-naphthalimide ring system with a piperidino ring at the C4 position of the naphthalene ring and a 2-methyl-prop-2-en-1-one substituent on the N1 atom of the dicarboxamide ring. The naphthalimide unit is planar with an rms deviation from the best fit meanplane through all 15 non-hydrogen atoms of 0.0494 Å. The C13 atom of the propenone headgroup and the N2 atom of the piperidine ring are both displaced slightly from this plane with deviations 0.170 (4) and 0.004 (3) Å respectively both in the same direction. Bond lengths within the dicarboxamide ring are normal (Allen et al., 1987) and consistent with a degree of delocalisation in the naphthalimide system. In keeping with previous observations (Easton et al., 1992; Batchelor et al., 1997; Tagg et al., 2008) the N1—C13 bond is relatively long, 1.486 (4) Å, suggesting that there is a node at the N1 atom. In contrast the C4—N2 bond is short, 1.404 (4) Å, indicating a degree of delocalisation between the naphthalene and piperidine units. The piperidine ring adopts a classical chair conformation with Cremer-Pople puckering parameters [Q(2) = 0.005 (4) Å, φ(2) = 154 (5)° and Q(3) = -0.575 (43) Å (Cremer & Pople, 1975). The N1, C13, (O1), C14, C15, C16 segment of the propenone is also planar (rms deviation 0.0920 Å) and subtends an angle of 84.44 (7)° to the naphthalimide plane. This orients the =C15H2 of the vinyl fragment towards the naphthalimide rings.
In the crystal structure intermolecular π–π contacts occur between the unsubstituted C5···C8, C9, C10 naphthalene ring of one molecule and the C1, C8, C9, C11, C12, N1 carboxamide and substituted C1···C4, C9, C10 rings of an adjacent molecule to form head to tail dimers, with centroid to centroid distances of 3.5351 (18) and 3.7794 (18) Å respectively, Fig 2. These contacts are supported by weak C25–H25B···O11 hydrogen bonds. The dimers are further aggregated by a range of additional C—H···O contacts, Table 2, to give an extensive three dimensional network structure with molecules stacked down the b axis, Fig. 3.
Experimental
4-Piperidine-1,8-naphthalic amide (200 mg, 0.72 mmol) was dissolved in distilled dichloromethane (DCM) (30 ml) under N2, triethylamine (3 ml) and methylacryoyl chloride (0.18 ml, 1.4 mmol) were added sequentially by syringe at 273 K. The mixture was stirred overnight. The mixture was then washed with distilled water (3 times) and the organic phase dried over MgSO4. The solvent was evaporated under reduced pressure and the crude product purified by silica gel column chromatography with hexane-ethyl acetate (50:50, v/v) to give an orange solid (168.3 mg, 0.48 mmol); yield of 67.2%. The compound was recrystallised by the slow diffusion of hexane into a concentrated solution of I in DCM to give very thin orange blades/needles of (I).
Refinement
All H-atoms were refined using a riding model with d(C—H) = 0.95 Å, Uiso=1.2Ueq (C) for aromatic, 0.99 Å, Uiso=1.2Ueq (C) for CH2 and 0.98 Å, Uiso = 1.5Ueq (C) for CH3 H atoms. Crystals were very thin and weakly diffracting and measurable reflection data could not be observed beyond θ = 21.2 °. This results in both low data resolution and a poor data/parameter ratio.
Figures
Fig. 1.

The structure of (I) showing displacement ellipsoids drawn at the 50% probability level.
Fig. 2.

Head to tail dimers of (I) formed by π–π contacts (black dotted lines) augmented by C—-H···O hydrogen bonds (blue dashed lines). The blue sphere represents the centroid of the the C5···C8,C9,C10 ring, the red and green spheres those of the C1, C8, C9, C11, C12, N1 and C1···C4, C9, C10 rings respectively of an adjacent molecule. The symmetry operation linking the two molecules is 3/2-x, 3/2-y, 1-z.
Fig. 3.

The crystal packing of (I) viewed down the b axis with hydrogen bonds drawn as blue dashed lines.
Crystal data
| C21H20N2O3 | F(000) = 1472 |
| Mr = 348.39 | Dx = 1.360 Mg m−3 |
| Monoclinic, C2/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -C 2yc | Cell parameters from 2032 reflections |
| a = 29.049 (3) Å | θ = 2.4–21.1° |
| b = 6.9852 (7) Å | µ = 0.09 mm−1 |
| c = 17.1503 (17) Å | T = 90 K |
| β = 102.013 (6)° | Blade, orange |
| V = 3403.7 (6) Å3 | 0.65 × 0.11 × 0.04 mm |
| Z = 8 |
Data collection
| Bruker APEXII CCD diffractometer | 1843 independent reflections |
| Radiation source: fine-focus sealed tube | 1440 reflections with I > 2σ(I) |
| graphite | Rint = 0.075 |
| ω scans | θmax = 21.2°, θmin = 2.4° |
| Absorption correction: multi-scan (SADABS; Bruker, 2006) | h = −29→29 |
| Tmin = 0.838, Tmax = 1.000 | k = −7→6 |
| 12211 measured reflections | l = −17→17 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.054 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.152 | H-atom parameters constrained |
| S = 1.12 | w = 1/[σ2(Fo2) + (0.0822P)2] where P = (Fo2 + 2Fc2)/3 |
| 1843 reflections | (Δ/σ)max < 0.001 |
| 236 parameters | Δρmax = 0.20 e Å−3 |
| 0 restraints | Δρmin = −0.21 e Å−3 |
Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| C1 | 0.70398 (11) | 0.5991 (4) | 0.6119 (2) | 0.0221 (9) | |
| C2 | 0.72082 (12) | 0.6631 (5) | 0.6879 (2) | 0.0237 (9) | |
| H2 | 0.6991 | 0.7017 | 0.7193 | 0.028* | |
| C3 | 0.76855 (12) | 0.6733 (5) | 0.7204 (2) | 0.0242 (9) | |
| H3 | 0.7788 | 0.7212 | 0.7730 | 0.029* | |
| C4 | 0.80161 (12) | 0.6150 (4) | 0.6777 (2) | 0.0217 (9) | |
| C5 | 0.81694 (12) | 0.5175 (4) | 0.5443 (2) | 0.0227 (9) | |
| H5 | 0.8499 | 0.5270 | 0.5640 | 0.027* | |
| C6 | 0.80025 (12) | 0.4695 (4) | 0.4661 (2) | 0.0255 (9) | |
| H6 | 0.8219 | 0.4450 | 0.4326 | 0.031* | |
| C7 | 0.75190 (12) | 0.4560 (4) | 0.4347 (2) | 0.0225 (9) | |
| H7 | 0.7408 | 0.4207 | 0.3805 | 0.027* | |
| C8 | 0.72057 (12) | 0.4939 (4) | 0.4825 (2) | 0.0217 (9) | |
| C9 | 0.73615 (12) | 0.5448 (4) | 0.5643 (2) | 0.0201 (9) | |
| C10 | 0.78588 (12) | 0.5534 (4) | 0.5964 (2) | 0.0218 (9) | |
| C11 | 0.66975 (13) | 0.4926 (4) | 0.4468 (2) | 0.0236 (9) | |
| C12 | 0.65327 (13) | 0.5993 (5) | 0.5773 (2) | 0.0236 (9) | |
| C13 | 0.58901 (13) | 0.5604 (5) | 0.4598 (2) | 0.0303 (10) | |
| C14 | 0.56012 (12) | 0.3865 (5) | 0.4482 (2) | 0.0294 (10) | |
| C15 | 0.57626 (13) | 0.2257 (5) | 0.4834 (2) | 0.0351 (10) | |
| H15A | 0.6068 | 0.2217 | 0.5166 | 0.042* | |
| H15B | 0.5574 | 0.1135 | 0.4757 | 0.042* | |
| C16 | 0.51218 (13) | 0.4093 (6) | 0.3979 (3) | 0.0469 (12) | |
| H16A | 0.4953 | 0.2871 | 0.3951 | 0.070* | |
| H16B | 0.5147 | 0.4489 | 0.3441 | 0.070* | |
| H16C | 0.4949 | 0.5068 | 0.4213 | 0.070* | |
| C21 | 0.87594 (12) | 0.4410 (5) | 0.7217 (2) | 0.0300 (10) | |
| H21A | 0.8647 | 0.3562 | 0.6754 | 0.036* | |
| H21B | 0.8694 | 0.3770 | 0.7697 | 0.036* | |
| C22 | 0.92823 (12) | 0.4714 (5) | 0.7318 (2) | 0.0346 (10) | |
| H22A | 0.9352 | 0.5248 | 0.6821 | 0.041* | |
| H22B | 0.9447 | 0.3471 | 0.7424 | 0.041* | |
| C23 | 0.94596 (13) | 0.6084 (5) | 0.8008 (2) | 0.0380 (11) | |
| H23A | 0.9424 | 0.5491 | 0.8516 | 0.046* | |
| H23B | 0.9798 | 0.6360 | 0.8043 | 0.046* | |
| C24 | 0.91787 (12) | 0.7929 (5) | 0.7872 (2) | 0.0350 (10) | |
| H24A | 0.9281 | 0.8790 | 0.8334 | 0.042* | |
| H24B | 0.9242 | 0.8580 | 0.7392 | 0.042* | |
| C25 | 0.86568 (12) | 0.7558 (5) | 0.7762 (2) | 0.0300 (9) | |
| H25A | 0.8588 | 0.6991 | 0.8254 | 0.036* | |
| H25B | 0.8483 | 0.8781 | 0.7658 | 0.036* | |
| N1 | 0.63959 (9) | 0.5398 (4) | 0.49807 (17) | 0.0237 (8) | |
| N2 | 0.85019 (10) | 0.6250 (4) | 0.70929 (16) | 0.0262 (8) | |
| O1 | 0.57432 (8) | 0.7182 (4) | 0.43997 (15) | 0.0387 (7) | |
| O11 | 0.65310 (8) | 0.4540 (3) | 0.37740 (14) | 0.0293 (7) | |
| O12 | 0.62306 (9) | 0.6481 (3) | 0.61341 (15) | 0.0331 (7) |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| C1 | 0.020 (2) | 0.018 (2) | 0.026 (2) | −0.0025 (15) | −0.0009 (18) | −0.0004 (16) |
| C2 | 0.028 (2) | 0.021 (2) | 0.024 (2) | 0.0006 (16) | 0.0074 (19) | 0.0028 (16) |
| C3 | 0.024 (2) | 0.024 (2) | 0.024 (2) | −0.0007 (16) | 0.0020 (18) | 0.0024 (16) |
| C4 | 0.021 (2) | 0.0138 (19) | 0.029 (2) | −0.0024 (15) | 0.0029 (19) | 0.0030 (15) |
| C5 | 0.021 (2) | 0.0160 (19) | 0.031 (2) | 0.0053 (15) | 0.0041 (18) | 0.0035 (16) |
| C6 | 0.029 (2) | 0.022 (2) | 0.027 (2) | 0.0027 (16) | 0.0106 (19) | 0.0023 (16) |
| C7 | 0.027 (2) | 0.0163 (19) | 0.022 (2) | 0.0003 (15) | 0.0016 (18) | −0.0002 (15) |
| C8 | 0.028 (2) | 0.0070 (19) | 0.029 (2) | 0.0007 (15) | 0.0031 (19) | 0.0015 (15) |
| C9 | 0.021 (2) | 0.0130 (19) | 0.026 (2) | −0.0017 (15) | 0.0034 (18) | 0.0024 (16) |
| C10 | 0.028 (2) | 0.0116 (18) | 0.024 (2) | −0.0012 (15) | 0.0005 (18) | 0.0025 (15) |
| C11 | 0.029 (2) | 0.016 (2) | 0.025 (2) | 0.0004 (16) | 0.003 (2) | 0.0036 (16) |
| C12 | 0.026 (2) | 0.020 (2) | 0.025 (2) | −0.0007 (17) | 0.008 (2) | 0.0005 (17) |
| C13 | 0.029 (2) | 0.028 (3) | 0.031 (2) | 0.0066 (19) | 0.0008 (19) | 0.0009 (18) |
| C14 | 0.027 (2) | 0.031 (3) | 0.028 (2) | −0.0014 (19) | 0.0020 (18) | −0.0012 (18) |
| C15 | 0.029 (2) | 0.032 (3) | 0.042 (3) | −0.0043 (19) | 0.002 (2) | −0.003 (2) |
| C16 | 0.030 (2) | 0.050 (3) | 0.053 (3) | 0.002 (2) | −0.011 (2) | −0.001 (2) |
| C21 | 0.027 (2) | 0.024 (2) | 0.036 (2) | 0.0026 (16) | −0.0004 (19) | 0.0035 (17) |
| C22 | 0.030 (2) | 0.037 (2) | 0.035 (2) | 0.0060 (18) | 0.0037 (19) | 0.0006 (18) |
| C23 | 0.024 (2) | 0.050 (3) | 0.038 (3) | 0.0006 (19) | 0.0013 (19) | −0.008 (2) |
| C24 | 0.028 (2) | 0.042 (3) | 0.033 (2) | −0.0062 (18) | 0.0034 (19) | −0.0139 (18) |
| C25 | 0.026 (2) | 0.034 (2) | 0.028 (2) | −0.0011 (17) | 0.0019 (18) | −0.0093 (18) |
| N1 | 0.0192 (18) | 0.0226 (17) | 0.0269 (19) | −0.0013 (13) | −0.0011 (15) | −0.0011 (13) |
| N2 | 0.0242 (19) | 0.0272 (18) | 0.0248 (18) | 0.0010 (14) | −0.0007 (14) | −0.0044 (14) |
| O1 | 0.0375 (17) | 0.0292 (17) | 0.0462 (18) | 0.0089 (13) | 0.0015 (14) | 0.0043 (13) |
| O11 | 0.0325 (17) | 0.0280 (15) | 0.0238 (16) | −0.0017 (11) | −0.0024 (13) | −0.0014 (11) |
| O12 | 0.0269 (16) | 0.0365 (16) | 0.0349 (17) | −0.0011 (12) | 0.0043 (14) | −0.0032 (12) |
Geometric parameters (Å, °)
| C1—C2 | 1.368 (5) | C13—N1 | 1.486 (4) |
| C1—C9 | 1.415 (5) | C14—C15 | 1.315 (5) |
| C1—C12 | 1.469 (5) | C14—C16 | 1.486 (5) |
| C2—C3 | 1.384 (5) | C15—H15A | 0.9500 |
| C2—H2 | 0.9500 | C15—H15B | 0.9500 |
| C3—C4 | 1.385 (5) | C16—H16A | 0.9800 |
| C3—H3 | 0.9500 | C16—H16B | 0.9800 |
| C4—N2 | 1.404 (4) | C16—H16C | 0.9800 |
| C4—C10 | 1.440 (5) | C21—N2 | 1.480 (4) |
| C5—C6 | 1.370 (4) | C21—C22 | 1.508 (5) |
| C5—C10 | 1.418 (5) | C21—H21A | 0.9900 |
| C5—H5 | 0.9500 | C21—H21B | 0.9900 |
| C6—C7 | 1.399 (5) | C22—C23 | 1.525 (5) |
| C6—H6 | 0.9500 | C22—H22A | 0.9900 |
| C7—C8 | 1.372 (5) | C22—H22B | 0.9900 |
| C7—H7 | 0.9500 | C23—C24 | 1.517 (5) |
| C8—C9 | 1.426 (5) | C23—H23A | 0.9900 |
| C8—C11 | 1.476 (5) | C23—H23B | 0.9900 |
| C9—C10 | 1.436 (5) | C24—C25 | 1.511 (5) |
| C11—O11 | 1.218 (4) | C24—H24A | 0.9900 |
| C11—N1 | 1.403 (4) | C24—H24B | 0.9900 |
| C12—O12 | 1.223 (4) | C25—N2 | 1.462 (4) |
| C12—N1 | 1.397 (4) | C25—H25A | 0.9900 |
| C13—O1 | 1.205 (4) | C25—H25B | 0.9900 |
| C13—C14 | 1.466 (5) | ||
| C2—C1—C9 | 119.3 (3) | H15A—C15—H15B | 120.0 |
| C2—C1—C12 | 121.0 (3) | C14—C16—H16A | 109.5 |
| C9—C1—C12 | 119.6 (3) | C14—C16—H16B | 109.5 |
| C1—C2—C3 | 122.0 (3) | H16A—C16—H16B | 109.5 |
| C1—C2—H2 | 119.0 | C14—C16—H16C | 109.5 |
| C3—C2—H2 | 119.0 | H16A—C16—H16C | 109.5 |
| C2—C3—C4 | 121.2 (3) | H16B—C16—H16C | 109.5 |
| C2—C3—H3 | 119.4 | N2—C21—C22 | 111.2 (3) |
| C4—C3—H3 | 119.4 | N2—C21—H21A | 109.4 |
| C3—C4—N2 | 122.2 (3) | C22—C21—H21A | 109.4 |
| C3—C4—C10 | 119.0 (3) | N2—C21—H21B | 109.4 |
| N2—C4—C10 | 118.7 (3) | C22—C21—H21B | 109.4 |
| C6—C5—C10 | 121.2 (3) | H21A—C21—H21B | 108.0 |
| C6—C5—H5 | 119.4 | C21—C22—C23 | 110.3 (3) |
| C10—C5—H5 | 119.4 | C21—C22—H22A | 109.6 |
| C5—C6—C7 | 121.0 (3) | C23—C22—H22A | 109.6 |
| C5—C6—H6 | 119.5 | C21—C22—H22B | 109.6 |
| C7—C6—H6 | 119.5 | C23—C22—H22B | 109.6 |
| C8—C7—C6 | 119.7 (3) | H22A—C22—H22B | 108.1 |
| C8—C7—H7 | 120.2 | C24—C23—C22 | 109.3 (3) |
| C6—C7—H7 | 120.2 | C24—C23—H23A | 109.8 |
| C7—C8—C9 | 121.4 (3) | C22—C23—H23A | 109.8 |
| C7—C8—C11 | 118.8 (3) | C24—C23—H23B | 109.8 |
| C9—C8—C11 | 119.6 (3) | C22—C23—H23B | 109.8 |
| C1—C9—C8 | 121.5 (3) | H23A—C23—H23B | 108.3 |
| C1—C9—C10 | 120.0 (3) | C25—C24—C23 | 111.5 (3) |
| C8—C9—C10 | 118.4 (3) | C25—C24—H24A | 109.3 |
| C5—C10—C9 | 118.2 (3) | C23—C24—H24A | 109.3 |
| C5—C10—C4 | 123.2 (3) | C25—C24—H24B | 109.3 |
| C9—C10—C4 | 118.4 (3) | C23—C24—H24B | 109.3 |
| O11—C11—N1 | 119.4 (3) | H24A—C24—H24B | 108.0 |
| O11—C11—C8 | 124.5 (3) | N2—C25—C24 | 110.0 (3) |
| N1—C11—C8 | 116.1 (3) | N2—C25—H25A | 109.7 |
| O12—C12—N1 | 119.1 (3) | C24—C25—H25A | 109.7 |
| O12—C12—C1 | 124.1 (3) | N2—C25—H25B | 109.7 |
| N1—C12—C1 | 116.8 (3) | C24—C25—H25B | 109.7 |
| O1—C13—C14 | 124.1 (3) | H25A—C25—H25B | 108.2 |
| O1—C13—N1 | 118.2 (3) | C12—N1—C11 | 126.2 (3) |
| C14—C13—N1 | 117.7 (3) | C12—N1—C13 | 117.1 (3) |
| C15—C14—C13 | 120.4 (3) | C11—N1—C13 | 115.8 (3) |
| C15—C14—C16 | 124.0 (3) | C4—N2—C25 | 117.0 (3) |
| C13—C14—C16 | 115.5 (3) | C4—N2—C21 | 116.7 (2) |
| C14—C15—H15A | 120.0 | C25—N2—C21 | 111.5 (3) |
| C14—C15—H15B | 120.0 |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C21—H21B···O12i | 0.99 | 2.54 | 3.485 (4) | 160 |
| C24—H24A···O12ii | 0.99 | 2.67 | 3.365 (4) | 127 |
| C21—H21A···O11iii | 0.99 | 2.36 | 3.258 (4) | 150 |
| C25—H25B···O11iv | 0.99 | 2.72 | 3.278 (4) | 117 |
Symmetry codes: (i) −x+3/2, y−1/2, −z+3/2; (ii) −x+3/2, y+1/2, −z+3/2; (iii) −x+3/2, −y+1/2, −z+1; (iv) −x+3/2, −y+3/2, −z+1.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: HB5457).
References
- Allen, F. H., Johnson, O., Shields, G. P., Smith, B. R. & Towler, M. (2004). J. Appl. Cryst.37, 335–338.
- Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor, R. (1987). J. Chem. Soc. Perkin Trans. 2, pp. S1–19.
- Batchelor, R. A., Hunter, C. A. & Simpson, J. (1997). Acta Cryst. C53, 1117–1119.
- Bruker (2006). APEX2, SAINT and SADABS Bruker AXS Inc., Madison, Wisconsin, USA.
- Cremer, D. & Pople, J. A. (1975). J. Am. Chem. Soc.97, 1354–1358.
- Dana, B. H., McAdam, C. J., Robinson, B. H., Simpson, J. & Wang, H. (2007). J. Inorg. Organomet. Polym. Mater.17, 547–559.
- Easton, C. J., Gulbis, J. M., Hoskins, B. F., Scharfbillig, I. M. & Tiekink, E. R. T. (1992). Z. Kristallogr.199, 249–254.
- Flood, A. H., McAdam, C. J., Gordon, K. C., Kjaergaard, H. G., Manning, A. R., Robinson, B. H. & Simpson, J. (2007). Polyhedron, 26, 448–455.
- Hunter, K. A. & Simpson, J. (1999). TITAN2000 University of Otago, New Zealand.
- Macrae, C. F., Bruno, I. J., Chisholm, J. A., Edgington, P. R., McCabe, P., Pidcock, E., Rodriguez-Monge, L., Taylor, R., van de Streek, J. & Wood, P. A. (2008). J. Appl. Cryst.41, 466–470.
- McAdam, C. J., Morgan, J. L., Robinson, B. H., Simpson, J., Rieger, P. H. & Rieger, A. L. (2003). Organometallics, 22, 5126– 5136.
- McAdam, C. J., Robinson, B. H., Simpson, J. & Tagg, T. (2010). Organometallics, doi: 10.1021/om1000452.
- Munro, N. H., Hanton, L. R., Robinson, B. H. & Simpson, J. (2008). React. Funct. Polym.68 671–678 .
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
- Tagg, T., McAdam, C. J., Robinson, B. H. & Simpson, J. (2008). Acta Cryst. C64, o388–o391. [DOI] [PubMed]
- Westrip, S. P. (2010). J. Appl. Cryst.43 Submitted.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536810018994/hb5457sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810018994/hb5457Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report