

Abstract
In the title compound, C6H10N2O, the 2,3-dihydro-1H-pyrazole ring is approximately planar, with a maximum deviation of 0.013 (1) Å. Pairs of intermolecular N—H⋯O hydrogen bonds link neighboring molecules into dimers, generating R 2 2(8) ring motifs. These dimers are further linked into two-dimensional arrays parallel to the bc plane by intermolecular N—H⋯O hydrogen bonds. The crystal structure is further stabilized by C—H⋯π interactions.
Related literature
For the background to and the biological activity of 3-ethyl-4-methyl-1H-pyrazol-5-ol, see: Brogden (1986 ▶); Coersmeier et al. (1986); Gursoy et al. (2000 ▶); Ragavan et al. (2009 ▶, 2010 ▶); Watanabe et al. (1984 ▶); Kawai et al. (1997 ▶); Wu et al. (2002 ▶). For related structures, see: Shahani et al. (2009 ▶, 2010 ▶). For hydrogen-bond motifs, see: Bernstein et al. (1995 ▶). For reference bond-length data, see: Allen et al. (1987 ▶). For the stability of the temperature controller used for the data collection, see: Cosier & Glazer (1986 ▶).
Experimental
Crystal data
C6H10N2O
M r = 126.16
Monoclinic,
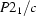
a = 8.374 (2) Å
b = 7.2881 (16) Å
c = 11.300 (3) Å
β = 109.955 (5)°
V = 648.3 (3) Å3
Z = 4
Mo Kα radiation
μ = 0.09 mm−1
T = 100 K
0.52 × 0.16 × 0.09 mm
Data collection
Bruker APEXII DUO CCD area-detector diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2009 ▶) T min = 0.954, T max = 0.992
10018 measured reflections
2745 independent reflections
2325 reflections with I > 2σ(I)
R int = 0.029
Refinement
R[F 2 > 2σ(F 2)] = 0.039
wR(F 2) = 0.123
S = 1.14
2745 reflections
122 parameters
All H-atom parameters refined
Δρmax = 0.52 e Å−3
Δρmin = −0.35 e Å−3
Data collection: APEX2 (Bruker, 2009 ▶); cell refinement: SAINT (Bruker, 2009 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXTL (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXTL; molecular graphics: SHELXTL; software used to prepare material for publication: SHELXTL and PLATON (Spek, 2009 ▶).
Supplementary Material
Crystal structure: contains datablocks global, I. DOI: 10.1107/S160053681001696X/wn2385sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S160053681001696X/wn2385Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
Cg1 is the centroid of the 1H-pyrazole ring (C1–C3/N1/N2).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1N1⋯O1i | 0.902 (15) | 1.829 (15) | 2.7267 (11) | 174.0 (16) |
| N2—H1N2⋯O1ii | 0.972 (14) | 1.715 (14) | 2.6777 (10) | 169.9 (13) |
| C5—H5A⋯Cg1iii | 1.013 (13) | 2.896 (15) | 3.6749 (14) | 134.2 (11) |
Symmetry codes: (i) 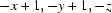 ; (ii)
; (ii) 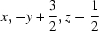 ; (iii)
; (iii) 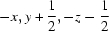 .
.
Acknowledgments
TSH and HKF thank Universiti Sains Malaysia (USM) for the Research University Golden Goose Grant (1001/PFIZIK/811012). VV is grateful to the DST-India for funding through the Young Scientist Scheme (Fast Track Proposal).
supplementary crystallographic information
Comment
Pyrazolone derivatives have a broad spectrum of biological activities as analgesic, antipyretic and anti-inflammatory therapeutical drugs (Brogden, 1986; Gursoy et al., 2000). A class of new pyrazolone compounds have been synthesized and reported to exhibit antibacterial and antifungal activities (Ragavan et al., 2010; Ragavan et al., 2009). A new pyrazolone derivative, edaravone (5-ethyl-4-methyl-1H-pyrazol-3(2H)-one), is being used as a drug in clinical practice for brain ischemia (Watanabe et al., 1984; Kawai et al., 1997) and it has also been found to be effective against myocardial ischemia (Wu et al., 2002).
In the crystal structure (Fig. 1), the 2,3-dihydro-1H-pyrazole ring (C1–C3/N1/N2) is approximately planar with a maximum deviation of 0.013 (1) Å for atoms N1 and N2 (but they are on opposite sides of the plane). The bond lengths (Allen et al., 1987) and angles are within normal ranges and comparable to those in closely related structures reported recently (Shahani et al., 2009; 2010).
In the crystal packing (Fig. 2), pairs of intermolecular N1—H1N1···O1 hydrogen bonds (Table 1) link neighboring molecules into dimers, generating R22(8) ring motifs (Bernstein et al., 1995). These dimers are further linked into 2D arrays parallel to the bc plane by intermolecular N2—H1N2···O1 hydrogen bonds (Table 1). The crystal structure is further stabilized by a C—H···π interaction (Table 1), involving the C1–C3/N1/N2 ring (centroid Cg1) .
Experimental
The compound 5-ethyl-4-methyl-1H-pyrazol-3(2H)-one has been synthesized using the method reported in the literature (Ragavan et al., 2009, 2010) and purified by column chromatography (MeOH: EtOAc, 1:99). It was recrystallised as a colourless solid, using ethanol. Mp: 496.4–507.1 K; MS calculated for C6H10N2O: 126.15. Found: 128.0 (M+).
Refinement
All hydrogen atoms were located in a difference map and were refined freely [N–H = 0.902 (14) – 0.972 (14) Å; C–H = 0.989 (13) – 1.015 (13) Å].
Figures
Fig. 1.

The molecular structure of the title compound, showing 30% probability displacement ellipsoids and the atom numbering scheme. Hydrogen atoms are shown as spheres of arbitrary radius.
Fig. 2.

The crystal packing of the title compound, showing a 2D array parallel to the bc plane. Hydrogen bonds are denoted by dashed lines. H atoms not involved in the hydrogen bond interactions have been omitted for clarity.
Crystal data
| C6H10N2O | F(000) = 272 |
| Mr = 126.16 | Dx = 1.293 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2ybc | Cell parameters from 3666 reflections |
| a = 8.374 (2) Å | θ = 2.6–34.5° |
| b = 7.2881 (16) Å | µ = 0.09 mm−1 |
| c = 11.300 (3) Å | T = 100 K |
| β = 109.955 (5)° | Plate, colourless |
| V = 648.3 (3) Å3 | 0.52 × 0.16 × 0.09 mm |
| Z = 4 |
Data collection
| Bruker APEXII DUO CCD area-detector diffractometer | 2745 independent reflections |
| Radiation source: fine-focus sealed tube | 2325 reflections with I > 2σ(I) |
| graphite | Rint = 0.029 |
| φ and ω scans | θmax = 34.6°, θmin = 3.4° |
| Absorption correction: multi-scan (SADABS; Bruker, 2009) | h = −13→13 |
| Tmin = 0.954, Tmax = 0.992 | k = −11→11 |
| 10018 measured reflections | l = −18→17 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.039 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.123 | All H-atom parameters refined |
| S = 1.14 | w = 1/[σ2(Fo2) + (0.0715P)2 + 0.0472P] where P = (Fo2 + 2Fc2)/3 |
| 2745 reflections | (Δ/σ)max < 0.001 |
| 122 parameters | Δρmax = 0.52 e Å−3 |
| 0 restraints | Δρmin = −0.35 e Å−3 |
Special details
| Experimental. The crystal was placed in the cold stream of an Oxford Cyrosystems Cobra open-flow nitrogen cryostat (Cosier & Glazer, 1986) operating at 100.0 (1) K. |
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| O1 | 0.42822 (7) | 0.62337 (8) | 0.11992 (5) | 0.01463 (13) | |
| N1 | 0.42529 (8) | 0.69441 (9) | −0.08076 (6) | 0.01353 (13) | |
| N2 | 0.35794 (9) | 0.82809 (9) | −0.16813 (6) | 0.01431 (13) | |
| C1 | 0.38533 (9) | 0.73007 (10) | 0.02374 (6) | 0.01110 (13) | |
| C2 | 0.29351 (9) | 0.89787 (9) | 0.00188 (6) | 0.01142 (13) | |
| C3 | 0.28136 (9) | 0.95309 (10) | −0.11791 (7) | 0.01249 (14) | |
| C4 | 0.19811 (10) | 1.11714 (10) | −0.19250 (7) | 0.01638 (15) | |
| C5 | 0.05452 (11) | 1.06785 (12) | −0.31308 (8) | 0.02089 (17) | |
| C6 | 0.22769 (10) | 0.99011 (11) | 0.09370 (7) | 0.01700 (15) | |
| H4A | 0.1538 (18) | 1.1950 (18) | −0.1386 (13) | 0.026 (3)* | |
| H4B | 0.2822 (16) | 1.1904 (17) | −0.2159 (11) | 0.019 (3)* | |
| H5A | −0.0064 (17) | 1.1779 (18) | −0.3632 (13) | 0.025 (3)* | |
| H5B | −0.0336 (19) | 0.991 (2) | −0.2946 (14) | 0.038 (4)* | |
| H5C | 0.0961 (19) | 0.994 (2) | −0.3704 (15) | 0.036 (4)* | |
| H6A | 0.3195 (17) | 1.0187 (18) | 0.1773 (13) | 0.027 (3)* | |
| H6B | 0.147 (2) | 0.9115 (19) | 0.1185 (14) | 0.033 (4)* | |
| H6C | 0.163 (2) | 1.103 (2) | 0.0557 (16) | 0.044 (4)* | |
| H1N1 | 0.4808 (19) | 0.5936 (19) | −0.0921 (14) | 0.028 (3)* | |
| H1N2 | 0.3762 (17) | 0.8332 (19) | −0.2486 (13) | 0.028 (3)* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O1 | 0.0221 (3) | 0.0147 (2) | 0.0089 (2) | 0.00498 (18) | 0.00754 (19) | 0.00296 (17) |
| N1 | 0.0211 (3) | 0.0125 (3) | 0.0092 (2) | 0.0048 (2) | 0.0080 (2) | 0.00233 (19) |
| N2 | 0.0221 (3) | 0.0130 (3) | 0.0098 (3) | 0.0033 (2) | 0.0080 (2) | 0.0027 (2) |
| C1 | 0.0144 (3) | 0.0120 (3) | 0.0078 (3) | 0.0006 (2) | 0.0050 (2) | −0.0002 (2) |
| C2 | 0.0142 (3) | 0.0109 (3) | 0.0096 (3) | 0.0009 (2) | 0.0047 (2) | −0.0003 (2) |
| C3 | 0.0159 (3) | 0.0106 (3) | 0.0110 (3) | 0.0000 (2) | 0.0047 (2) | 0.0000 (2) |
| C4 | 0.0213 (3) | 0.0118 (3) | 0.0143 (3) | 0.0012 (2) | 0.0039 (3) | 0.0027 (2) |
| C5 | 0.0212 (3) | 0.0193 (3) | 0.0178 (3) | 0.0024 (3) | 0.0010 (3) | 0.0032 (3) |
| C6 | 0.0208 (3) | 0.0187 (3) | 0.0132 (3) | 0.0048 (3) | 0.0079 (3) | −0.0015 (3) |
Geometric parameters (Å, °)
| O1—C1 | 1.2839 (9) | C4—C5 | 1.5209 (12) |
| N1—C1 | 1.3578 (9) | C4—H4A | 0.993 (14) |
| N1—N2 | 1.3645 (9) | C4—H4B | 0.989 (13) |
| N1—H1N1 | 0.902 (14) | C5—H5A | 1.013 (13) |
| N2—C3 | 1.3459 (10) | C5—H5B | 1.003 (15) |
| N2—H1N2 | 0.972 (14) | C5—H5C | 0.992 (16) |
| C1—C2 | 1.4206 (10) | C6—H6A | 1.015 (13) |
| C2—C3 | 1.3823 (10) | C6—H6B | 0.994 (15) |
| C2—C6 | 1.4908 (10) | C6—H6C | 1.000 (16) |
| C3—C4 | 1.4916 (11) | ||
| C1—N1—N2 | 109.19 (6) | C5—C4—H4A | 109.8 (8) |
| C1—N1—H1N1 | 124.9 (9) | C3—C4—H4B | 110.2 (7) |
| N2—N1—H1N1 | 125.8 (9) | C5—C4—H4B | 107.8 (7) |
| C3—N2—N1 | 108.49 (6) | H4A—C4—H4B | 107.7 (11) |
| C3—N2—H1N2 | 128.1 (8) | C4—C5—H5A | 114.0 (8) |
| N1—N2—H1N2 | 123.1 (8) | C4—C5—H5B | 111.0 (9) |
| O1—C1—N1 | 122.64 (7) | H5A—C5—H5B | 106.9 (12) |
| O1—C1—C2 | 130.32 (6) | C4—C5—H5C | 111.5 (9) |
| N1—C1—C2 | 107.04 (6) | H5A—C5—H5C | 106.6 (12) |
| C3—C2—C1 | 105.99 (6) | H5B—C5—H5C | 106.3 (12) |
| C3—C2—C6 | 128.98 (7) | C2—C6—H6A | 113.4 (8) |
| C1—C2—C6 | 125.03 (6) | C2—C6—H6B | 112.5 (9) |
| N2—C3—C2 | 109.23 (6) | H6A—C6—H6B | 103.1 (11) |
| N2—C3—C4 | 120.16 (7) | C2—C6—H6C | 110.4 (10) |
| C2—C3—C4 | 130.59 (7) | H6A—C6—H6C | 110.9 (12) |
| C3—C4—C5 | 113.02 (7) | H6B—C6—H6C | 106.1 (13) |
| C3—C4—H4A | 108.2 (8) | ||
| C1—N1—N2—C3 | 2.59 (8) | N1—N2—C3—C4 | 179.19 (6) |
| N2—N1—C1—O1 | 177.88 (7) | C1—C2—C3—N2 | 0.73 (8) |
| N2—N1—C1—C2 | −2.09 (8) | C6—C2—C3—N2 | −179.69 (7) |
| O1—C1—C2—C3 | −179.13 (7) | C1—C2—C3—C4 | 179.34 (7) |
| N1—C1—C2—C3 | 0.84 (8) | C6—C2—C3—C4 | −1.08 (13) |
| O1—C1—C2—C6 | 1.27 (12) | N2—C3—C4—C5 | 60.72 (10) |
| N1—C1—C2—C6 | −178.76 (7) | C2—C3—C4—C5 | −117.76 (9) |
| N1—N2—C3—C2 | −2.03 (8) |
Hydrogen-bond geometry (Å, °)
| Cg1 is the centroid of the 1H-pyrazole ring (C1–C3/N1/N2). |
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1N1···O1i | 0.902 (15) | 1.829 (15) | 2.7267 (11) | 174.0 (16) |
| N2—H1N2···O1ii | 0.972 (14) | 1.715 (14) | 2.6777 (10) | 169.9 (13) |
| C5—H5A···Cg1iii | 1.013 (13) | 2.896 (15) | 3.6749 (14) | 134.2 (11) |
Symmetry codes: (i) −x+1, −y+1, −z; (ii) x, −y+3/2, z−1/2; (iii) −x, y+1/2, −z−1/2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: WN2385).
References
- Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor, R. (1987). J. Chem. Soc. Perkin Trans. 2, pp. S1–19.
- Bernstein, J., Davis, R. E., Shimoni, L. & Chang, N.-L. (1995). Angew. Chem. Int. Ed. Engl.34, 1555–1573.
- Brogden, N. R. (1986). Drugs, 32, 60–70. [DOI] [PubMed]
- Bruker (2009). APEX2, SAINT and SADABS. Bruker AXS Inc., Madison, Wiscosin, USA.
- Coersmeier, C., Wittenberg, H. R., Aehringhaus, U., Dreyling, K. W., Peskar, B. M., Brune, K. & Pesker, B. A. (1986). Agents Actions Suppl.19, 137–153. [PubMed]
- Cosier, J. & Glazer, A. M. (1986). J. Appl. Cryst.19, 105–107.
- Gursoy, A., Demirayak, S., Capan, G., Erol, K. & Vural, K. (2000). Eur. J. Med. Chem.35, 359–364. [DOI] [PubMed]
- Kawai, H., Nakai, H., Suga, M., Yuki, S., Watanabe, T. & Saito, K. I. (1997). J. Pharmacol. Exp. Ther.281, 921–927. [PubMed]
- Ragavan, R. V., Vijayakumar, V. & Kumari, N. S. (2009). Eur. J. Med. Chem.44, 3852–3857.
- Ragavan, R. V., Vijayakumar, V. & Kumari, N. S. (2010). Eur. J. Med. Chem.45, 1173–1180. [DOI] [PubMed]
- Shahani, T., Fun, H.-K., Ragavan, R. V., Vijayakumar, V. & Sarveswari, S. (2009). Acta Cryst. E65, o3249–o3250. [DOI] [PMC free article] [PubMed]
- Shahani, T., Fun, H.-K., Ragavan, R. V., Vijayakumar, V. & Sarveswari, S. (2010). Acta Cryst. E66, o142–o143. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
- Watanabe, T., Yuki, S., Egawa, M. & Nishi, H. (1984). J. Pharmacol. Exp. Ther.268, 1597–1604. [PubMed]
- Wu, T. W., Zeng, L. H., Wu, J. & Fung, K. P. (2002). Life Sci.71, 2249–2255. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks global, I. DOI: 10.1107/S160053681001696X/wn2385sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S160053681001696X/wn2385Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report