

Abstract
In the crystal structure of the title compound, C7H4Cl2O, the molecules form a network of weak C—H⋯O interactions involving the aldehyde O atom and the ortho-H atom on the benzene ring together with C—H⋯O interactions between the formyl groups. Together, these connect the molecules into (10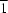 ) layers, which are stabilized additionally by π–π stacking interactions of the benzene rings [centroid–centroid distance = 3.772 (1) Å]. The aldehyde group is twisted relative to the benzene ring by 7.94 (13)°.
) layers, which are stabilized additionally by π–π stacking interactions of the benzene rings [centroid–centroid distance = 3.772 (1) Å]. The aldehyde group is twisted relative to the benzene ring by 7.94 (13)°.
Related literature
For applications of the title compound, see: Katagi (1988 ▶); Wang et al. (2004 ▶). For a related structure, see: Gawlicka-Chruszcz et al. (2006 ▶).
Experimental
Crystal data
C7H4Cl2O
M r = 175.01
Monoclinic,
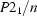
a = 13.100 (1) Å
b = 3.772 (1) Å
c = 15.332 (1) Å
β = 113.797 (2)°
V = 693.2 (3) Å3
Z = 4
Mo Kα radiation
μ = 0.85 mm−1
T = 100 K
0.40 × 0.10 × 0.10 mm
Data collection
Rigaku R-AXIS RAPID diffractometer
Absorption correction: multi-scan (Otwinowski et al., 2003 ▶) T min = 0.90, T max = 0.92
6924 measured reflections
3737 independent reflections
3221 reflections with I > 2σ(I)
R int = 0.063
Refinement
R[F 2 > 2σ(F 2)] = 0.036
wR(F 2) = 0.114
S = 1.10
3737 reflections
107 parameters
All H-atom parameters refined
Δρmax = 0.67 e Å−3
Δρmin = −0.41 e Å−3
Data collection: HKL-2000 (Otwinowski & Minor, 1997 ▶); cell refinement: HKL-2000; data reduction: HKL-2000; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶) and HKL-3000SM (Minor et al., 2006 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶) and HKL-3000SM; molecular graphics: HKL-3000SM, ORTEPIII (Burnett & Johnson, 1996 ▶), ORTEP-3 (Farrugia, 1997 ▶), Mercury (Macrae et al., 2006 ▶) and POV-RAY (The POV-RAY Team, 2004 ▶); software used to prepare material for publication: HKL-3000SM.
Supplementary Material
Crystal structure: contains datablocks I, global. DOI: 10.1107/S160053680905435X/gk2246sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S160053680905435X/gk2246Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C7—H7⋯O1i | 0.946 (17) | 2.533 (17) | 3.4289 (11) | 158.0 (14) |
| C6—H6⋯O1ii | 0.950 (19) | 2.512 (17) | 3.2774 (11) | 137.8 (12) |
Symmetry codes: (i) 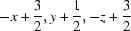 ; (ii)
; (ii) 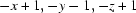 .
.
Acknowledgments
This work was supported by contract GI11496 from HKL Research, Inc.
supplementary crystallographic information
Comment
2,4-Dichlorobenzaldehyde is primarily used in the preparation of dyes, insecticides, herbicides, antiseptics and disinfectants (Wang et al., 2004). It is also used as an intermediate of organic synthesis of fungicide diniconazole (Katagi, 1988).
In the crystal structure of 2,4-dichlorobenzaldehyde (Fig. 1), the aldehyde group is twisted relative to the benzene ring with torsion angles C6—C1—C7—O1 and C2—C1—C7—O1 being -7.94 (13)° and 170.86 (9)°. These torsion angles are significantly smaller in comparison to the corresponding angles in 2,6-dichlorobenzaldehyde (Gawlicka-Chruszcz et al., 2006) which are -27.3° and 152.6° respectively. Significantly bigger twist of the aldehyde group in the case of 2,6-dichlorobenzaldehyde is caused by presence of the chlorine atoms in ortho positions.
The change of the position of chlorine atom causes that interactions in which chlorine atoms are involved in 2,4-dichlorobenzaldehyde and 2,6-dichlorobenzaldehyde differ significantly. In the case of 2,6-dichlorobenzaldehyde Cl2 was involved in weak interaction with hydrogen atom from neighboring benzene ring, while in 2,4-dichlorobenzaldehyde structure such interactions are not observed for any of the chlorine atoms. However, in the case of 2,4-dichlorobenzaldehyde, the chlorine atoms from neighboring molecules form short contacts with Cl1···Cl2 (1/2 + x,1/2 - y,1/2 + z) distance being 3.442Å (Fig. 2).
The weak O···H—C interactions (Table 1) between the aldehyde oxygen and the benzene hydrogen atoms connect molecules to form layers, which are additionally stabilized by stacking of benzene rings (Fig. 2). The oxygen atom from the aldehyde group plays a central role in the formation of weak interactions, and O1···H6—C6 (1 - x,-1 - y,1 - z) and O1···H7—C7 (1,5 - x,-1/2 + y,1.5 - z) distances are 2.51Å and 2.53Å respectively.
Experimental
2,4-dichlorobenzaldehyde was purchased from ALDRICH (99% purity, lot 08722CD). The compound was provided in crystalline form.
Refinement
All hydrogen atoms were localized using the difference density Fourier map. Their positions and isotropic displacement parameters were refined.
Figures
Fig. 1.

The asymmetric unit of the reported structure. Displacement ellipsoids are drawn at the 50% probability level and hydrogen atoms are drawn as grey spheres of an arbitrary radius.
Fig. 2.

The molecular packing of 2,4-dichlorobenzaldehyde. Weak interactions, in which the oxygen atom participates, are shown as blue, dashed lines.
Crystal data
| C7H4Cl2O | F(000) = 352 |
| Mr = 175.01 | Dx = 1.677 Mg m−3 |
| Monoclinic, P21/n | Mo Kα radiation, λ = 0.71074 Å |
| Hall symbol: -P 2yn | Cell parameters from 31891 reflections |
| a = 13.100 (1) Å | θ = 1.0–37.8° |
| b = 3.772 (1) Å | µ = 0.85 mm−1 |
| c = 15.332 (1) Å | T = 100 K |
| β = 113.797 (2)° | Block, colorless |
| V = 693.2 (3) Å3 | 0.40 × 0.10 × 0.10 mm |
| Z = 4 |
Data collection
| Rigaku R-AXIS RAPID diffractometer | 3737 independent reflections |
| Radiation source: fine-focus sealed tube | 3221 reflections with I > 2σ(I) |
| graphite | Rint = 0.063 |
| Detector resolution: 10 pixels mm-1 | θmax = 37.8°, θmin = 1.0° |
| ω scans | h = −22→22 |
| Absorption correction: multi-scan (Otwinowski et al., 2003) | k = −6→6 |
| Tmin = 0.90, Tmax = 0.92 | l = −26→24 |
| 6924 measured reflections |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.036 | Hydrogen site location: difference Fourier map |
| wR(F2) = 0.114 | All H-atom parameters refined |
| S = 1.10 | w = 1/[σ2(Fo2) + (0.0708P)2 + 0.0197P] where P = (Fo2 + 2Fc2)/3 |
| 3737 reflections | (Δ/σ)max = 0.001 |
| 107 parameters | Δρmax = 0.67 e Å−3 |
| 0 restraints | Δρmin = −0.41 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Cl2 | 0.138241 (15) | 0.17792 (6) | 0.579292 (15) | 0.02422 (7) | |
| Cl1 | 0.558151 (16) | 0.15135 (6) | 0.840216 (14) | 0.02526 (7) | |
| C1 | 0.49878 (6) | −0.1166 (2) | 0.66074 (6) | 0.01968 (13) | |
| C3 | 0.35318 (6) | 0.1385 (2) | 0.70157 (6) | 0.02006 (14) | |
| C2 | 0.46410 (6) | 0.0484 (2) | 0.72565 (5) | 0.01980 (13) | |
| C6 | 0.41880 (6) | −0.1866 (2) | 0.56890 (6) | 0.02060 (14) | |
| C5 | 0.30765 (6) | −0.0964 (2) | 0.54240 (6) | 0.02080 (13) | |
| C4 | 0.27652 (6) | 0.0634 (2) | 0.60987 (5) | 0.01992 (13) | |
| O1 | 0.64561 (5) | −0.4059 (2) | 0.63526 (5) | 0.02975 (14) | |
| C7 | 0.61622 (6) | −0.2245 (2) | 0.68671 (6) | 0.02340 (14) | |
| H3 | 0.3319 (13) | 0.260 (4) | 0.7450 (12) | 0.038 (3)* | |
| H5 | 0.2576 (13) | −0.150 (4) | 0.4813 (13) | 0.042 (4)* | |
| H6 | 0.4423 (13) | −0.293 (5) | 0.5239 (12) | 0.046 (4)* | |
| H7 | 0.6686 (14) | −0.131 (4) | 0.7449 (13) | 0.041 (4)* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Cl2 | 0.01697 (10) | 0.02944 (12) | 0.02558 (11) | 0.00285 (6) | 0.00789 (8) | 0.00236 (6) |
| Cl1 | 0.02130 (11) | 0.02926 (12) | 0.02104 (11) | −0.00059 (6) | 0.00422 (8) | −0.00518 (6) |
| C1 | 0.0165 (3) | 0.0207 (3) | 0.0213 (3) | −0.0005 (2) | 0.0071 (2) | −0.0007 (2) |
| C3 | 0.0190 (3) | 0.0209 (3) | 0.0206 (3) | 0.0003 (2) | 0.0083 (3) | −0.0003 (2) |
| C2 | 0.0182 (3) | 0.0203 (3) | 0.0195 (3) | −0.0014 (2) | 0.0062 (2) | −0.0017 (2) |
| C6 | 0.0189 (3) | 0.0226 (3) | 0.0206 (3) | −0.0002 (2) | 0.0082 (2) | −0.0008 (2) |
| C5 | 0.0185 (3) | 0.0229 (3) | 0.0195 (3) | −0.0007 (2) | 0.0061 (2) | −0.0008 (2) |
| C4 | 0.0168 (3) | 0.0209 (3) | 0.0215 (3) | 0.0001 (2) | 0.0071 (2) | 0.0016 (2) |
| O1 | 0.0214 (3) | 0.0378 (3) | 0.0304 (3) | 0.0045 (2) | 0.0108 (2) | −0.0046 (3) |
| C7 | 0.0178 (3) | 0.0266 (3) | 0.0251 (3) | −0.0001 (3) | 0.0080 (3) | −0.0013 (3) |
Geometric parameters (Å, °)
| Cl2—C4 | 1.7327 (7) | C3—H3 | 0.939 (17) |
| Cl1—C2 | 1.7343 (8) | C6—C5 | 1.3869 (11) |
| C1—C2 | 1.3961 (11) | C6—H6 | 0.950 (19) |
| C1—C6 | 1.3999 (11) | C5—C4 | 1.3930 (11) |
| C1—C7 | 1.4820 (11) | C5—H5 | 0.923 (18) |
| C3—C4 | 1.3877 (11) | O1—C7 | 1.2180 (11) |
| C3—C2 | 1.3893 (11) | C7—H7 | 0.946 (17) |
| C2—C1—C6 | 118.32 (7) | C1—C6—H6 | 118.6 (9) |
| C2—C1—C7 | 122.14 (7) | C6—C5—C4 | 118.43 (7) |
| C6—C1—C7 | 119.53 (7) | C6—C5—H5 | 118.4 (10) |
| C4—C3—C2 | 118.11 (7) | C4—C5—H5 | 123.2 (10) |
| C4—C3—H3 | 121.2 (10) | C3—C4—C5 | 122.11 (7) |
| C2—C3—H3 | 120.6 (10) | C3—C4—Cl2 | 118.26 (6) |
| C3—C2—C1 | 121.73 (7) | C5—C4—Cl2 | 119.62 (6) |
| C3—C2—Cl1 | 116.99 (6) | O1—C7—C1 | 123.05 (8) |
| C1—C2—Cl1 | 121.28 (6) | O1—C7—H7 | 121.4 (10) |
| C5—C6—C1 | 121.30 (7) | C1—C7—H7 | 115.5 (10) |
| C5—C6—H6 | 120.0 (9) | ||
| C4—C3—C2—C1 | −0.72 (12) | C1—C6—C5—C4 | −0.68 (12) |
| C4—C3—C2—Cl1 | 179.11 (6) | C2—C3—C4—C5 | −0.19 (12) |
| C6—C1—C2—C3 | 0.90 (12) | C2—C3—C4—Cl2 | −179.59 (6) |
| C7—C1—C2—C3 | −177.92 (8) | C6—C5—C4—C3 | 0.88 (12) |
| C6—C1—C2—Cl1 | −178.92 (6) | C6—C5—C4—Cl2 | −179.73 (6) |
| C7—C1—C2—Cl1 | 2.26 (11) | C2—C1—C7—O1 | 170.86 (9) |
| C2—C1—C6—C5 | −0.18 (12) | C6—C1—C7—O1 | −7.94 (13) |
| C7—C1—C6—C5 | 178.67 (7) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C7—H7···O1i | 0.946 (17) | 2.533 (17) | 3.4289 (11) | 158.0 (14) |
| C6—H6···O1ii | 0.950 (19) | 2.512 (17) | 3.2774 (11) | 137.8 (12) |
Symmetry codes: (i) −x+3/2, y+1/2, −z+3/2; (ii) −x+1, −y−1, −z+1.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: GK2246).
References
- Burnett, M. N. & Johnson, C. K. (1996). ORTEPIII Report ORNL-6895. Oak Ridge National Laboratory, Tennessee, USA.
- Farrugia, L. J. (1997). J. Appl. Cryst.30, 565.
- Gawlicka-Chruszcz, A., Zheng, H., Hyacinth, M., Cymborowski, M., Sabat, M. & Minor, W. (2006). Z. Kristallogr. New Cryst. Struct. 221, 545–546.
- Katagi, T. (1988). J. Agric. Food Chem.36, 344–349.
- Macrae, C. F., Edgington, P. R., McCabe, P., Pidcock, E., Shields, G. P., Taylor, R., Towler, M. & van de Streek, J. (2006). J. Appl. Cryst.39, 453–457.
- Minor, W., Cymborowski, M., Otwinowski, Z. & Chruszcz, M. (2006). Acta Cryst. D62, 859–866. [DOI] [PubMed]
- Otwinowski, Z., Borek, D., Majewski, W. & Minor, W. (2003). Acta Cryst. A59, 228–234. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods in Enzymology, Vol. 276, Macromolecular Crystallography, Part A, edited by C. W. Carter Jr & R. M. Sweet, pp. 307–326. New York: Academic Press.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- The POV-RAY Team (2004). POV-RAY http://www.povray.org/download/.
- Wang, S.-X., Tan, Z.-C., Di, Y.-Y., Xu, F., Zhang, H.-T., Sun, L.-X. & Zhang, T. (2004). J. Chem. Thermodyn.36, 393–399.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks I, global. DOI: 10.1107/S160053680905435X/gk2246sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S160053680905435X/gk2246Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report