

Abstract
Previously, we identified a novel calcium-independent phospholipase, designated calcium-independent phospholipase A2 γ (iPLA2γ), which possesses dual mitochondrial and peroxisomal subcellular localization signals. To identify the roles of iPLA2γ in cellular bioenergetics, we generated mice null for the iPLA2γ gene by eliminating the active site of the enzyme through homologous recombination. Mice null for iPLA2γ display multiple bioenergetic dysfunctional phenotypes, including 1) growth retardation, 2) cold intolerance, 3) reduced exercise endurance, 4) greatly increased mortality from cardiac stress after transverse aortic constriction, 5) abnormal mitochondrial function with a 65% decrease in ascorbate-induced Complex IV-mediated oxygen consumption, and 6) a reduction in myocardial cardiolipin content accompanied by an altered cardiolipin molecular species composition. We conclude that iPLA2γ is essential for maintaining efficient bioenergetic mitochondrial function through tailoring mitochondrial membrane lipid metabolism and composition.
Mitochondria transduce chemical energy from multiple dietary substrates into ATP and heat and are important participants in cellular lipid metabolism and signaling (1, 2). Through the precisely regulated partitioning of energy derived from substrates between ATP synthesis and heat production, a finely tuned dynamic balance is maintained to promote organism survival. In addition, excess energy derived from substrates, not immediately needed to fulfill chemical or thermodynamic demands, is directed into the production of lipid synthetic intermediates for energy storage (e.g. mitochondrial synthesis of lysophosphatidic acid, a precursor of triglycerides). Moreover, mitochondria generate diverse chemical signals that reflect the bioenergetic status of the cell (e.g. NO and cytochrome c) and thus integrate multiple energetic, metabolic, and signaling cascades (1–5).
Mitochondrial phospholipases are important participants in the regulation of mitochondrial function and signaling (6–11). Mitochondrial phospholipases catalyze the production of non-esterified fatty acids that regulate UCP function, release lipid second messengers, such as 2-arachidonoyl lysophosphatidylcholine (LPC),2 and modulate membrane molecular dynamics (12). Maladaptive activation of phospholipases has deleterious metabolic effects in many systems, and previous studies have implicated calcium-independent phospholipases A2 (iPLA2s) as the enzymic mediators of membrane dysfunction in diabetic cardiomyopathy (13–16). Metabolic flexibility is dependent on the efficient coordinated transitions of substrate flux that occur during alterations in energy sources (e.g. glucose versus fatty acid) or energy demand (e.g. exercise). Obesity and type II diabetes are thought to result from disruption of these transitions (17). Diabetic cardiomyopathy is characterized by altered lipid metabolism (i.e. greatly increased utilization of fatty acids) that results in changes in the chemical composition of cardiac membranes, modifying their biophysical properties and function (13, 18–20). Many of the metabolites used in mitochondrial energy metabolism possess dual roles in modulating signaling cascades (e.g. acyl-CoA, acylcarnitines, fatty acids, and lysophosphatidic acid), thereby integrating lipid energy metabolism and cellular signaling functions (21–25). Ultimately, the disproportionate utilization of fatty acids in diabetic myocardium leads to a profound metabolic imbalance resulting in bioenergetic inefficiency, resulting, at least in part, from alterations of cardiolipin (CL) molecular species that occur in diabetic myocardium (18, 20, 26, 27).
Previously, we cloned and characterized iPLA2γ, a major phospholipase activity in murine myocardium based on activity assays, mRNA content, and kinetic analyses using enantiomeric selective mechanism-based inhibitors (28). Subsequently, iPLA2γ was found to possess dual mitochondrial and peroxisomal localization signals, suggesting its importance in regulating myocardial bioenergetics and lipid metabolism. Next, we demonstrated that cardiac myocyte-specific overexpression of iPLA2γ results in dramatic reductions of cardiac phospholipid mass and mitochondrial dysfunction (29). Remarkably, fasting of cardiac myocyte-restricted iPLA2γ overexpressing mice resulted in large increases in myocardial triglyceride content that precipitated hemodynamic dysfunction (29).
To gain further insight into the role of iPLA2γ in cardiac function, we inactivated the iPLA2γ gene in mice by targeted deletion of exon 5 encoding the lipase active site (GVSTG). Genetic ablation of iPLA2γ resulted in growth retardation, cold intolerance, and dramatically reduced exercise endurance. The importance of iPLA2γ in the ability of myocardium to respond to stress was identified by the dramatically reduced survival of iPLA2γ knock-out mice following transverse aortic constriction. Finally, mitochondrial function assays in mice null for iPLA2γ demonstrated markedly decreased complex IV function that was associated with decreases in tetra-18:2 cardiolipin content. Collectively, these studies identify mitochondrial dysfunction resulting from alterations in mitochondrial lipid metabolism as the mechanistic basis for the phenotypic features of mice null for iPLA2γ.
EXPERIMENTAL PROCEDURES
Materials
Radiolabeled nucleotides and 1-palmitoyl-2-[1-14C]arachidonoyl phosphatidylcholine were purchased from PerkinElmer Life Sciences. ECL reagents were purchased from GE Life Sciences. PCR reagents were purchased from Applied Biosystems (Foster City, CA) and used with an ABI GeneAmp PCR System 9700 thermocycler. Racemic BEL was purchased from Calbiochem. A rabbit antibody to a peptide derived from the human iPLA2γ sequence was used in these studies and prepared by affinity chromatography as previously described in detail (28). Normal rabbit serum was affinity-purified in an identical manner for use as a control primary antibody. Synthetic phospholipids used as internal standards in mass spectrometric analyses were purchased from Avanti Polar Lipids (Alabaster, AL), Nu-Chek Prep, Inc. (Elysian, MN), and Cambridge Isotope Laboratories, Inc. (Cambridge, MA) and used as described previously (30). Solvents for sample preparation and for mass spectrometric analysis were purchased from Burdick and Jackson (Honeywell International Inc., Burdick and Jackson, Muskegon, MI). Most other reagents were obtained from Sigma.
Ablation of the Active Site of iPLA2γ
Mouse genomic DNA was utilized as a template for PCR amplification with primers P1 and P2 (Table 1) to generate a 4917-nt fragment containing sequence 5′ of exon 5 and flanked by EcoRI restriction sites. This fragment was cloned into the corresponding site of vector pPNT (31) upstream of the neomycin gene sequence. Similarly, a 1055-nt PCR fragment amplified utilizing primers P3 and P4 (Table 1) and corresponding to sequence 3′ of exon 6 was cloned into XhoI and NotI restriction sites downstream of the neomycin gene sequence. This generated a construct containing iPLA2γ genomic sequence, for which exon 5 was replaced with the positive selection marker (neomycin in the opposite orientation) and leaving regions of homology on either side of the neomycin gene. An SfiI restriction site within the plasmid construct containing the iPLA2γ was used for linearization. The linearized construct was electroporated into male SCC#10 ES cells (derived from 129X1Sv/J mice; Jackson Laboratories (Bar Harbor, ME)) for subsequent neomycin selection using G418. Homologous recombinants were identified by Southern blot analysis and tail PCR. For genomic Southern blot analysis, PstI restriction site differences between wild type (WT) and KO mouse genomic DNA sequence were utilized for detection of 7.4-kb WT and 5.9-kb KO iPLA2γ bands by hybridization with a 459-nt genomic probe generated by PCR amplification with primers P5 and P6 (Table 1). Two clones of 238 screened by Southern analysis were positive for the homologous recombination. These clones were injected into C57BL/6 blastocysts, followed by implantation into pseudopregnant mice to generate the chimeric mouse founder line utilizing the Neurosciences Transgenic Core facility at Washington University. Chimeras were mated to C57BL6/J mice, and germ line transmission to heterozygous offspring was confirmed by Southern blotting of tail DNA. Interbreeding of heterozygous offspring was used to generate homozygous KO and WT littermates used in all studies. In addition to genomic Southern analysis, tail PCR was routinely performed for genotyping. “Neoforward” and “Common REV” primers (Table 1) were utilized for amplification of a 354-nt product from the knock-out allele, whereas paired “iPLA2 FOR” and “Common REV” primers (Table 1) were utilized to amplify a 181-nt product for detection of the wild type allele. All experiments were performed using sex- and age-matched littermate controls as noted.
TABLE 1. Primers for construction and genotyping of the iPLA2γ knock-out.
iPLA2γ KO primers P1–P4 were used in PCR to prepare the KO construct. PCR primers P5 and P6 were used to generate the 459-nt genomic probe for genotyping by genomic Southern blot analyses. The tail PCR primer set was used for genotyping. “Neo forward” paired with the “Common REV” amplified a 354-nt product from KO DNA whereas “iPLA2γ FOR” paired with “Common REV” amplified a 181-nt product from WT DNA. Reverse transcription-PCR primers 1–6 were used to demonstrate an absence of iPLA2γ message expression from the KO, whereas “GAPDH F” and “GAPDH R” PCR primers were used as positive controls for amplification of GAPDH PCR product from both WT and KO cDNA.
| Primers | Sequence (5′–3′) |
|---|---|
| iPLA2γ KO primers | |
| P1 | AAAAGAATTCAGGCAGAGGAGAAAAAGCGTGTGCTACTTC |
| P2 | AAAAGAATTCGCTCACAACCATCCGTAATGAGATCTGGATG |
| P3 | AAAACTCGAGCCAAGAACATGCAGCACAGGCTTGAGGCCAT |
| P4 | AAAAGCGGCCGCTTGTTGACATCATAAAAACCATAATT |
| P5 | CTCTATCGAAAGTTGGGCTCAGATGT |
| P6 | AATAGGTAAATAAATACTAAAATGGA |
| Tail PCR primer set | |
| NEO forward | AAAGGCCTACCCGCTTCCATTGCTCA |
| Common REV | GCCTCAAGCCTGTGCTGCATGTTCTT |
| iPLA2γ FOR | CTGTTCACAGAGGTGTGGTTGC |
| KO band = 354 base pairs | |
| WT band = 181 base pairs | |
| Reverse transcription-PCR primers | |
| 1 | CAAGAAGGCAGAGGAGAAAAAGC |
| 2 | CCCAAAGCTCTGTATTACTAGGG |
| 3 | GATTACATCTGTGGAGTAAGCACAGGG |
| 4 | CGTGAAAGGCCAAGGCCGAAGGG |
| 5 | AGTGAGCATTCATATGTCCCG |
| 6 | CGGAGAATGACTCTCCAAATCTG |
| GAPDH F | TGTTGCCATCAATGACCCCTTCA |
| GAPDH R | TGATGATCTTGAGGCTGTTGTC |
General Animal Studies
Animal protocols were in strict accordance with the National Institutes of Health guidelines for humane treatment of animals and were reviewed and approved by the Animal Care Committee of Washington University. Conscious animals fed ad libitum were weighed at various time points after weaning up to 10 months of age. Following euthanasia, tissues were dissected from male and female mice, weighed, and expressed as a fraction of total body weight. Rectal temperature was measured to the nearest 0.1°C by inserting the probe of a thermocouple (Fisher) 20 mm into the rectum until a stable reading was obtained. For these temperature studies, mice were singly housed at ambient temperature with food and water provided ad libitum, and the mean of temperatures taken at 8 a.m. on three consecutive days was reported. For cold tolerance studies, male and female WT and KO mice were singly housed at 4°C for up to 24 h with food and water provided ad libitum. For cold adaptation studies, core body temperatures were monitored utilizing a rectal probe at time 0 and hourly thereafter with constant monitoring of mice for signs of lethargy. Mice were removed from the study when their core temperature dropped below 28°C. For food intake studies, ad libitum food intake of individually housed mice was measured at 1, 1.5, 4, and 9 months of age (n = 4) over a period of 3–5 consecutive days.
RNA, DNA, and Protein Analyses
For Northern blotting, total RNA was isolated utilizing an RNeasy tissue kit, and hybridizations were performed as previously described (8), utilizing labeled ~1-kb iPLA2γ probe derived from IMAGE clone 6591346 (ATCC, Manassas, VA). For quantitative PCR analyses, total RNA was isolated from tissues using Trizol (Invitrogen) according to the manufacturer’s directions. RNA was quantified by spectrophotometry (NanoDrop, Wilmington, DE). cDNA was synthesized from RNA using Superscript III Reverse Transcriptase (Invitrogen). cDNA samples were amplified using Power SYBR Green PCR Master Mix (Applied Biosystems) on the ABI 7500 real time PCR system (Applied Biosystems). Acidic ribosomal protein 36B4 message was used for normalization, and results were analyzed using the comparative Ct method. Primers used for quantitative PCR were as follows (5′ –3′): UCP1, CTCACTCAGGATTGGCCTCTA (forward) and TCTGACCTTCACGACCTCTGTA (reverse); UCP2, TCCACGCAGCCTCTACAAT (forward) and GACCTTTACCACATCTGTAGGC (reverse); UCP3, CAGAGGGACTATGGATGCCTAC (forward) and AGGTGAGACTCCA-GCAACTTCT (reverse); 36B4, GCAGACAACGTGGGCTCCAAGCAGAT (forward) and GGTCCTCCTTGGTGAACACGAAGCCC (reverse). Results were analyzed by comparing the threshold crossing (Ct) of each sample after normalization to the control genes (dCt). The changes in the threshold crossing (dCt) were used to calculate the relative levels of each mRNA from the various samples using the formula 2−dCt. Genomic Southern blot studies were performed by PstI digestion of 10 mg of genomic DNA, followed by electrophoreses on a 0.8% TAE gel and transfer to a Hybond-XL (GE Life Sciences) membrane for hybridization. Western blotting was performed as previously described (28) utilizing an ECL detection system (GE Life Sciences).
Treadmill Studies
Treadmill studies were performed utilizing the Mouse Cardiovascular Phenotyping Core at Washington University School of Medicine. WT and knock-out mice (ages ~2–9 months) were run to exhaustion utilizing a motorized, speed-controlled, modular treadmill system (Columbus Instruments, Columbus, OH) equipped with an electric shock stimulus and an adjustable inclination angle as previously described (32). Runs were performed on a level inclination, and running velocity was initiated at 10 m/min and then incrementally increased at a rate of 3 m/min until exhaustion, defined as the inability to remain on the treadmill for 5 s.
Transverse Aortic Constriction
Transverse Aortic Constriction studies were performed by the Mouse Cardiovascular Phenotyping Core at the Washington University School of Medicine. The protocol for transverse aortic constriction (TAC) has been previously described (33, 34). In brief, surgery was performed on mice anesthetized with a xylazine (16 mg/kg) and ketamine (80 mg/kg) mixture, and a 7–0 silk suture was placed around the transverse aorta and tied around a 25-gauge blunt needle, which was subsequently removed. Following constriction, the gradient across the band was measured to ensure that adequate stenosis had been achieved. Subsequently, the thorax was closed, and mice were allowed to recover on a heating pad until responsive to stimuli. Surgeons were blinded to mouse genotypes. As controls, sham-operated animals underwent identical surgery but without aortic constriction.
Mitochondrial Respiration Studies
Mitochondria were prepared essentially as previously described (35, 36). Hearts were removed from WT and knock-out animals and immediately placed at 10% (w/v) into ice-cold pH 7 HEPES buffer containing 1 mM EDTA and 250 mM sucrose. Hearts were homogenized with a loose fitting dounce homogenizer. Next, the homogenate was centrifuged at 500 × g for 10 min to remove cellular debris, nuclei, and connective tissue. The supernatant solution was centrifuged at 9,000 × g for 35 min to isolate mitochondria, which were resuspended in homogenization buffer. Protein concentration was determined using the micro-BCA protein assay kit (Pierce). Oxygen consumption was measured polarographically using a dual channel Instech dissolved oxygen measuring system. In the presence of malate and succinate, O2 consumption was initiated with ADP to determine substrate-supported O2 consumption and ability to generate ATP. Cytochrome oxidase activity, after the addition of rotenone, was determined by the addition of ascorbate and TMPD. The addition of azide completely abolished oxygen uptake in the rote-none-inhibited preparation.
Mass Spectrometric Analysis of CL
Enhanced shotgun lipidomics analyses of CL were performed on a QqQ mass spectrometer (Thermo-Fisher Scientific, San Jose, CA) equipped with an electrospray ion source as previously described (26). All electrospray ionization mass spectrometric analyses of lipids were conducted by direct infusion employing a Harvard syringe pump at a flow rate of 4 μl/min. Typically, a 1-min period of signal averaging was employed for each mass spectrum, and a 2-min period of signal averaging was employed for each tandem mass spectrometric spectrum. A mass resolution of 0.4 Thomson was employed for acquisition of mass spectra with a QqQ instrument.
Statistical Analysis
Data were analyzed using a two-tailed Student’s t test, χ test, one-way analysis of variance (for treadmill studies), and the log-rank test (for TAC studies). Differences were regarded as significant at p < 0.05. All data are reported as the mean ± S.E. unless otherwise noted.
RESULTS
Targeted Disruption of the iPLA2γ Gene
Since iPLA2γ has multiple promoters and splice variants (8, 28), we employed a knock-out strategy targeting the ablation of exon 5, which contains the active site. A neomycin-based gene-targeting vector was designed to undergo homologous recombination, leading to deletion of exon 5 of the murine iPLA2γ gene. The strategy for the targeting event employed to effect homologous recombination and the multiple methods used for its identification are shown in Fig. 1A. First, a linearized vector was used to electroporate SCC#10 ES cells (derived from 129X1/SvJ mice) as described under “Experimental Procedures.” After neomycin selection, colonies were screened for targeted disruption of exon 5 by Southern blot analyses. Of the 238 ES clones identified possessing the neomycin resistance cassette, two independent clones (clones 7 and 48) exhibited evidence of the desired insertion/recombination event. These clones were separately injected into C57BL/6 blastocysts and implanted into pseudopregnant females as described under “Experimental Procedures.” Male chimeras (10 chimerics from clone 7 and 1 chimeric from clone 48) whose coat color ranged from 50 to 100% agouti were subsequently bred with C57BL/6 mice. Heterozygotes originating from clone 7 were used for the subsequent generation of all mice. Breeding of C57BL/6 mice containing the insert from clone 7 led to the generation of viable mice null for the iPLA2γ gene as demonstrated by both tail PCR analyses and genomic Southern analyses (Fig. 1B).
FIGURE 1. Strategy for iPLA2γ gene ablation and genetic and expression analyses.

A, schematic representation of the iPLA2γ gene-targeting strategy. The region of the wild type murine iPLA2γ gene containing exons 2– 6 is shown schematically (top) along with the predicted knock-out region resulting from homologous recombination (below). Relevant restriction endonuclease sites are also shown. The neomycin cassette is indicated by a shaded arrow (labeled Neo). The regions between exons 3 and 4 and between exons 5 and 6 were targeted for homologous recombination. The locations of PCR primers utilized for tail PCR genetic analyses are indicated with arrows (labeled PCR). The region corresponding to the probe used for genomic Southern blot analyses is indicated as a shaded box (labeled probe), whereas relevant restriction fragments resulting from PstI restriction digestion of the WT and KO are indicated. B, genomic Southern analysis of wild type and iPLA2γ knock-out mice. Southern blot analyses of genomic DNA from wild type and iPLA2γ null mice were performed utilizing tail DNA digested with PstI and hybridized with the probe indicated in Fig. 1A. Autoradiography of the genomic Southern blots revealed the presence of an expected 7.4-kb wild type allelic band labeled as W in lanes 3 and 6, whereas analyses of iPLA2γ knockouts labeled as K in lanes 1, 2, 4, 5, 7, and 8 revealed the presence of the predicted 5.9-kb knock-out allele band. Results are representative of separate Southern analyses of genomic DNA from over 30 animals. C, PCR analyses of different regions of the iPLA2γ coding sequence. Primers 1– 6 (represented by arrows below the schematic of the iPLA2γ coding sequence (top) surrounding the lipase active site) were paired as indicated (5′-primer/3′-primer underlined) to amplify by PCR different regions of the sequence utilizing cDNA prepared from either WT or KO heart RNA as template. Agarose-gel electrophoresis demonstrated the presence of amplified PCR products of the expected size (in kb) utilizing only wild type (W) and not knock-out (K) template with primer pairs 1/4, 2/4, 3/4, and 5/6. PCR products amplified from both wild type and knock-out template utilizing a control GAPDH primer pair are shown at the right. Data are representative of those obtained from reverse transcription-PCR analyses of RNA from the myocardium of three separate animals. D, expression of iPLA2γ mRNA in wild type but not iPLA2γ knock-out tissues. Total RNA was isolated from heart (H), liver (L), skeletal (S), kidney (K), lung (N), and brain (B) tissues wild type (W) and iPLA2γ knock-out (K) prior to separation by agarose-gel electrophoresis and Northern blot analysis using a 1-kb probe complementary to the region of iPLA2γ cDNA (iPLA2γ) flanking the sequence encoding the lipase site. Ethidium bromide staining of 28 S ribosomal RNA (28S RNA) in each corresponding lane is shown below. Results are representative of separate Northern analyses of RNA extracted from multiple tissues from three WT and three KO animals.
Further characterization of the targeting strategy for the disruption of the iPLA2γ gene demonstrated that the ablation of exon 5 resulted in an unstable transcript, since multiple different regions of the gene could not be detected by reverse transcription-PCR analyses (Fig. 1C) and Northern blot analyses of
mRNA purified from heart and other tissues in iPLA2γ KO mice did not demonstrate any message corresponding to the altered transcript (Fig. 1D). Targeted disruption of the iPLA2γ active site resulted in over a 60% reduction in the production of 2-[14C]arachidonoyl LPC from 1- palmitoyl 2-[14C]arachidonoyl-PC in incubations with mitochondrial homogenates prepared from mice null for iPLA2γ (Fig. 2). The marked reduction in this “signature” reaction of iPLA2γ substantiates the targeted ablation of iPLA2γ activity. The remaining measurable mitochondrial phospholipase activity was probably largely due to iPLA2β, since it was inhibitable by BEL, and substantive amounts of iPLA2β are known to be present in murine mitochondria (7, 9). In addition, iPLA2β can hydrolyze the sn-1-position of 1-palmitoyl-2-arachidonoyl choline glycerophospholipids, albeit at much slower rates and with less regiospecificity in comparison with iPLA2γ (12). It should be noted that small amounts of a calcium-independent activity that was not inhibitable by BEL were also present in both WT and iPLA2γ−/− murine mitochondria. This activity probably results from cPLA2γ that is localized in the mitochondrial compartment and is both calcium-independent and not inhibitable by BEL (37). Taken together, these results support the conclusion that the gene-targeting event resulted in an iPLA2γ null allele and that iPLA2γ−/− mice were viable.
FIGURE 2. iPLA2γ activity in wild type and iPLA2γ knock-out mouse myocardial mitochondria.

Calcium-independent phospholipase A2γ has been previously demonstrated to selectively hydrolyze 1-palmitoyl-2-[1-14C]arachidonoyl phosphatidylcholine to generate 2-[1-14C]arachidonoyl LPC (12). To selectively measure iPLA2γ activity among other cellular mitochondrial phospholipases, mitochondria were isolated from WT and KO hearts, sonicated, and incubated in the presence of 1-palmitoyl-2-[1-14C]arachidonoyl phosphatidylcholine as described under “Experimental Procedures.” Remaining radiolabeled substrate and reaction products were extracted into butanol and resolved by TLC, and the amount of 2-[14C]arachidonoyl LPC was determined by scintillation spectrometry counting and expressed as nmol of 2-[14C]arachidonoyl LPC min−1 mg of mitochondrial protein−1. A dramatic 60% reduction in the production of 2-arachidonoyl LPC was observed in KO mitochondria in comparison with their WT counterparts (*, p < 0.05). All data were averaged from analyses of duplicate determinations from three mice and are shown as mean ± S.D.
General Phenotypic Features of Mice Null for iPLA2γ
Subsequent large scale breeding of heterozygous mice resulted in normal litter sizes with no unexpected deaths of offspring. Genotypic analyses of 751 offspring from heterozygous crosses resulted in 201 WT, 395 heterozygous, and 155 homozygous KO offspring, indicating a small (17%) reduction in the expected Mendelian inheritance of the homozygous KO (0.89: 2.26:1.15 versus the expected 1:2:1 ratio (p < 0.02 by the χ test). Most of the reduction was due to the lower proportion of female KO mice (71 versus the expected 94), a 24% reduction in anticipated female offspring (p < 0.05). Although iPLA2γ knock-out mice appeared normal and healthy after birth and weaning by visual inspection, body weights of iPLA2γ KO mice were consistently 9% lower compared with sex-matched WT littermates (8.78 ± 0.4 versus 10.95 ± 0.2 g, respectively, at 28 days of age (p < 0.01)) and remained lower through approximately day 50 (Fig. 3A). In contrast to their WT littermates, which progressively increased in weight, mice null for iPLA2γ did not gain significant weight after adolescence. By day 288, KO male animals had a mean weight of 22.2 ± 0.4 g versus 34.1 ± 3.1 g for WT males (i.e. 35% lower weight). Female KO animals weighed 21.4 ± 1.1 versus 31.2 ± 5.2 g for WT females at day 255 (31% lower weight) (Fig. 3B). Along with a reduction in body mass, by 9 months of age, KO male mice had a significantly shorter body length by ~8% (measured from the tip of the nose to the anus). KO mice were 9.3 ± 0.1 cm versus 10.0 ± 0.1 cm for WT littermates (p < 0.001, n = 11 WT and 13 KO). A slightly reduced food intake (9%) was identified for the 2–3-month-old iPLA2γ KO animals, which was not statistically significant. In contrast, a 24% reduction in food intake was observed in 9-month-old male KO animals (3.7 g/day for KO versus 4.7 g/day for WT, p < 0.06, five separate measurements of n = 6 per group). This reduction was not significant after adjusting for differences in body weight. Both male and female KOs (between 3 and 12 months of age) had a significantly lower basal core body temperature by nearly 0.5°C. The mean core body temperatures were 36.3 ± 0.1°C (WT males) versus 35.7 ± 0.1°C (KO males, p < 0.02) and 36.8 ± 0.1°C (WT females) versus 36.4 ± 0.1°C (KO females) based on n of ~19 per genotype and an average of 50 readings per genotype. The weights of brain, liver, kidney, skeletal muscle, brain, WAT, and BAT relative to total body weight were not significantly different compared with wild type littermates. Other measured parameters, including fasting blood glucose levels (116.5 ± 11.3 mg/dl; average blood pressure measured by five consecutive tail cuff readings (99.4 ± 8.16/86.2 ± 5.7 mm Hg for iPLA2γ knockout mice versus 113.4 ± 1.97/96.9 ± 3.1 mm Hg for wild type littermates)) and glucose tolerance (blood glucose measurements following 16 h of fasting and intraperitoneal glucose administration), were within the normal range and were not significantly different in 2–4-month-old KO and WT littermates. Echocardiographic analyses of iPLA2γ mice at ages of 2–3 months did not reveal any significant differences in chamber sizes or ventricular function compared with WT control mice fed ad libitum, after fasting, or after high fat feeding for 6 weeks. No differences were found between iPLA2γ knock-out mice and iPLA2γ controls in general ambulatory activity studies (data not shown).
FIGURE 3. Severely reduced growth rate of iPLA2γ KO mice in comparison with WT controls.

Total average body weight (g) was measured and plotted versus age (in days) for male (A) and female (B) WT and iPLA2γ KO mice. The results represent 155 measurements of 43 wild type and 33 knock-out males and 25 wild type and 33 knock-out females between the ages of 28 and 282 days. Values represent the mean ± S.E. for each data point. Both male and female iPLA2γ KO animals (dashed lines) had significantly decreased growth (*, p < 0.05) as early as day 28 (by Student’s t test) in comparison with WT littermates (solid lines). KO animals demonstrated severe growth retardation after approximately day 50 with very little weight gain through day 280.
Compromised Thermal Adaptation in iPLA2γ KO Mice
To compare the ability of 4–6-month-old WT and iPLA2γ KO animals to adapt to cold, decreases in core body temperature were examined as a function of time following exposure to 4°C with ready access to food and water as described under “Experimental Procedures.” All WT littermates were able to maintain core body temperatures for 24 h at 4°C. In sharp contrast, cold intolerance was manifest in both male and female mice null for iPLA2γ (15.3 ± 2.7 and 11.5 ± 3.4 h mean endurance times at 4°C, respectively) (Fig. 4). Similarly, aged mice (9–11 months) null for iPLA2γ also demonstrated cold intolerance, whereas their WT littermates did not. The mean endurance times for KO male and female animals were 8 ± 0.4 and 7.1 ± 0.6 h, respectively, whereas both WT male and female animals maintained their body temperatures throughout the entire experimental interval. Collectively, these results demonstrated that both young and old iPLA2γ knock-out mice had defects in cold adaptation.
FIGURE 4. Cold intolerance of iPLA2γ KO mice.

For cold tolerance tests, mice (4 – 6 months) were individually housed at 4°C in standard breeding cages containing bedding with free access to food and water. Rectal core body temperatures were measured using a thermocouple probe inserted 2 cm into the rectum until a stable reading was obtained. Measurements were taken every 60 min for up to 24 h or until the mice became torpid or the core temperature dropped below ~28°C, after which mice were removed for recovery. All WT mice maintained their body temperature between 30 and 36°C during the entire course of the studies (24 h). The bar graph shows mean cold tolerance time ± S.E. for male and female WT (solid bars) and KO (open bars) animals. The mean time at which male KO mice were able to maintain their core body temperature was 15.3 ± 2.7 h (n = 6 per group, p < 0.05 by Student’s t test) compared with WT controls (which maintained their body temperatures for the entire experimental interval). For female KOs, the corresponding mean endurance time at 4°C was 11.5 ± 3.4 h (n = 6 per group, p < 0.02) compared with WT controls, which maintained their body temperatures throughout the entire experimental interval.
Uncoupling of electron transport and conversion of the proton-motive force across the inner membrane into heat by uncoupling proteins (UCPs) is a well described mechanism for heat production. In particular, mitochondrial uncoupling in brown adipose tissue mediated by UCP1 has been previously shown to support body temperature after exposure to cold (reviewed in Ref. 38). Accordingly, we examined levels of mRNA encoding UCP1 levels in brown adipose tissue by reverse transcription-PCR during normothermic and hypothermic conditions (Fig. 5A). Although the basal UCP1 message level of the KO is slightly higher than that of WT mice (1.5-fold), the data indicate that both WT and KO mice responded to cold with similar (~2-fold) increases in brown adipose UCP1 mRNA expression after exposure to cold. We also measured levels of UCP2 and UCP3 in cardiac muscle in both WT and KO animals. Although no alterations in UCP2 levels were detected (Fig. 5B), iPLA2γ KO mice had 8-fold lower levels of UCP3 mRNA as determined by reverse transcription-PCR in comparison with their WT counterparts (Fig. 5C).
FIGURE 5. Expression of UCP1 mRNA in brown adipose tissue and also UCP2 and UCP3 mRNA in heart tissue from WT and KO mice.
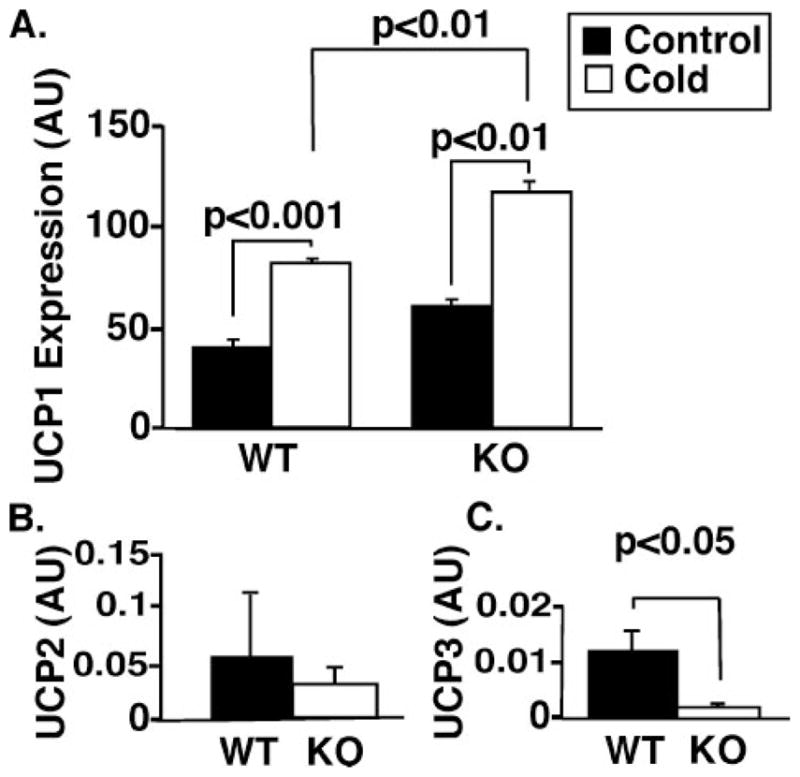
Expression levels of UCP1 (A), UCP2 (B), and UCP3 (C) mRNA were measured using real time PCR as described under “Experimental Procedures.” A, UCP1 message levels in brown adipose tissue were measured under ambient conditions (control) and during exposure to cold (4°C). Animals were exposed to 4°C until their core body temperature dropped to hypothermic levels (below ~28°C). The mean cold tolerance time for KO animals was 8 h, and WT controls (still maintaining the core body temperatures) were sacrificed at the same mean time. B, no significant difference was observed for UCP2 expression levels in WT and KO hearts. C, a significant reduction in UCP3 expression was observed in the hearts of iPLA2γ KO mice (p < 0.05). n = 4 per group. Values represent arbitrary units (AU) after normalization to a ribosomal 36B4 internal standard. Data are presented as means ± S.E. Statistical significance was performed using the Student’s t test, and significant differences are indicated by brackets.
Decreased Exercise Endurance of iPLA2γ KO Mice
Considering the defects in cold adaptation in the iPLA2γ KO mice, we next focused on the ability of these mice to tolerate prolonged exercise on a treadmill in comparison with their WT counter-parts. Interestingly, 53-day-old mice null for iPLA2γ exhibited a dramatic 50% decrease in exercise endurance (19.1 ± 1.3 min) compared with their WT littermates (39.3 ± 1.5 min, p < 0.001) when the animals were run to exhaustion on a treadmill (Fig. 6A). This exercise deficit was progressive, since the endurance time of 86-day-old KO mice relative to WT decreased to less than 25% relative to WT littermates (9.6 ± 0.7 versus 32 ± 3.2 min for the KO versus WT, respectively, p < 0.001) (Fig. 6B). By day 270, the KO treadmill time had further decreased to 5 min (5.3 ± 0.7 min) (Fig. 6B).
FIGURE 6. Reduced exercise capacity of the iPLA2γ knock-out.
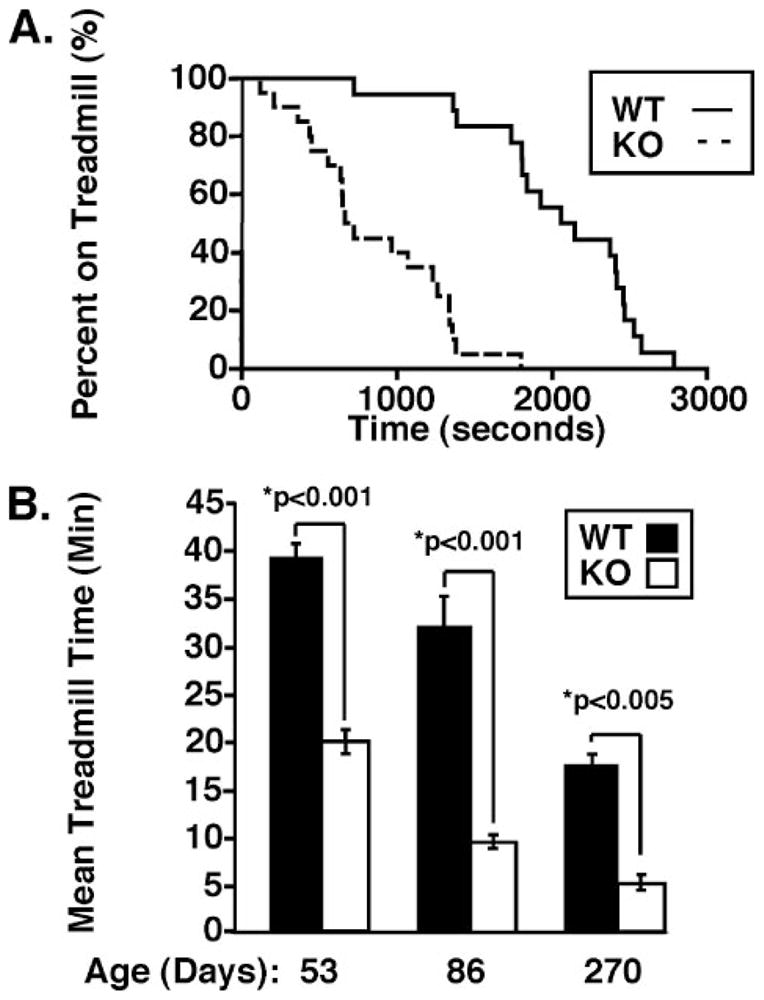
A, Kaplan-Meier survival curve analysis of iPLA2γ KO and WT treadmill performance plotted as the percentage of animals remaining on the treadmill versus time (s). iPLA2γ WT (n = 20) and iPLA2γ KO (n = 22) male and female mice aged 2– 4 months were subjected to an exercise-to-exhaustion protocol on a motorized treadmill, as described under “Experimental Procedures.” Median exercise times were 2103 and 692 s for WT and KO animals, respectively, p < 0.001 by one-way analysis of variance. B, a significant reduction in exercise endurance was revealed for mice null for iPLA2γ when the above data were replotted as treadmill time versus mean age in days comparing mice of ~53, 86, and 270 days. Treadmill times for iPLA2γ KO mice were approximately half those for WT animals at 53 days but dropped to nearly 25% of WT treadmill times by day 86. *, p < 0.001 by Student’s t test.
Compromised Myocardial Functional Reserve in iPLA2γ KO Mice
TAC with careful monitoring of pressure gradients has been a useful model in the evaluation of myocardial metabolic reserve in response to hemodynamic stress. Mice null for iPLA2γ were dramatically more susceptible to sudden death following transverse aortic constriction. Six of 8 knock-out mice (ages 2–4 months) died within 24 h following surgery (25% survival), whereas all of the sham operated mice or wild type littermates survived. No significant alterations in pressure gradients generated by banding between the two groups were present. These results demonstrate that iPLA2γ KO mice are unable to successfully compensate for the increased myocardial energy demands generated by hemodynamic-induced increases in ventricular afterload.
Decreased Mitochondrial Function in iPLA2γ KO Mice Due to Inefficient Complex IV-mediated O2 Reduction
The markedly decreased survival of mice null for iPLA2γ in response to pressure overload suggested an iPLA2γ-induced defect in cardiac mitochondrial function. To determine if this contributed to the increased TAC mortality rate and decreased exercise endurance observed in iPLA2γ knock-out mice, mitochondria were isolated from WT and iPLA2γ KO myocardium for measurement of respiratory function as described under “Experimental Procedures.” Initially, the ability of WT and iPLA2γ KO mitochondria to support cytochrome oxidase (Complex IV)-mediated reduction of molecular oxygen in the presence of a surrogate electron donor (ascorbate) was measured. Remarkably, the rate of O2 consumption by iPLA2γ KO mitochondria was decreased by 65% compared with that of WT controls (Fig. 7A). To demonstrate the functional integrity of the isolated mitochondria, ADP, succinate, and malate were added in separate experiments to the mitochondria, which resulted in robust O2 consumption in both WT and iPLA2γ KO mitochondria with only minimal differences in the rate of O2 reduction (Fig. 7B). After correction for azide inhibition, a replot of ascorbate-TMPD oxygen consumption (relative site IV Vmax) more dramatically demonstrated the reduced functional activity of complex IV in both young and old iPLA2γ KOs versus WT controls (Fig. 7C).
FIGURE 7. Functional analysis of wild type and knock-out iPLA2γ isolated myocardial mitochondria.

Mitochondria were isolated, and oxygen consumption was measured under different experimental conditions as described under “Experimental Procedures.” A, a representative TMPD-ascorbate-supported oxygen consumption by mitochondria from WT and KO animals is shown. The calculated rates of oxygen consumption were normalized for protein concentration. Also shown is the effect of azide upon the TMPD-ascorbate oxygen consumption. B, a representative graph of the malate-ADP-supported oxygen consumption by mitochondria from WT and KO animals. The calculated rates of oxygen consumption are normalized for protein concentration. C, ascorbate-TMPD oxygen consumption (plotted as relative site IV Vmax) is corrected for azide inhibition and summarized for all animals in WT and KO groups. n = 6 young (4 – 6 months) and 3 old (9 –11 months) animals/group. The numbers within each bar indicate the total number of mitochondrial measurements. Analyses of these results indicate that for both age groups, the KO reduces the observed azide-inhibitable oxygen consumption (p < 0.05).
Alterations in Cardiolipin Content and Molecular Species Distribution in iPLA2γ KO Mice
To determine if genetic ablation of iPLA2γ resulted in changes in myocardial lipid composition, shotgun lipidomics was performed on heart tissue from WT and KO animals aged 4–6 months. No statistically different alterations in major lipid classes (e.g. phosphatidylcholine, phosphatidylethanolamine, phosphatidylglycerol, triacylglycerol, etc.) or molecular species examined were noted with the exception of a decrease in CL content and an altered CL molecular species distribution (Fig. 8). Specifically, the total CL content was reduced by 15 ± 0.2 mol % in iPLA2γ KO hearts in comparison with their wild type littermates (Fig. 9A). Moreover, a more marked decrease in the symmetric CL species tetra-18:2 CL was present in mice null for iPLA2γ in comparison with their WT littermates (33 ± 0.6 mol %, p < 0.02) (Fig. 9B). Similar results were obtained from analyses of hearts from animals 9 –11 months of age (data not shown). The peak at m/z 747.5 was composed of tri-18:2–22:6 CL and was not substantially changed in WT versus iPLA2γ KO mice. Collectively, these results demonstrate that iPLA2γ participates in mitochondrial cardiolipin metabolism, presumably by supplying the lysocardiolipin acceptor substrate for acylation/transacylation remodeling reactions. Thus, although iPLA2γ is not obligatory for the production of tetra-18:2 CL (i.e. tetra-18:2 CL is present in the iPLA2γ KO), the absence of iPLA2 cannot be completely compensated for by other enzymes.
FIGURE 8. Expanded negative ion electrospray ionization mass spectra of myocardial lipid extracts from iPLA2γ KO mice and their wild type littermates.

Myocardial lipid extracts of wild type mice (A) and iPLA2γ KO mice (B) were prepared by a modified Bligh and Dyer procedure. Negative ion electrospray ionization mass spectra were acquired using a QqQ mass spectrometer as described under “Experimental Procedures.” Spectra have been normalized to a CL internal standard (see insets). The asterisks indicate the doubly charged CL plus-one isotopologues whose ion peak intensities were utilized to quantify individual CL molecular species as described previously (26). The results indicate the paucity of tetra-18:2 CL molecular species in the iPLA2γ KO in comparison with WT (compare peaks at m/z 723.5 in control versus iPLA2γ KO). The insets on the right of each panel show the extended mass spectra that display additional internal standards and other major anionic phospholipids.
FIGURE 9. Cardiolipin content of iPLA2γ knock-out mouse myocardium.
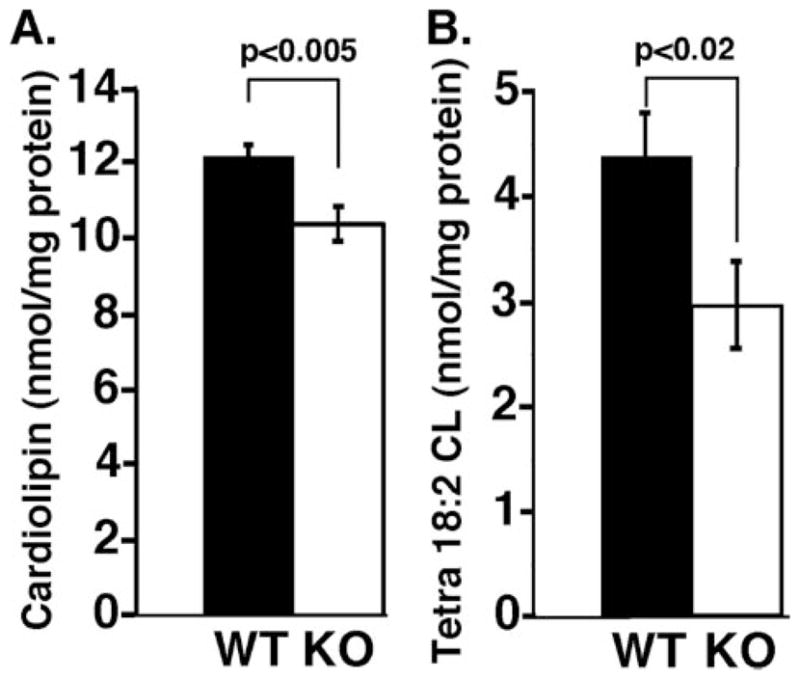
Mouse heart tissues were obtained from 3–4-month-old wild type (WT) or iPLA2γ KO (KO) mice fed ad libitum. Mouse myocardial lipids were extracted by a modified Bligh and Dyer method and subjected to electrospray ionization/mass spectrometry analyses in the negative ion mode as described under “Experimental Procedures.” The levels of individual cardiolipin molecular species after identification were calculated in comparison with the selected internal standard after 13C deisotoping as described previously (26). A, alterations in total cardiolipin content (nmol of CL/mg of protein) between WT and iPLA2γ KO mice. B, alterations in the content of the major cardiolipin species, tetra-18:2 CL (nmol/mg protein) in iPLA2γ KO versus WT mice. Experiments were performed with three WT and five KO animals between 3 and 6 months in age. Significant decreases in both total cardiolipin (p < 0.005) and tetra-18:2 CL species (p < 0.02) were consistently observed in iPLA2γ KO hearts.
DISCUSSION
Through the use of targeted genetic ablation of iPLA2γ, we have demonstrated the fundamental role of this enzyme in cellular bioenergetics that cannot be compensated for by alterations in other enzymic activities. Previously, we hypothesized that iPLA2γ had an important role in modulating cellular bioenergetics based on identification of its mitochondrial localization (8, 29). The findings in this study that iPLA2γ KO mice have attenuated growth, decreased exercise endurance, cold intolerance, and the inability to compensate for increased cardiac stress after hemodynamic overload collectively provide strong evidence of mitochondrial dysfunction induced by iPLA2γ deficiency. The biochemical mechanisms leading to mitochondrial dysfunction were localized to defects in Complex IV function demonstrated by the inability to increase O2 reduction provoked by ascorbate. In addition, lipidomics analyses indicated that the dysfunction was probably related to alterations in the content of tetra-18:2 cardiolipin molecular species. It has been demonstrated previously by multiple techniques, including co-crystallization and blue native gel electrophoresis, that CL is tightly associated with Complex IV (39–42). Moreover, the CL molecular species specifically associated with cytochrome oxidase is symmetric tetra-18:2 CL, which occupies a tightly associated and highly symmetric position in the complex (43). The absence of iPLA2γ in murine myocardium probably results in a paucity of CL lysocardiolipin acceptor, which limits CL remodeling and the resultant synthesis of tetra-18:2 CL (Fig. 9) (44). The deficiency of tetra-18:2 CL was anticipated based upon the known importance of phospholipases in the remodeling of newly synthesized CL molecular species. This deficiency in symmetric CL molecular species probably results in inefficient Complex IV function, as demonstrated by mitochondrial function assays in vitro. The inability of mitochondria to adapt effectively to a variety of stressors under pathophysiologic conditions underscores the importance of specific CL molecular species in mitochondrial bioenergetic efficiency and the important role of iPLA2γ in generating the lysocardiolipin acceptor necessary for the generation of symmetric tetra-18:2 CL molecular species. We specifically point out that other alterations, in addition to the changes in CL content and molecular species composition, could contribute to the dysfunctional mitochondrial adaptations to stress that were demonstrated in this study, including cryptic changes in lipid-protein interactions, alterations in membrane dynamics, and changes in membrane surface charge and distribution.
The iPLA2γ KO phenotype is consistent with an extensive literature demonstrating the association between mitochondrial dysfunction and deficiencies in growth as well as cold and exercise tolerance (3, 45–48). Previously, it has been demonstrated in many models that mitochondrial integrity is essential for cold adaptation (38, 49–52). Thus, the ability to adapt to cold has previously been utilized to identify mitochondrial dysfunction in many different types of mice with genetic alterations in energy-producing pathways (e.g. MCAD (53), UCP1 (54), GPAT1 (55), orphan nuclear receptor estrogen (56), and PGC-1γ (57, 58)). Mutations in mitochondrial proteins encoded by both mitochondrial DNA and nuclear DNA have been implicated in a wide range of degenerative diseases, which include clinical features of myopathy, cardiomyopathy, growth retardation, movement disorders, and diabetes. Many animal models have confirmed that alterations in mitochondrial energy generation, ROS production, and apoptosis can all contribute to the pathophysiology of mitochondrial dysfunction (59 – 62).
Recent investigations into the role of mitochondrial membrane dynamics have uncovered the critical role of mitochondrial membrane fusion and fission in facilitating mitochondrial function (63–65). Although the precise mechanisms that mediate fusion and fission are not known with certainty, it is clear that CL, which has a propensity for HII phase formation, plays a critical role in this process (66). The large difference in the volume occupied by the four highly unsaturated aliphatic chains of CL compared with that of the polar head groups probably promotes prominent curvature of the inner membrane (67). In addition, the doubly anionic charge present in CLs has profound effects on membrane surface charge and facilitates specific lipid-protein interactions. Although the structural features of polyunsaturated CLs have yet to be assigned specific mechanistic roles, the unique characteristics of this molecule and its highly symmetric configuration are probably critical to mitochondrial function on many levels. The production of symmetric CL species is facilitated by iPLA2γ and, as shown in the genetic knock-out, cannot be compensated for by the other phospholipases present in the mitochondrial compartment.
The iPLA2γ KO mice had significantly reduced cold tolerance, yet there were no large differences in expression of UCP1 under basal conditions or after cold induction. Considering the cold intolerance and lower basal core body temperature of the KO (probably due to mitochondrial inefficiency in heat generation), the small increase in KO UCP1 is not unexpected and may represent a compensatory mechanism for heat generation. Prior work has demonstrated the obligatory role of fatty acids in facilitating the function of uncoupling proteins (68–71). Thus, by providing fatty acids locally, mitochondrial phospholipases could function in the acute regulation of inner membrane UCP activity. The significance of the large down-regulation of UCP3 expression in the myocardium of iPLA2γ KO mice is unknown, as is the precise functional role of UCP3 itself.
Phospholipases could also play a key role in regulating the relative proportion of Gibbs free energy in substrate converted into either chemical energy (e.g. ATP) or heat in the mitochondria. In many systems previously studied, the role of phospholipases is to regulate signaling functions. In this regard, we point out that iPLA2γ selectively generates 2-arachidonyl (AA) (20:4) LPC, which serves as a node in lipid second messenger signaling pathways (12, 72, 73). Support for the role of iPLA2γ in the generation of 2-AA LPC as well as 2-docosahexaenoyl (2-DHA) (22:6)-LPC in vivo has come from transgenic mice overexpressing iPLA2γ in a cardiac restricted manner, where dramatic increases in 2-AA LPC and 2-DHA LPC have been demonstrated (29).
A fourth function of phospholipase activity in mitochondria may be to regulate the binding and dynamics of cytochrome c to the inner membrane, which is highly dependent on CL content, inner membrane surface charge, and membrane molecular dynamics (74). Thus, abnormal CL content in conjunction with alterations in the physical properties of the mitochondrial inner membrane could significantly change mitochondrial responses to physiologic and pathophysiologic perturbations. Some of the sequelae of altered CL content have been uncovered in the study of the genetic disease Barth syndrome, where a defect in tafazzin synthesis precludes the production of physiologic amounts of CL mass and molecular species (75–78). Notably, CL remodeling by tafazzin requires the generation of lysocardiolipin for subsequent transacylation/reacylation (44). Through the genetic knock-out of iPLA2γ, it appears that physiologic CL remodeling does not occur effectively, precipitating mitochondrial dysfunction. It is intriguing to note that the phenotype of Barth syndrome includes growth retardation and cardiomyopathy (79 – 81), two features recapitulated in the present study.
Genetic ablation of iPLA2γ results in many readily discernable phenotypic alterations. These stand in sharp contrast to the normal growth and development of cPLA2α and iPLA2β KO mice in which discernable alterations were either limited to the reproductive system or required careful manipulations of specialized cells to identify alterations (82–85). Recent studies have demonstrated the close structural similarity and evolutionary relationship between the active sites of the iPLA2 and cPLA2 families of enzymes (86). The results of this study clearly identify the role of iPLA2γ in mitochondrial bioenergetic function and underscore its importance in cellular energy metabolism and homeostasis. Based on the importance of mitochondrial efficiency in energy metabolism, it is likely that the alterations described in the present work represent only the major and most easily identifiable abnormalities apparent after genetic ablation. Future studies will probably uncover additional roles for iPLA2γ in mammalian biology as our understanding of the roles of phospholipases and their contribution to the regulation of mitochondrial function expands. The iPLA2γ KO mouse should provide a valuable model toward this goal.
Footnotes
This research was supported by National Institutes of Health Grant 5PO1HL57278-10 and by Clinical Nutrition Research Unit Grant DK56351.
The abbreviations used are: LPC, Lysophosphatidylcholine; iPLA2, calcium-independent phospholipase A2; iPLA2γ, calcium-independent phospholipase A2γ (AF263613); KO, knock-out; BEL, (E)-6-(bromomethylene)-3-(1-naphthalenyl)-2H-tetrahydropyran-2-one; TAC, transverse aortic constriction; CL, cardiolipin; cPLA2, cytosolic phospholipase A2; WT, wild type; WAT, white adipose tissue; BAT, brown adipose tissue; UCP, uncoupling protein; nt, nucleotide; TMPD, tetramethyl p-phenylene diamine.
References
- 1.Goldenthal MJ, Marin-Garcia J. Mol Cell Biochem. 2004;262:1–16. doi: 10.1023/b:mcbi.0000038228.85494.3b. [DOI] [PubMed] [Google Scholar]
- 2.Marin-Garcia J, Goldenthal MJ. J Mol Med. 2004;82:565–578. doi: 10.1007/s00109-004-0567-7. [DOI] [PubMed] [Google Scholar]
- 3.Vercesi AE, Kowaltowski AJ, Oliveira HC, Castilho RF. Front Biosci. 2006;11:2554–2564. doi: 10.2741/1990. [DOI] [PubMed] [Google Scholar]
- 4.Garrido C, Galluzzi L, Brunet M, Puig PE, Didelot C, Kroemer G. Cell Death Differ. 2006;13:1423–1433. doi: 10.1038/sj.cdd.4401950. [DOI] [PubMed] [Google Scholar]
- 5.Duchen MR. Diabetes. 2004;53(Suppl 1):96–102. doi: 10.2337/diabetes.53.2007.s96. [DOI] [PubMed] [Google Scholar]
- 6.Kinsey GR, McHowat J, Beckett CS, Schnellmann RG. Am J Physiol. 2007;292:F853–F860. doi: 10.1152/ajprenal.00318.2006. [DOI] [PubMed] [Google Scholar]
- 7.Gadd ME, Broekemeier KM, Crouser ED, Kumar J, Graff G, Pfeiffer DR. J Biol Chem. 2006;281:6931–6939. doi: 10.1074/jbc.M510845200. [DOI] [PubMed] [Google Scholar]
- 8.Mancuso DJ, Jenkins CM, Sims HF, Cohen JM, Yang J, Gross RW. Eur J Biochem. 2004;271:4709–4724. doi: 10.1111/j.1432-1033.2004.04435.x. [DOI] [PubMed] [Google Scholar]
- 9.Williams SD, Gottlieb RA. Biochem J. 2002;362:23–32. doi: 10.1042/0264-6021:3620023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brustovetsky T, Antonsson B, Jemmerson R, Dubinsky JM, Brustovetsky N. J Neurochem. 2005;94:980–994. doi: 10.1111/j.1471-4159.2005.03248.x. [DOI] [PubMed] [Google Scholar]
- 11.Seleznev K, Zhao C, Zhang XH, Song K, Ma ZA. J Biol Chem. 2006;281:22275–22288. doi: 10.1074/jbc.M604330200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yan W, Jenkins CM, Han X, Mancuso DJ, Sims HF, Yang K, Gross RW. J Biol Chem. 2005;280:26669–26679. doi: 10.1074/jbc.M502358200. [DOI] [PubMed] [Google Scholar]
- 13.Su X, Han X, Mancuso DJ, Abendschein DR, Gross RW. Biochemistry. 2005;44:5234–5245. doi: 10.1021/bi047773a. [DOI] [PubMed] [Google Scholar]
- 14.Turk J, Ramanadham S. Can J Physiol Pharmacol. 2004;82:824–832. doi: 10.1139/y04-064. [DOI] [PubMed] [Google Scholar]
- 15.Ramanadham S, Hsu FF, Zhang S, Jin C, Bohrer A, Song H, Bao S, Ma Z, Turk J. Biochemistry. 2004;43:918–930. doi: 10.1021/bi035536m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Z, Ramanadham S, Ma ZA, Bao S, Mancuso DJ, Gross RW, Turk J. J Biol Chem. 2005;280:6840–6849. doi: 10.1074/jbc.M405287200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huss JM, Kelly DP. J Clin Invest. 2005;115:547–555. doi: 10.1172/JCI200524405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han X, Abendschein DR, Kelley JG, Gross RW. Biochem J. 2000;352:79–89. [PMC free article] [PubMed] [Google Scholar]
- 19.Han X, Gross RW. Mass Spectrom Rev. 2005;24:367–412. doi: 10.1002/mas.20023. [DOI] [PubMed] [Google Scholar]
- 20.Han X, Yang J, Yang K, Zhao Z, Abendschein DR, Gross RW. Biochemistry. 2007;46:6417–6428. doi: 10.1021/bi7004015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mingrone G. Ann N Y Acad Sci. 2004;1033:99–107. doi: 10.1196/annals.1320.009. [DOI] [PubMed] [Google Scholar]
- 22.Budnik LT, Mukhopadhyay AK. Biol Reprod. 2002;67:935–944. doi: 10.1095/biolreprod.101.003087. [DOI] [PubMed] [Google Scholar]
- 23.Budnik LT. Reprod Biol Endocrinol. 2003;1:37–38. doi: 10.1186/1477-7827-1-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haber EP, Ximenes HM, Procopio J, Carvalho CR, Curi R, Carpinelli AR. J Cell Physiol. 2003;194:1–12. doi: 10.1002/jcp.10187. [DOI] [PubMed] [Google Scholar]
- 25.MacDonald MJ, Fahien LA, Brown LJ, Hasan NM, Buss JD, Kendrick MA. Am J Physiol. 2005;288:E1–E15. doi: 10.1152/ajpendo.00218.2004. [DOI] [PubMed] [Google Scholar]
- 26.Han X, Yang K, Yang J, Cheng H, Gross RW. J Lipid Res. 2006;47:864–879. doi: 10.1194/jlr.D500044-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Han X, Gross RW. Expert Rev Proteomics. 2005;2:253–264. doi: 10.1586/14789450.2.2.253. [DOI] [PubMed] [Google Scholar]
- 28.Mancuso DJ, Jenkins CM, Gross RW. J Biol Chem. 2000;275:9937–9945. doi: 10.1074/jbc.275.14.9937. [DOI] [PubMed] [Google Scholar]
- 29.Mancuso DJ, Han X, Jenkins CM, Lehman JJ, Sambandam N, Sims HF, Yang J, Yan W, Yang K, Green K, Abendschein DR, Saffitz JE, Gross RW. J Biol Chem. 2007;282:9216–9227. doi: 10.1074/jbc.M607307200. [DOI] [PubMed] [Google Scholar]
- 30.Han X, Cheng H, Mancuso DJ, Gross RW. Biochemistry. 2004;43:15584–15594. doi: 10.1021/bi048307o. [DOI] [PubMed] [Google Scholar]
- 31.Tybulewicz VL, Crawford CE, Jackson PK, Bronson RT, Mulligan RC. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- 32.Leone TC, Lehman JJ, Finck BN, Schaeffer PJ, Wende AR, Boudina S, Courtois M, Wozniak DF, Sambandam N, Bernal-Mizrachi C, Chen Z, Holloszy JO, Medeiros DM, Schmidt RE, Saffitz JE, Abel ED, Semenkovich CF, Kelly DP. PLoS Biol. 2005;3:e101. doi: 10.1371/journal.pbio.0030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rockman HA, Ross RS, Harris AN, Knowlton KU, Steinhelper ME, Field LJ, Ross J, Jr, Chien KR. Proc Natl Acad Sci U S A. 1991;88:8277–8281. doi: 10.1073/pnas.88.18.8277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xing H, Zhang S, Weinheimer C, Kovacs A, Muslin AJ. EMBO J. 2000;19:349–358. doi: 10.1093/emboj/19.3.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ricci JE, Gottlieb RA, Green DR. J Cell Biol. 2003;160:65–75. doi: 10.1083/jcb.200208089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nicholls DG, Ferguson SJ. Bioenergetics. Vol. 3. Academic Press, Inc; London: 2001. [Google Scholar]
- 37.Stewart A, Ghosh M, Spencer DM, Leslie CC. J Biol Chem. 2002;277:29526–29536. doi: 10.1074/jbc.M204856200. [DOI] [PubMed] [Google Scholar]
- 38.Mozo J, Emre Y, Bouillaud F, Ricquier D, Criscuolo F. Biosci Rep. 2005;25:227–249. doi: 10.1007/s10540-005-2887-4. [DOI] [PubMed] [Google Scholar]
- 39.Robinson NC. J Bioenerg Biomembr. 1993;25:153–163. doi: 10.1007/BF00762857. [DOI] [PubMed] [Google Scholar]
- 40.Gomez B, Jr, Robinson NC. Biochemistry. 1999;38:9031–9038. doi: 10.1021/bi990603r. [DOI] [PubMed] [Google Scholar]
- 41.Zhang M, Mileykovskaya E, Dowhan W. J Biol Chem. 2002;277:43553–43556. doi: 10.1074/jbc.C200551200. [DOI] [PubMed] [Google Scholar]
- 42.Tsukihara T, Shimokata K, Katayama Y, Shimada H, Muramoto K, Aoyama H, Mochizuki M, Shinzawa-Itoh K, Yamashita E, Yao M, Ishimura Y, Yoshikawa S. Proc Natl Acad Sci U S A. 2003;100:15304–15309. doi: 10.1073/pnas.2635097100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shinzawa-Itoh K, Aoyama H, Muramoto K, Terada H, Kurauchi T, Tadehara Y, Yamasaki A, Sugimura T, Kurono S, Tsujimoto K, Mizushima T, Yamashita E, Tsukihara T, Yoshikawa S. EMBO J. 2007;26:1713–1725. doi: 10.1038/sj.emboj.7601618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu Y, Malhotra A, Ren M, Schlame M. J Biol Chem. 2006;281:39217–39224. doi: 10.1074/jbc.M606100200. [DOI] [PubMed] [Google Scholar]
- 45.Filosto M, Mancuso M. Acta Neurol Scand. 2007;115:211–221. doi: 10.1111/j.1600-0404.2006.00777.x. [DOI] [PubMed] [Google Scholar]
- 46.Ritz P, Dumas JF, Ducluzeau PH, Simard G. Curr Opin Clin Nutr Metab Care. 2005;8:415–418. doi: 10.1097/01.mco.0000172582.86890.19. [DOI] [PubMed] [Google Scholar]
- 47.Siciliano G, Volpi L, Piazza S, Ricci G, Mancuso M, Murri L. Biosci Rep. 2007;27:53–67. doi: 10.1007/s10540-007-9037-0. [DOI] [PubMed] [Google Scholar]
- 48.McFarland R, Taylor RW, Turnbull DM. Curr Top Dev Biol. 2007;77:113–155. doi: 10.1016/S0070-2153(06)77005-3. [DOI] [PubMed] [Google Scholar]
- 49.Ricquier D. Proc Nutr Soc. 2005;64:47–52. doi: 10.1079/pns2004408. [DOI] [PubMed] [Google Scholar]
- 50.Silva JE. Physiol Rev. 2006;86:435–464. doi: 10.1152/physrev.00009.2005. [DOI] [PubMed] [Google Scholar]
- 51.Klingenspor M. Exp Physiol. 2003;88:141–148. doi: 10.1113/eph8802508. [DOI] [PubMed] [Google Scholar]
- 52.Nedergaard J, Golozoubova V, Matthias A, Shabalina I, Ohba K, Ohlson K, Jacobsson A, Cannon B. Biochem Soc Trans. 2001;29:756–763. doi: 10.1042/0300-5127:0290756. [DOI] [PubMed] [Google Scholar]
- 53.Tolwani RJ, Hamm DA, Tian L, Sharer JD, Vockley J, Rinaldo P, Matern D, Schoeb TR, Wood PA. PLoS Genet. 2005;1:e23. doi: 10.1371/journal.pgen.0010023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Monemdjou S, Hofmann WE, Kozak LP, Harper ME. Am J Physiol. 2000;279:E941–E946. doi: 10.1152/ajpendo.2000.279.4.E941. [DOI] [PubMed] [Google Scholar]
- 55.Gonzalez-Baro MR, Lewin TM, Coleman RA. Am J Physiol. 2007;292:G1195–G1199. doi: 10.1152/ajpgi.00553.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Villena JA, Hock MB, Chang WY, Barcas JE, Giguere V, Kralli A. Proc Natl Acad Sci U S A. 2007;104:1418–1423. doi: 10.1073/pnas.0607696104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sonoda J, Mehl IR, Chong LW, Nofsinger RR, Evans RM. Proc Natl Acad Sci U S A. 2007;104:5223–5228. doi: 10.1073/pnas.0611623104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lelliott CJ, Medina-Gomez G, Petrovic N, Kis A, Feldmann HM, Bjursell M, Parker N, Curtis K, Campbell M, Hu P, Zhang D, Litwin SE, Zaha VG, Fountain KT, Boudina S, Jimenez-Linan M, Blount M, Lopez M, Meirhaeghe A, Bohlooly YM, Storlien L, Stromstedt M, Snaith M, Oresic M, Abel ED, Cannon B, Vidal-Puig A. PLoS Biol. 2006;4:e369. doi: 10.1371/journal.pbio.0040369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wallace DC. Methods Mol Biol. 2002;197:3–54. doi: 10.1385/1-59259-284-8:003. [DOI] [PubMed] [Google Scholar]
- 60.Wallace DC. Am J Med Genet. 2001;106:71–93. doi: 10.1002/ajmg.1393. [DOI] [PubMed] [Google Scholar]
- 61.Wallace DC. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wallace DC. Ment Retard Dev Disabil Res Rev. 2001;7:158–166. doi: 10.1002/mrdd.1023. [DOI] [PubMed] [Google Scholar]
- 63.Chen JL, Peacock E, Samady W, Turner SM, Neese RA, Hellerstein MK, Murphy EJ. J Biol Chem. 2005;280:25396–25402. doi: 10.1074/jbc.M413948200. [DOI] [PubMed] [Google Scholar]
- 64.Heath-Engel HM, Shore GC. Biochim Biophys Acta. 2006;1763:549–560. doi: 10.1016/j.bbamcr.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 65.McBride HM, Neuspiel M, Wasiak S. Curr Biol. 2006;16:R551–560. doi: 10.1016/j.cub.2006.06.054. [DOI] [PubMed] [Google Scholar]
- 66.Siegel DP. Biophys J. 1984;45:399–420. doi: 10.1016/S0006-3495(84)84164-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dahlberg M. J Phys Chem B. 2007;111:7194–7200. doi: 10.1021/jp071954f. [DOI] [PubMed] [Google Scholar]
- 68.Skulachev VP. FEBS Lett. 1991;294:158–162. doi: 10.1016/0014-5793(91)80658-p. [DOI] [PubMed] [Google Scholar]
- 69.Garlid KD, Orosz DE, Modriansky M, Vassanelli S, Jezek P. J Biol Chem. 1996;271:2615–2620. doi: 10.1074/jbc.271.5.2615. [DOI] [PubMed] [Google Scholar]
- 70.Breen EP, Gouin SG, Murphy AF, Haines LR, Jackson AM, Pearson TW, Murphy PV, Porter RK. J Biol Chem. 2006;281:2114–2119. doi: 10.1074/jbc.M511575200. [DOI] [PubMed] [Google Scholar]
- 71.Porter RK. Biochim Biophys Acta. 2006;1757:446–448. doi: 10.1016/j.bbabio.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 72.Di Marzo V, De Petrocellis L, Sugiura T, Waku K. Biochem Biophys Res Commun. 1996;227:281–288. doi: 10.1006/bbrc.1996.1501. [DOI] [PubMed] [Google Scholar]
- 73.Pete MJ, Exton JH. J Biol Chem. 1996;271:18114–18121. doi: 10.1074/jbc.271.30.18114. [DOI] [PubMed] [Google Scholar]
- 74.Pinheiro TJ, Watts A. Biochemistry. 1994;33:2459–2467. doi: 10.1021/bi00175a014. [DOI] [PubMed] [Google Scholar]
- 75.Vreken P, Valianpour F, Nijtmans LG, Grivell LA, Plecko B, Wanders RJ, Barth PG. Biochem Biophys Res Commun. 2000;279:378–382. doi: 10.1006/bbrc.2000.3952. [DOI] [PubMed] [Google Scholar]
- 76.Schlame M, Towbin JA, Heerdt PM, Jehle R, DiMauro S, Blanck TJ. Ann Neurol. 2002;51:634–637. doi: 10.1002/ana.10176. [DOI] [PubMed] [Google Scholar]
- 77.Xu Y, Kelley RI, Blanck TJ, Schlame M. J Biol Chem. 2003;278:51380–51385. doi: 10.1074/jbc.M307382200. [DOI] [PubMed] [Google Scholar]
- 78.Schlame M, Ren M, Xu Y, Greenberg ML, Haller I. Chem Phys Lipids. 2005;138:38–49. doi: 10.1016/j.chemphyslip.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 79.Spencer CT, Bryant RM, Day J, Gonzalez IL, Colan SD, Thompson WR, Berthy J, Redfearn SP, Byrne BJ. Pediatrics. 2006;118:e337–346. doi: 10.1542/peds.2005-2667. [DOI] [PubMed] [Google Scholar]
- 80.Bolhuis PA, Hensels GW, Hulsebos TJ, Baas F, Barth PG. Am J Hum Genet. 1991;48:481–485. [PMC free article] [PubMed] [Google Scholar]
- 81.Schlame M, Ren M. FEBS Lett. 2006;580:5450–5455. doi: 10.1016/j.febslet.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 82.Bonventre JV, Huang Z, Taheri MR, O’Leary E, Li E, Moskowitz MA, Sapirstein A. Nature. 1997;390:622–625. doi: 10.1038/37635. [DOI] [PubMed] [Google Scholar]
- 83.Fujishima H, Sanchez Mejia RO, Bingham CO, III, Lam BK, Sapirstein A, Bonventre JV, Austen KF, Arm JP. Proc Natl Acad Sci U S A. 1999;96:4803–4807. doi: 10.1073/pnas.96.9.4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rosenberger TA, Villacreses NE, Contreras MA, Bonventre JV, Rapoport SI. J Lipid Res. 2003;44:109–117. doi: 10.1194/jlr.m200298-jlr200. [DOI] [PubMed] [Google Scholar]
- 85.Bao S, Miller DJ, Ma Z, Wohltmann M, Eng G, Ramanadham S, Moley K, Turk J. J Biol Chem. 2004;279:38194–38200. doi: 10.1074/jbc.M406489200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wilson PA, Gardner SD, Lambie NM, Commans SA, Crowther DJ. J Lipid Res. 2006;47:1940–1949. doi: 10.1194/jlr.M600185-JLR200. [DOI] [PubMed] [Google Scholar]