

Abstract
The brain cannabinoid (CB1) receptor that mediates numerous physiological processes in response to marijuana and other psychoactive compounds is a G protein coupled receptor (GPCR) and shares common structural features with many rhodopsin class GPCRs. For the rational development of therapeutic agents targeting the CB1 receptor, understanding of the ligand-specific CB1 receptor interactions responsible for unique G protein signals is crucial. For a more than a decade, a combination of mutagenesis and computational modeling approaches has been successfully employed to study the ligand-specific CB1 receptor interactions. In this review, after a brief discussion about recent advances in understanding of some structural and functional features of GPCRs commonly applicable to the CB1 receptor, the CB1 receptor functional residues reported from mutational studies are divided into three different types, ligand binding (B), receptor stabilization (S) and receptor activation (A) residues, to delineate the nature of the binding pockets of anandamide, CP55940, WIN55212-2 and SR141716A and to describe the molecular events of the ligand-specific CB1 receptor activation from ligand binding to G protein signaling. Taken these CB1 receptor functional residues, some of which are unique to the CB1 receptor, together with the biophysical knowledge accumulated for the GPCR active state, it is possible to propose the early stages of the CB1 receptor activation process that not only provide some insights into understanding molecular mechanisms of receptor activation but also are applicable for identifying new therapeutic agents by applying the validated structure-based approaches, such as virtual high throughput screening (HTS) and fragment-based approach (FBA).
Keywords: G protein coupled receptor (GPCR), the brain cannabinoid (CB1) receptor, functional residues, mechanism of receptor activation, structure-based drug design
Introduction
G-protein coupled receptors (GPCRs), composed of seven transmembrane (TM) spanning helices (H1-H7) interconnected by three intracellular loops (I1-I3) and three extracellular loops (E1-E3) [1,2], are known to be among the most important drug targets [3,4] (for reviews, see [5-7]). For the rational development of therapeutic agents targeting a specific GPCR, it is crucial to understand the ligand-receptor interactions that determine the effectiveness of the ligand for modulating receptor activity toward unique G protein signals. If the ligand binding affinity is defined by how strongly a ligand binds to a receptor and the receptor efficacy is defined by how efficiently a receptor activates the coupled G protein, a ligand as a drug candidate should exhibit high binding affinity and more importantly desirable binding efficacy. Thus, it should be at the center of the rational development of GPCR drugs to understand the ligand-initiated receptor conformational change responsible for GPCR signaling [8,9].
Understanding of binding efficacy in GPCRs is challenging due to the conformational complexity of GPCRs [10,11]. Even in the absence of ligands GPCRs exhibit basal activity, suggesting that GPCRs are in motion with inherent conformational flexibility. Conformational equilibrium of a GPCR between the inactive state and the active state can be modified by ligand binding [11]. By definition, agonists activate the receptor and produce signaling activity, inverse agonists stabilize the receptor and inhibit basal activity, and antagonists block the receptor and produce no activity: Binding of an agonist or a partial agonist would lower the energy barrier from the inactive state to the active state and/or stabilize the active state, shifting the equilibrium toward the active state, while binding of an inverse agonist would enhance the energy barrier and/or stabilize the inactive state, shifting the equilibrium toward the inactive state. Not only ligand binding but also coupling to cognate G proteins would modify the equilibrium between the inactive state and the active state [11]. In addition to ligands or G proteins that contribute to receptor stability, altering receptor residues involved in receptor activation (e.g., constitutively active mutations (CAMs) that increase the basal activity of the receptor in the absence of a ligand (for reviews, see [12-14]) is an alternative way to modify the equilibrium toward the active state.
It should be noted that structurally diverse ligand classes of a GPCR can induce the ligand-specific conformational changes in the receptor, determining different receptor states that are capable of activating specific subtypes of cognate G protein, as proposed in the protein ensemble theory describing proteins as a collection of conformational states [15]. In fact, evidence from many biophysical studies supports the existence of multiple, ligand-specific conformational states of GPCRs [16-19]. In addition to ligand-specific receptor conformations, it has been indicated that the agonist bound GPCRs can have multiple, state-specific conformations corresponding to different signaling states [20,21]. For example, it is known that two distinct forms of the photoactivated rhodopsin, MII and MIIa/MIIb have been reported [22-24] and that the agonist-bound β2 adrenergic receptor (β2AR) exists in at least two different conformational states [25,26].
To date, the X-ray structures available include rhodopsin [27-37], β1AR [38], β2AR [39-41], and the adenosine A2A receptor (AA2AR) [42]. In spite of the overall same topology, these X-ray structures show several interesting differences in local regions [43], as illustrated in the comparison of β2AR with rhodopsin: 1) H1 of rhodopsin is packed closely to the helical bundle because of a kink in the middle of the helix, while H1 of β2AR is loosely packed to the helical bundle without the equivalent kink; 2) The N-terminal of rhodopsin is ordered with β-sheets stacked on the top of E2 covering the ligand binding pocket, while that of β2AR is disordered; 3) The binding pocket in rhodopsin with a β-sheet alignment of E2 is totally blocked, while that in β2AR with an α-helical segment within E2 is only partially blocked for easy solvent or ligand access [40]; 4) The X-ray structure of rhodopsin [46] represents the inactive state, while that of β2AR-T4L fusion protein [40] represents an active-like state [44,45].
It appears that a receptor conformational change suitable for G protein activation does not require a dramatic change in the whole receptor but a change in the cytoplasmic side. For example, the X-ray structure of β2AR-T4L fusion protein [40], an active-like state, shows little difference from the X-ray structure of rhodopsin (inactive state) [46]. Similarly, compared with the inactive form of rhodopsin [46], the photoactivated state metarhodopsin II (MII) [32] showed surprisingly little change, except an increased disorder in I3. This result suggests that photoactivation results in I3 flexibility. Evidence not only from the X-ray structures of GPCRs but also from many biophysical studies supports the conformational change in the intracellular side [47-51], including the breakage of the salt bridge between R3.50 and E(D)6.30 of the receptor, proposed as the ionic lock [52], that maintains the inactive state of the rhodopsin class GPCRs mainly through H6 stabilization but needs to be cleaved upon receptor activation. Elling et al. [50] proposed a global toggle switch activation model, where upon activation the receptor changes conformation such that the intracellular segments of H6/H7 move outward and the highly conserved TM Pro in the middle of H6 and H7 act as the pivot for the vertical seesaw movements. (In this review, a numbering system similar to Ballesteros-Weinstein system [53] is used for all the amino acids. For example, R1353.50 represents Arg135 with the highest conservation, indicated by the number 50, in the TM helix 3, indicated by the number 3. Similarly, V173E2 represents Val 173 of the second extracellular loop.)
All the X-ray structures are the antagonist or inverse agonist bound forms: for rhodopsin, retinal (inverse agonist) [28]; for β2AR, carazolol (partial inverse agonist) [40]; for β1AR, cyanopindolol (antagonist) [38]; and for AA2AR, ZM241385 (antagonist) [43]. In considering the roles of the antagonist in blocking the receptor and the inverse agonist in stabilizing the inactive form of the receptor, it would be natural to have the X-ray structures of antagonist or inverse agonist bound GPCRs compared to those with agonist bound GPCRs, which is short-lived for G protein signaling. It has been argued that the X-ray structures of GPCRs are unsuitable for screening partial or full agonists [54-56]. Thus, if the screening of novel compounds is directed by using a receptor structure that is from its complex with an antagonist or inverse agonist, one may end up with antagonists or inverse agonists rather than with agonists. For example, Kolb et al., [57] used the X-ray structure of β2AR to dock approximately 1 million commercially available small molecules and identified some compounds with relatively high affinities, most of which showed inverse agonist activity, suggesting that the receptor conformation of the reported X-ray structure of β2AR is adapted to mostly recognize the inverse agonist.
Thus, it seems apparent that the receptor structure of the agonist-bound form, at least, is necessary for drug design of agonists, though without knowing the protein structure of the high-affinity state GPCRs in complex with a G-protein [58,59] the picture is incomplete to fully understand the receptor in its active state to which an agonist preferentially binds. However, the agonist bound GPCR structures, are extremely difficult to obtain [56]. Theoretically it is possible to obtain the receptor in its active state by starting from the inactive state of the receptor along the reaction path allowed for this conversion. However, the current state of the art MD simulations reported in the microsecond time scale [60,61] are not sufficient to provide the molecular details of the active state of a GPCR that would form in the millisecond scale [44]. In addition, this path, largely unknown, contains multiple binding motifs for diverse ligands and multiple active conformers of the receptor. In this regard, recent studies [62-71] reported possible early events along this path from the inactive state toward the active state.
The Cannabinoid Receptors Belong to Gpcrs
Brain CB1 cannabinoid receptors [72] are GPCRs and belong to the rhodopsin-like subfamily [2]. The CB1 receptor is coupled to G-proteins for signal transduction pathways that inhibit adenylyl cyclase activity and regulate ion channels [72-76]. As shown in Fig. (1), the sequence alignment of the CB1 receptor with some GPCRs whose X-ray structures are available indicates that they share common features, including the seven TM helices and the highly conserved functional motifs, suggesting that the CB1 receptor has a similar molecular mechanism for receptor activation as for other rhodopsin class GPCRs. Based upon the phylogenetic study by Joost and Methner [77], where 241 human GPCRs were divided into 19 subgroups (A1 through A19), it was shown that the CB1 receptor belonged to subgroup A13 next to subgroup A17 from which AA2AR and βARs were branched and that these receptors altogether belonged to a larger cluster different from subgroup A16 to which rhodopsin belonged. Similarly, Fredriksson et al. [78] performed phylogenetic analyses of 342 unique functional non-olfactory human GPCRs to obtain five main families, including glutamate, rhodopsin (α, β, γ, and δ groups), adhesion, frizzled/taste2, and secretin. Further, the α-group of the rhodopsin family was classified into several distinct clusters, including the prostaglandin cluster, the amine receptor cluster, the opsin receptor cluster, the melatonin receptor cluster and the MECA (melanocortin, endothelial differentiation, cannabinoid and adenosine binding) receptor cluster to which cannabinoid receptors belong. Interestingly, both phylogenetic analyses [77,78] revealed that some of the closest receptors to the cannabinoid receptors are the melanocortin (MC) receptors. Both receptors, commonly lacking the conserved disulfide linkage between H3 and E2, common to most rhodopsin class GPCRs [79,80] and important for receptor structure and activity [81], exhibit a high degree of basal activity [82,83], suggesting a role of the flexibility of E2 in basal activity. Interestingly, the MC-4 receptor (MC4R), which exhibits the highest sequence homology to the CB receptors (approximately 80 %) among the MC receptors, is known to be associated with 6 % of early onset obesity [84], indicating their common roles in regulating obesity. In addition to the traditional cannabinoid receptors by which agonist activation is mediated through Gi/o protein [85], a novel putative cannabinoid receptor GPR55 has recently been characterized [86,87]. According to the GPCR classification by Fredriksson et al. [78], GPR55 belonged to the δ-group of the rhodopsin family within the purine receptor cluster, which is different from the α-group to which the cannabinoid receptors belong.
Fig. (1).

Sequence alignment by T-COFFEE (V7.71, mode: expresso) (http://www.tcoffee.org) [211] of CB1 and CB2 with the GPCRs whose structures have been determined by the X-ray crystallography, including AA2AR (PDB code: 3EML) [42], β1AR (PDB code: 2VT4) [38], β2AR (PDB code: 2RH1) [40], and rhodopsin (PDB code: 1U19) [46]. The TM helical boundaries for the CB1 and CB2 receptors are from the respective homology models [136,182], while the TM helical boundaries for AA2AR, β1AR, β2AR, and rhodopsin are from the respective X-ray structures. Conservancy of the aligned sequence by CLUSTALW (http://www.ebi.ac.uk/Tools/clustalw/) [212] is represented by consensus symbols: “*” for identical residues; “:” for conserved substitutions; and “.” for semi-conserved substitutions. Highly conserved residues in the rhodopsin family of GPCRs reported by Baldwin et al (1997) are in bold. Color codes for TM helices: H1 (in red); H2 (in orange); H3 (in yellow); H4 (in green); H5 (in cyan); H6 (in blue); H7 (in purple); and H8 (in dark green).
Three Types of Cannabinoid Receptor Functional Residues
Mutagenesis data for the cannabinoid receptors have been accumulated for more than a decade. The mutagenesis analysis, often combined with the computational modeling analysis, provides invaluable insights into characterizing the role of the CB1 receptor residues in ligand binding and receptor activity. However, it is not always straightforward to interpret the effects of the mutated residue due to the conformational complexity inherent to GPCRs [11] as described earlier. This is especially true for the case of the cannabinoid receptors that exhibit a high level of basal activity [82,88-90]. Thus, Picone et al. [91] writes, “Introduction of a point mutation within a receptor binding site has the potential of altering ligand binding properties. However, it could also confer undesirable and frequently unpredictable consequences on the global conformation of the receptor that may affect ligand binding in a manner unrelated to binding site structure.” In the same context, Beukers and Ijzerman [92] describe, “By the technique of site-directed mutagenesis by which point mutations are introduced, the role of specific residues in receptor structure and function can be easily studied. However, careful analysis of mutagenesis data is required because the replacement of an amino acid can result in a local effect as a result of a gain or loss of interaction with neighboring residues or in a global effect such as an alteration of protein folding or protein stability”.
In this review, I divide the CB1 receptor functional residues into three different types, ligand binding (B), receptor stabilization (S) and receptor activation (A) residues, according to their roles as suggested from the mutational studies. Effects of the mutations of these residues are schematically shown in Fig. (2). It is interesting to note that all these residues can contribute to receptor activation. It should be noted that it is often difficult to define the function of a residue as one specific type over the others due to the fact that their roles are often closely associated with each other.
Fig. (2).

The functional residues of the CB1 receptor are divided into three different types, receptor stabilization (S) (in red circle), ligand binding (B) (in blue circle) and receptor activation (A) (in yellow circle). The partial overlaps of the circles indicate those residues with more than one function. Among the residues that affect ligand binding, the type Bd residues directly contact with the ligand, while the types BiS and BiA residues indirectly alter the ligand binding site geometry (see text). Color code of mutational effects: upon the receptor structure (in magenta); upon ligand binding (in cyan); and signaling (in orange).
The type Bd residues form the binding pocket and directly contact with the ligand (Fig. (2)). Thus, mutations of the type Bd residues alter binding site structure and affect ligand binding by disrupting specific ligand interactions. This type of residues are useful not only for describing the binding pockets distinct to the individual classes of ligands but also for delineating the early stages of receptor activation (see below).
The type S residues stabilize the receptor structure [93] (Fig. (2)). Mutations of this type of residues may cause the failure of receptor expression due to misfolding and abolish the ligand binding affinity and receptor activation. An example of the type S residue is illustrated by W4.64 (i.e., the Trp residue at the 4.64 position [53]) of the cannabinoid receptors that is known to be critical for ligand binding and signaling [94]. It was reported that the mutation of W2554.64 of the CB1 receptor resulted in the failure of receptor expression [95,96], suggesting that W4.64 is important for receptor structure. Combining these results with the finding that the highly conserved W4.64 in many GPCRs is known to be important for receptor folding [97], W2554.64 of the CB1 receptor is a type S residue important for receptor structure.
If a type S residue affects ligand binding by indirectly modifying the binding pocket topology, it can be further classified into BiS residues important for the binding of inverse agonists that stabilize the receptor. Thus, mutations of the type BiS residues result in alteration in ligand binding and impair ligand binding and receptor activation (Fig. (2)). In fact, many residues reported to be important for ligand binding belong to this type of residues (see below).
The type A residues play an important role in receptor activation by being directly involved in receptor activation (Fig. (2)). Thus, mutations of the type A residues directly alter G protein signaling. Examples of the type A residues are seen in CAMs which decrease the inverse agonist binding and increase basal activity [13,98]. Another example is seen from those residues associated with the D(E)RY motif known to be crucial for G-protein activation [99], by the formation of the ionic lock [52], without participating in ligand binding. Thus, Song and his colleagues [100] reported that D6.30N mutation of the CB receptors maintained the ligand binding affinity but exhibited the greatly reduced signaling activity, suggesting that these residues are the type A residues.
If the type A residues indirectly affect ligand binding through modifying the binding pocket topology, they can be further classified into BiA (Fig. (2)). An example of the type BiA residue was illustrated by Song and Feng [101] in their site-directed mutagenesis study showing that Y2095.58A mutation of the peripheral cannabinoid (CB2) receptor [102] reduced the binding affinity of 2,3-dihydro-5-methyl-3-[(4-morpholinyl)methyl]pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl] (1-naphthyl)methanone (WIN55212-2), (6aR,10aR)-9-(Hydroxymethyl)-6,6-dimethyl-3-(2-methyloctan-2-yl)-6a,7,10,10a-tetrahydrobenzo[c]chromen-1-ol (HU210) and N-arachidonoylethanolamine (anandamide) by 5- to 8-fold but abolished signaling activity. Although this study was done on the CB2 receptor, considering that Y5.58 is important for breaking the ionic lock [103] and located deep down the core and not involved directly in ligand binding, it is likely that the conserved Y2945.58 of the CB1 receptor is a type BiA residue for cannabinoid agonists.
It was shown that the L3416.33A/A3426.34L double mutation of the CB1 receptor exhibited decreased (1R,3R,4R)-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-4-(3-hydroxypropyl)cyclo-hexan-1-ol (CP55940) binding by 4-fold an partial constitutive activation (not Gi but Gs) [159] and that D1303.49A, R1313.50A and A2446.34E mutations of the CB2 receptor abolished agonist-induced G-protein activation with retaining the binding affinity of anandamide, HU210 and WIN55212-2 only by R1313.50A mutation [104]. Considering all these residues are conserved in both the CB1 and CB2 receptors (Fig. (1)), it is likely that R1313.50 is a type A residue without interfering in ligand binding, while D2133.49 and A3426.34 of the CB1 receptor are BiA residues for anandamide, CP55940 and WIN55212-2.
It was shown that T2103.46I and L2073.43A mutations of the CB1 receptor increased the binding affinity of methanandamide, a synthetic analog of anandamide known to be metabolically stable, by 6-fold and 3-fold, respectively [105,106]. It was shown that these mutations increased CP55940 binding affinity by 3-fold, decreased WIN55212-2 binding by 3-fold and decreased N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (SR141716A) binding by about 30-fold, suggesting that these residues are CAMs. With the location of these residues near the DRY motif far away from the binding site, these residues are less likely to directly interact with the ligand but play an important role in regulating the receptor conformation in association with G protein activation. Thus, it is likely that they are BiA residues for anandamide, CP55940, WIN55212-2 and SR141716A.
CB1 Receptor Residues Important for Anandamide Binding
It was shown from a recent study combining computational and mutational analyses that anandamide exhibited an approximately 13-fold decrease in binding affinity by the Y2755.39F mutation of the CB1 receptor and that the Y2755.39I mutation abolished ligand binding and receptor signaling [107]. These results suggest that Y2755.39 is important for anandamide binding, possibly through H-bonding. The authors concluded that the Y2755.39I mutation altered ligand interactions within the binding site. Thus, it is likely that Y2755.39 of the CB1 receptor is a Bd residue for anandamide. However, it is possible that the Y2755.39I mutation may cause a global effect [92] resulting in a significant modification in the ligand binding pocket. In support, it was reported that Y2755.39 of the CB1 receptor and Y1915.39 of the CB2 receptor were not direct ligand contact sites [108,109] and that the Y2755.39S and Y2755.39A mutations resulted in failure of receptor expression [110].
No mutations of F2003.36A, W2795.43A and W3566.48A, but F1893.25A moderately (approximately 6-fold) decreased anandamide binding affinity [95]. Illustrated by an anandamide docking model developed from these obtained mutation data, the C5-C6 double bond of the ligand interacted with F1893.25 via aromatic/π interaction [95]. These results indicate that F1893.25 is a Bd residue for anandamide.
CB1 Receptor Residues Important for CP55940 Binding
McAllister et al. [107] showed that CP55940 binding to the CB1 receptor was retained by the Y5.39F mutation but abolished by the Y5.39I mutation, suggesting that the aromaticity of the residue at the 5.39 position is crucial for CP55940 binding. The authors concluded that altered interactions caused by the Y5 39I mutation resulted in alteration in ligand binding. Thus, it is likely that Y2755.39 of the CB1 receptor is a Bd residue for CP55940.
It was shown that the Ala mutations of the E1 residues (H181, R182, K183 and D184) and the H3 proximal residues (V1883.24 and F1893.25) resulted in reduced binding affinities of CP55940, but not of SR141716A [111]. With decreases in CP55940 binding less than expected by direct ligand contact residues, the results suggest that the effects are not due to direct contact with the ligand but due to loop structural alteration that indirectly affects ligand binding through modifying the binding pocket topology. Thus, these residues on the extacellular side of the receptor are BiS residues for CP55940. Interestingly, for the binding affinity of CP55940 by the F1893.25A mutation of the CB1 receptor, Murphy and Kendall [111] reported an approximately 60-fold decrease, but McAllister et al. [112] reported a 3-fold decrease.
It has been shown from a combined study of affinity labeling, site-directed mutagenesis, and ligand docking studies [91] that a classical cannabinoid derivative having the reactive electrophilic isothiocyanate moiety at the end of the C3 side chain formed a covalent bond to the nucleophilic C3556.47 and retained the binding affinities to the C3556.47A and C3556.47S mutants but had a reduced affinity to the C3556.47L mutant. No change in WIN55212-2 binding affinity for any of mutant receptors was shown. These results suggest that C3556.47 of the CB1 receptor is part of the binding site of the classical and non-classical cannabinoids, distinct from that of WIN55212-2, and is involved in receptor activation as part of the highly conserved functional CWxP motif [112,113]. Thus, C3556.47 of the CB1 receptor is a Bd residue for CP55940.
In contrast to the profound effect the S3837.39A mutation of the CB1 receptor produced upon CP55940 binding, the binding of WIN55212-2 and SR141716A was unchanged by this mutation [114]. Rhee observed that the S2857.39A mutation of the CB2 receptor resulted in dropping HU243 binding approximately 13-fold [94], suggesting that the conserved S7.39 of the CB receptors are involved in CP55940 binding. Demonstrating by computational modeling studies that the S3837.39A mutation reduced the H7 kink, Kapur et al. [114] proposed that S3837.39 induces a bend in the extracellular side of H7 required for CP55940 binding. It is interesting to note that the mutation of the equivalent residue A2927.39 of rhodopsin resulted in a constitutively active receptor [115]. Taken together, it is likely that S3837.39 is a BiA residue for CP55940.
In a very recent Ala scanning mutagenesis study, Kendall and her colleagues [96] comprehensively analyzed the E2 of the CB1 receptor and demonstrated that the Ala mutations of both the N-terminal and C-terminal residues abolished CP55940 binding and G protein signaling. Interestingly, Ala mutations of P269E2, H270E2 and I271E2 were insensitive for SR141716A binding. As shown from the accompanied homology molecular model, these hydrophobic residues were deeply inserted into the core and formed part of the ligand binding site, suggesting that they are CP55940-specific residues. Together, W255E2 and N256E2 are S residues, while F268E2, P269E2, H270E2 and I271E2 are Bd residues for CP55940.
CB1 Receptor Residues Important for WIN55212-2 Binding
From a mutagenesis study [116], it was shown that the CB1 D1632.50N mutation reduced WIN55212-2 binding by approximately 400-fold, but not for other cannabinoid ligands. It was also shown that the mutated receptor exhibited greatly attenuated inhibition of cyclic AMP production by both CP55940 and WIN55212-2. These results strongly suggest that D1632.50, a highly conserved residue in many GPCRs, is important in the CB1 receptor for WIN55212-2 binding and receptor activation. As the authors discussed, the D1632.50N mutation may cause the receptor to alter the binding pocket uniquely defined for WIN55212-2. Thus, it is likely that D1632.50, without directly interacting with the ligand, is a BiA residue for WIN55212-2. However, it should be noted that Mackie and his colleagues, in contrast, reported that D1642.50 in the rat CB1 receptor had no effect on ligand binding or inhibition of cyclic AMP production but did alter signaling via Gi by WIN55212-2 [117].
It was shown from the CB1 chimeric receptor formed by the replacement of H3 with that of the CB2 receptor [118] that H3 residues were important for WIN55212-2 binding and that the G1953.31S mutated CB1 receptor exhibited an enhanced WIN55212-2 binding by about 5-fold. The authors proposed that the enhanced WIN55212-2 binding affinity was attributed to H-bonding between the carboxyl oxygen of the ligand and S3.31 of the mutated receptor, based upon the report that AAI analogs lacking the carboxyl oxygen displayed a 7- to 10-fold decrease in binding affinity [119]. Thus, it is likely that G1953.31 of the CB1 receptor is a Bd residue for WIN55212-2.
It was shown from a study combining molecular modeling and site-directed mutagenesis approaches [108] that the V2825.46F mutation of the CB1 receptor enhanced the WIN55212-2 binding affinity by 12-fold; this mutation did not change the binding affinity of cannabinoid compounds and anandamide, suggesting that V2825.46 is a Bd residue for WIN55212-2 binding [95].
McAllister et al. [107] showed that WIN55212-2 binding was retained by the Y2755.39F mutation of the CB1 receptor but abolished by the Y2755.39 I mutation, suggesting that the aromaticity of the residue at the 5.39 position is crucial for WIN55212-2 binding. Thus, it is likely that Y2755.39 of the CB1 receptor is a Bd residue for WIN55212-2.
It was shown that the F2003.36A and W2795.43A mutations of the CB1 receptor reduced the binding of WIN55212-2 by 9-fold and 16-fold, respectively, but did not affect CP55940 binding [95], leading to proposing the importance of an aromatic cluster within the CB1 receptor aromatic microdomain formed by H3-H4-H5-H6 for WIN55212-2. Similarly, the F2003.36A mutation of CB1 showed a moderate decrease in WIN55212-2 binding but no change in CP55940 binding [120]. Thus, these residues are Bd residues for WIN55212-2.
CB1 Receptor Residues Important for SR141716A Binding
It was shown that the W2795.43A or W3566.48A mutation of the CB1 receptor reduced significantly the binding of SR141716A [95], suggesting the important of these residues for the binding of SR141716A. Thus, these residues are type Bd residues for SR141716A.
It was shown that the mutation of E2 Cys residues C257E2 and C264E2 to Ala abolished the binding affinity of SR141716A [121], implying a significant conformational change, possibly due to breaking the intra-loop disulfide bond and a modification of the ligand binding pocket. In the same study, it was also shown that the introduction of a bulky group on C3867.42 inhibited SR141716A binding [121], suggesting that C3867.42 directly interacts with SR141716A. Thus, C257 and C264 are BiS residues, while C3867.42 is a Bd residue for SR141716A.
A Role of K1923.28 in Ligand Binding
From site-directed mutagenesis studies of the human CB1 receptor [122,123], it was shown that the K1923.28A mutation resulted in a complete loss of binding and a significant reduction in receptor activity for HU210, CP55940 and anandamide, but retained binding and receptor activity for WIN55212-2. It was shown that a similar binding affinity decrease occurred (17-fold) for SR141716A in the K1923.28A mutant as was seen (14-fold) for a SR141716 analog lacking the C3 carboxamide oxygen binding in the wild type receptor [124,125]. Thus, the current understanding of ligand binding to the CB1 receptor is heavily dependent upon the generally accepted role of K1923.28 as one of the key residues that directly interact with CP55940 [122,126] and SR141716A [124,125] but not with WIN55212-2. Thus, most of the computational docking studies of the CB1 receptor [91,95,114,128-133] employed K1923.28 as the primary interaction site for the phenolic OH of the cannabinoid ligands through H-bonding.
However, as opposed to the generally accepted hypothesis that K1923.28 residue directly contacts with the ligand (i.e., as a Bd residue), there have been a number of findings indicative of a role of K1923.28 in indirectly modifying the binding pocket geometry (i.e., as a BiS residue). First, the effect of the K1923.28A mutation of the CB1 receptor on ligand binding was, in fact, universal to all the ligands: anandamide and CP55244 were very sensitive and exhibited no binding, SR141716A binding was moderately sensitive to exhibit a 17-fold decrease [124], and WIN55212-2 was less sensitive and exhibited about 2-fold decrease [122,123]. This can be interpreted as follows: K1923.28A mutation leads to an indirect modification of the ligand binding pocket geometry such that the binding of anandamide and CP55244 is more severely disrupted than the binding of WIN55212-2. Second, it was observed from the mutation study by Chin et al. [123] that CP55940 showed no difference in binding affinity for the wild type and the K1923.28R CB1 receptor. This finding suggests that K1923.28 is not a Bd residue for CP55940, for if K1923.28 directly interacts with CP55940 the binding should be less favored by the bulkier replacement. Third, the removal of the phenolic OH of CP55940 showed only a 35-fold drop in ligand binding but a 200-fold decrease in receptor activity [134]. This drop in ligand binding is much lower than expected [135] for deletion of the direct H-bond between the charged N of K1923.28 and the phenolic OH of the ligand. Fourth, according to a recent CB1 receptor homology model embedded in a lipid bilayer [136], it was shown that K1923.28, as the charged form at physiological conditions, was snorkeling [137-139] by stretching out its long side chain N atom for the formation of a strong salt bridge with D184E1 near the polar membrane surface, while continuing to surround the side chain hydrocarbon moiety with several hydrophobic residues from H2 and H3 inside the lipophilic membrane. It is conceivable, however, that K1923.28 can directly interact with the ligand if the ligand contains a suitable functional group, such as a carboxylate or phenoxy moiety, or a π-electron rich moiety (i.e., an aromatic ring) that satisfies the positively charged N side chain of K1923.28 in the hydrophobic lipid environment by forming a H-bond or π-cation interaction [140,141] by the replacement of the existing salt bridge with D184E1.
Taken altogether, it is possible that K1923.28 can play roles in ligand binding not by directly contacting with the ligand but by indirectly modifying the binding pocket geometry. With its role remaining to be further confirmed for a better understanding of the CB1 receptor-ligand interaction, which is crucial not only for studying receptor activation but also for developing CB1 receptor structure-based drugs, K1923.28 is considered in this review as a BiS residue for the CB1 receptor ligands.
Emerging Binding Sites for Structurally Diverse Cannabinoid Ligands
The functional residues of the CB1 receptor involved in ligand binding (Bd,BiS and BiA) are listed in Table 1. Based upon the CB1 receptor residues defined as the types Bd that are directly involved in contacting with the ligand, the binding pockets of anandamide, CP55940, WIN55212-2 and SR141716A can be approximately delineated. Among these residues identified as key binding site residues, it is interesting to note that F1893.25 [95] is uniquely defined for anandamide, F268E2-I271E2 [96] and C3556.47 [91] for CP55940, G1953.31 [118] and V2825.46 [108] for WIN55212-2, and W3566.48 and C3867.42 [121] for SR141716A (Table 1 and Fig. (3)). These results indicate that, with some overlap in common structural features for CB1 receptor binding, each class of structurally diverse ligands exhibits unique interactions with the receptor [142] for ligand-specific effects on signaling activity. Considering the fact that an agonist preferentially binds to the high-affinity state receptor (i.e., an active state receptor), where the rigid-body movements occur mainly in H6 and H7 according to the global toggle switch model [50], the Bd residues on H6 and H7 of the inactive receptor are not included as initial contacts for agonist binding but become available when the receptor is fully activated by the inward movements of H6 and H7 [50,103]. Thus, it is likely that C3556.47 [91] is not an initial contact residue of the receptor in the inactive state with CP55940 (Fig. (3B)). In contrast, the Bd residues on H6 and H7 for SR141716A (e.g., W3566.48 [95] and C3867.42 [121]), positioned toward the binding core without rigid-body movements of H6 and H7, are directly involved in the ligand binding (Fig. (3D)) and stabilize the inactive receptor. In this regard, the competitive ligand binding between the CB1 receptor agonists and SR141716A should be viewed in terms of not only competing with the ligand binding pocket but also competing with shifting the equilibrium between the inactive state and the active state.
Table 1.
Key residues of the CB1 receptor involved in ligand binding (i.e., Bd, BiS and BiA residues) of anandamide, CP55940, WIN55212-2 and SR141716A
| Ligand | Residue | Position | Residue type | Reference |
|---|---|---|---|---|
| anandamide | F1893.25 | H3 | Bd | [95] |
| K1923.28 | H3 | BiS | [122] | |
| L2073.43 | H3 | BiA | [106] | |
| T2103.46 | H3 | BiA | [105] | |
| D2133.49,a) | H3 | BiA | [104] | |
| Y2755.39 | H5 | Bd | [107] | |
| Y2945.58,a) | H5 | BiA | [101] | |
| A3426.34,a) | H6 | BiA | [104] | |
| CP55940 | H181E1 | E1 | BiS | [111] |
| R182E1 | E1 | BiS | [111] | |
| D184E1 | E1 | BiS | [111] | |
| V1883.24 | H3 | BiS | [111] | |
| F1893.25 | H3 | BiS | [95,111] | |
| K1923.28 | H3 | BiS | [122,123] | |
| L2073.43 | H3 | BiA | [106] | |
| T2103.46 | H3 | BiA | [105] | |
| D2133.49,a) | H3 | BiA | [104] | |
| F268E2 | E2 | Bd | [96] | |
| P269E2 | E2 | Bd | [96] | |
| H270E2 | E2 | Bd | [96] | |
| I271E2 | E2 | Bd | [96] | |
| Y2755.39 | H5 | Bd | [107] | |
| Y2945.58,a) | H5 | BiA | [104] | |
| A3426.34,a) | H6 | BiA | [104,159] | |
| C3556.47 | H6 | Bd | [91] | |
| S3837.39 | H7 | BiA | [114] | |
| WIN55212-2 | D1632.50 | H2 | BiA | [116] |
| K1923.28 | H3 | BiS | [122,123] | |
| G1953.31 | H3 | Bd | [118] | |
| F2003.36 | H3 | Bd | [95,120] | |
| L2073.43 | H3 | BiA | [106] | |
| T2103.46 | H3 | BiA | [105] | |
| D2133.49,a) | H3 | BiA | [104] | |
| Y2755.39 | H5 | Bd | [95] | |
| W2795.43 | H5 | Bd | [95] | |
| V2825.46 | H5 | Bd | [108] | |
| Y2945.58,a) | H5 | BiA | [101] | |
| A3426.34,a) | H6 | BiA | [104] | |
| SR141716A | K1923.28 | H3 | BiS | [124] |
| F2003.36 | H3 | Bd | [95] | |
| L2073.43 | H3 | BiA | [106] | |
| T2103.46 | H3 | BiA | [105] | |
| C257E2 | E2 | BiS | [121] | |
| C264E2 | E2 | BiS | [121] | |
| W2795.43 | H5 | Bd | [95] | |
| W3566.48 | H6 | Bd | [95] | |
| C3867.42 | H7 | Bd | [121] | |
inferred from the results of the CB2 receptor.
Fig. (3).
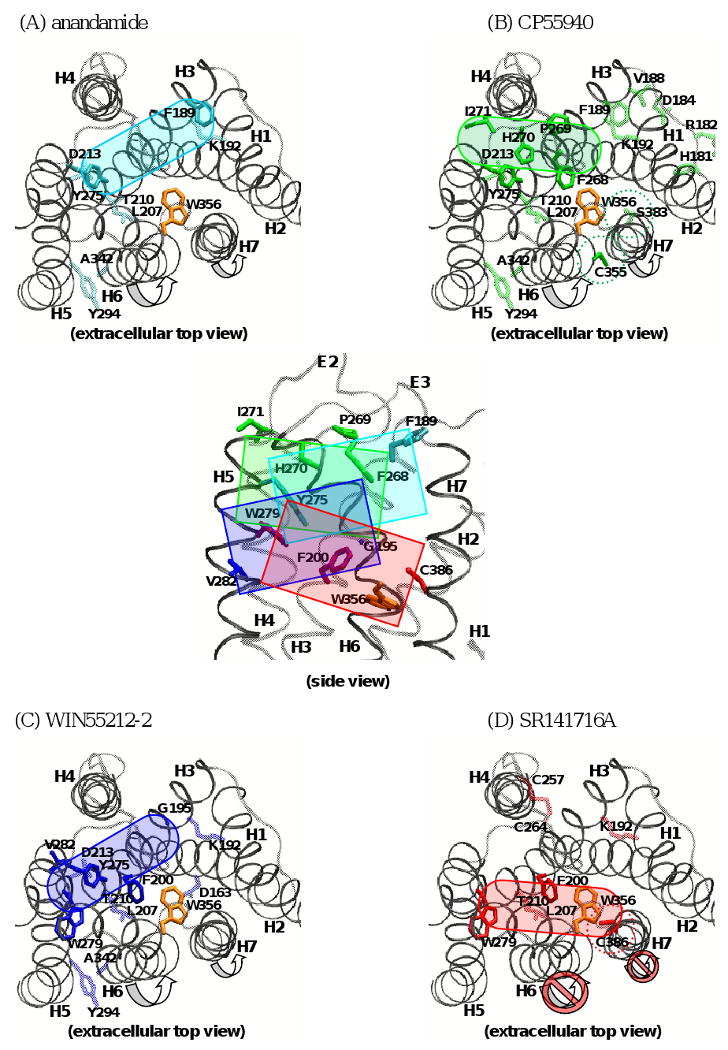
Extracellular top view of the key initial CB1 receptor binding contacts (by ellipsoids), using the recently developed homology model of the CB1 receptor (apo) in the inactive state [136], of (A) anandamide (in cyan), (B) CP55940 (in green), (C) WIN55212-2 (in blue) and (D) SR141716A (in red) are delineated by the type Bd residues (in stick). The type BiS and BiA residues are also represented in transparent stick. At the center of the figure, the side views of the ligand binding sites (by rectangles) of anandamide (in cyan), CP55940 (in green), WIN55212-2 (in blue) and SR141716A (in red) are depicted by the type Bd residues (in stick). Only the TM helical domain (in black ribbon) and E1/E2/E3 (in gray ribbon) is shown and other segments are omitted for clarity. For residues, only side chains are shown. W3566.48 (in orange) of the proposed toggle switch [112,165] is also represented. Considering the fact that an agonist preferentially binds to the active state receptor, where the rigid-body movements occur in H6 and H7 [50,103] (by arrows), the Bd residues on H6 or H7 are not included as initial contacts for agonist binding. For example, C3556.47 [91] & S3837.39 [114] (in green dotted circles), Bd residue for CP55940, become fully engaged in ligand binding only when the receptor is fully activated. In contrast, Bd residues for SR141716A, including those in H6 and H7 (in red dotted circles), are fully engaged in ligand binding without a significant change in the receptor conformation. It appears that WIN55212-2 interaction with F2003.36 disrupts W3566.48, leading to the H6 displacement, while SR141716A interaction with F2003.36 protects W3566.48 from the H6 displacement. In this regard, the competitive ligand binding between the CB1 receptor agonists and SR141716A should be viewed in terms of not only competing with the ligand binding pocket but also competing with shifting the equilibrium between the inactive state and the active state.
Only a few residues, including F1893.25 [95] and Y2755.39 [107,110], are known to directly interact with anandamide. Taken the H2/H3/H6/H7 region proposed to be important for anandamide binding [95] together with the exclusion of H6 and H7 due to their involvement in agonist binding at the late stage of receptor activation, the key initial contacts for anandamide binding would be in the H2-H3-H5 region (Fig. (3A)).
For CP55940, F268E2/P269E2/H270E2/I271E2 [96], Y2755.39 [107] and C3556.47 [91] are directly contacting with the ligand. A very recent substituted-cysteine accessibility method (SCAM) [143] study on H6 of the CB2 receptor [100] showed that V2616.51, L2626.52, L2646.54, M2656.55, L2696.59 and T2726.62 were on the solvent-accessible surface of the binding site crevice of CP55940, suggesting that the equivalent residues in the CB1 receptor are accessible to CP55940. Taken the region formed by H3-H5-H6-H7 proposed to be important for CP55940 binding [114,144] together with the exclusion of H6-H7 due to their involvement in agonist binding at the late stage of receptor activation, the key initial contacts for CP55940 binding would be in the H3-E2-H5 region (Fig. (3B)).
For WIN55212-2, G1953.31 [118], F2003.36 [95,120], Y2755.39 [107], W2795.43 [95] and V2825.46 [108] are the residues in direct contact. Taken the proposed H3-H4-H5-H6 region for WIN55212-2 binding [95,145] together with the exclusion of H6/H7 due to their involvement in agonist binding at the late stage of receptor activation, the key initial contacts for WIN55212-2 binding would be in the H3-H4-E2-H5 region (Fig. (3C)).
For SR141716A, F2003.36 [95], W2795.43 [95], W3566.48 [95] and C3867.42 [121] are the residues in direct contact. It has been suggested that E1 and H3 (proximal to the extracellular side) [111] and E2 [96,110] are not sensitive to SR141716A binding. Taken the proposed H3-H4-H5-H6 region for SR141716A binding [95] together with the inclusion of H6/H7, the key contacts for SR141716A binding would be in the H3-H5-H6-H7 region (Fig. (3D)). It is interesting to note that the binding site of SR141716A is located at W3566.48 [95], in the region deep in the receptor core and relatively distal to the extracellular side (see Fig. (3)), possibly protecting the receptor from activation.
Integrity of E2 of the CB1 Receptor Critical for Ligand Binding and Receptor Activation
The second extracellular loop (E2) connecting H4 and H5 appears to play an important role in receptor stabilization, ligand binding and receptor activation [16,17,79,146-148]. It is shown from the GPCR X-ray structures that E2 residues form part of the binding site for the ligand. It has been suggested that different E2 residues are involved in different functions [149,150], possibly with distinct conformations [16,19]. It has been suggested from several studies [96,110,121,145,151] that E2 of the cannabinoid receptors are important for ligand binding and receptor activity. It is known that E2 Cys residues C257E2 and C264E2 of the CB1 receptor are required for receptor stabilization [110,121]. It was shown from an early study by Martin and his colleagues [152] that CP55940 binding was inhibited competitively by sulfhydryl blocking agents but not by a disulfide reducing agent, suggesting that at least one reactive sulfhydryl group exists at the CP55940 binding site and that a disulfide bond, whose reduction impacts on CP55940 binding, exists away from the CP55940 binding site. Among the Cys residues conserved in the cannabinoid receptors, C3556.47 would be the best candidate to provide the reactive sulfhydryl group with the following reasons: First, C3556.47 is known to form part of the CP55940 binding site [91]; Second, it was shown from a mutational analysis [121] that the CB1 receptor mutations of C257E2 and C264E2 did not affect CP55940 binding; Third, it was shown that sulfhydryl blocking of C3867.42 did not affect CP55940 binding [121]. Accordingly, it can be argued that although the CB1 receptor lacks the inter-disulfide linkage between H3 and E2, two E2 Cys residues form an intra-loop disulfide bond [110,121,145] important for CP55940 binding [152] as well as receptor stabilization [110,121].
It has been shown that the C-terminal of E2 of many GPCRs is important for ligand binding and receptor function [146,150,153]. It is also shown from the GPCR X-ray structures of rhodopsin [46] and β2AR [40,154] that an aromatic residue of the Cys-X-X-X-Ar motif, where Cys is tied to another Cys from H3, conserved in many GPCRs [153], plays a role in receptor stabilization and ligand binding. The importance of the C-terminal region of E2 shown by many GPCRs has also been demonstrated in the CB1 receptor [96]. Thus, it was shown from a recent Ala scanning mutagenesis of E2 of the CB1 receptor [96] that the C-terminal residues F268E2-I271E2 were critical for CP55940 but not for SR141716A binding, suggesting that the C-terminal end of E2 plays an important role not only in ligand binding but also in receptor activation.
It has been suggested that E2 plays an important role in receptor activation, primarily by coupling to the TM helical domain [8,155,156]. It appears that E2 rearrangement triggered by the ligand is coupled to the TM helical domain, especially the segments of H5 [156] and H7 [8] to achieve receptor activation. For the CB1 receptor, it was suggested from a recent study [96] that the C-terminal end of E2 of the CB1 receptor played an important role in receptor activation, presumably through the coupling to H5 similar to rhodopsin [156]. It is possible that the Bd residues in the extracellular side of the H5 through H7 region [95,144], including Y2755.39 [107], W2795.43 [95], V2825.46 [108] and C3556.47 [91] of the CB1 receptor (Table 1), would be involved in providing the necessary movement in the TM helical region for receptor activation.
Emerging CB1 Receptor Activation Mechanism Involving E2
Activation of a GPCR is a multistep process [11,157] that is initiated by binding of an agonist to the inactive state of the receptor. Ahuja and Smith [103] proposed a mechanism of GPCR activation by multiple sequential switches, including a switch of E2 displacement, a switch of the H5 motion coupled to E2, a rotamer toggle switch of the CWxP motif on H6, and an internal switch of the NPxxY motif on H7, the final switch that eventually breaks the ionic lock between R3.50 and E6.30 on the intracellular sides of H3 and H6 to relay the molecular signal to the coupled G protein. Thus, according to this mechanism, the molecular signal from the extracellular side passes through the receptor in the following order: E2 -> H5 -> H6 (the rotamer toggle switch) -> H7 (the internal switch) -> H3/H6 (the ionic lock). As shown in Fig. (4A), most of these residues involved in receptor activation (i.e., A and BiA residues) identified from the mutational studies of the CB1 receptor are, in fact, part of the functional motifs conserved in many GPCRs [158] (see Fig. (1)). R2143.50 [104], D3386.30 [100], L3416.33/A3426.34 [159] and T2103.46/L2073.43 [105,106] are associated with the DRY motif; and D1632.50 [116,160] of the SLAxAD motif. Although C3556.47 [112,113] and W3566.48 [95] of the CWxP motif are classified as Bd residues, they also play an important role in receptor activation. It was shown in a recent CB1 receptor helical bundle model [136] that most of the functional motifs were involved in maintaining the inactive state of the CB1 receptor, suggesting that disruption of these functional motifs is necessary in order to achieve a conformational change of the receptor necessary for G-protein signaling [49,161].
Fig. (4).
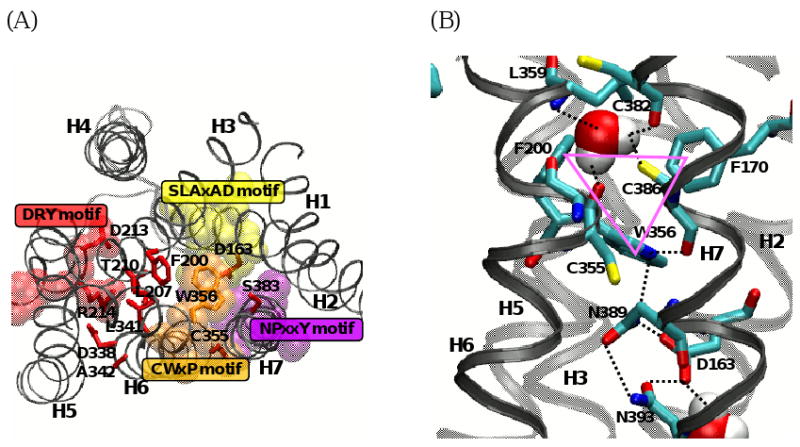
(A) Functional motifs, represented by space-filling, including the SLAxAD motif (in yellow) on H2, the DRY motif (in red) on H3, the CWxP motif (in orange) on H6, and NPxxY motif (in purple) on H7 of the CB1 receptor are represented along with the CB1 receptor types A and BiA residues (in red stick) important for G protein signaling. It appears that disruption of these functional motifs is necessary for receptor activation [49,161]. W3566.48 (in orange) of the proposed toggle switch [112,165] is also represented. It is noted that all the A and BiA residues are located in the interface of H6 and its surrounding helices H2/H3/H5/H7, suggesting that the rigid-body movement of H6 is important for receptor activation (see text). (B) Molecular constraints of W3566.48 in the inactive state of the CB1 receptor [136], including the aromatic stacking interactions (in magenta triangle) between W3566.48 and F2003.36/F1702.57, a direct H-bond network (in black dotted lines) by W3566.48 and C3867.42/N3897.45/D163/N393, a water-mediated H-bond network (in black dotted lines) by C3556.47/L3596.51/C3827.38/C3867.42. The water molecule coordinated to the CWxP motif was quite stable during 105 ns duration of molecular dynamics (MD) simulations in a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayer [136]. The water molecules coordinated to the CWxP and the SLAxAD motifs are represented by space-filling. Color coding: C, cyan; O, red; N, blue; and S, yellow. H6 and H7 are represented in black ribbon, while other helices are represented in gray ribbon. Hydrogen atoms are omitted for clarity.
It has been proposed that upon receptor activation an outward movement of the extracellular side of H5, coupled to an outward displacement of E2 [156], releases the molecular constraints on W6.48 [49], while an inward movement of the intracellular H5 replaces E6.30 by Y5.58, disrupting the ionic lock [103]. It has also been proposed that an outward movement of the intracellular side of H6, as a result of the disruption of the rotamer toggle switch [52], is one of the key steps in receptor activation [103]. The X-ray structure of rhodopsin [28] reveals that W2656.48 is stabilized by aromatic stacking with F2616.44 and Y2686.51, highly homologous residues in rhodopsin class GPCRs [162], and by the water-mediated H-bond interaction with N3027.49 of the NPxxY motif [163]. It has been suggested that the breakage of the H-bond between W2656.48 and N3027.49 allows M2576.40 to move away and Y3067.53 to come in contact with H6, contributing to breaking the ionic lock [164]. Thus, it appears to be crucial to see how W3566.48 is stabilized in the CB1 receptor inactive state for understating the mechanism of the CB1 receptor activation. The CB1 receptor lacks aromatic residues at the 6.44 and 6.51 positions, and consequently is expected to have unique interaction patterns for W3566.48. As shown in Fig. (4B), it was revealed from a recent homology model of the CB1 receptor in the inactive state [136] that W3566.48 formed aromatic stacking with F2003.36 and F1702.57 and was involved in an extensive H-bond network by direct H-bonds with C3867.42/N3897.45. It was also shown that W3566.48 was conserved by a water-mediated H-bond network by C3556.47/L3596.51/C3827.38/C3867.42, located just above W3566.48 (Fig. (4B)). The importance of the aromatic stacking between W3566.48 and F2003.36 is supported by the proposed rotamer toggle switch by W3566.48/F2003.36 of the CB1 receptor [165; 112], similar to W2866.48/F2906.52 of β2AR [113] and also by the finding that the F200A mutation of the CB1 receptor resulted in higher constitutive activity compared with the wild type receptor [120]. Thus, alternative ways to release the molecular constraints on W3566.48 of the CB1 receptor to achieve CB1 receptor activation include: 1) the breakage of aromatic stacking between W3566.48 and F2003.36/F1702.57; and 2) the breakage of a water-mediated H-bond network by C3556.47/L3596.51/C3827.38/C3867.42.
It was also shown in a recent CB1 receptor model [136] that N3937.49 of the NPxxY motif formed direct H-bonds to N3897.45 and D1632.50 (Fig. (4B)). Thus, similar to rhodopsin, the breakage of the N3897.45-mediated H-bond between W3566.48 and N3937.49 contributes to breaking the ionic lock in the CB1 receptor. It has been proposed that L2073.43, one of the highly conserved residues among many GPCRs [158], maintains the receptor in the inactive state by locking the movement of H6. In support of this, L2073.43 of the CB1 receptor has been reported to be a CAM residue [106]. It is possible that an outward movement of the intracellular side of H6 allows I3486.40, similar to M2576.40 in rhodopsin [164], to move away from L2073.43, thereby causing N3937.49 to come in contact with H6 (Fig. (5)). Taking into consideration the role of D1632.50 in receptor activation [116], it is possible that the breakage of the interaction between N3937.49 and D1632.50, which is proposed as an activation switch in other GPCRs [166-168], would contribute to breaking the ionic lock. It also appears that upon receptor activation Y2945.58 and Y3977.52 of the CB1 receptor, both highly conserved in many GPCRs, come closer and contribute to the breakage of the ionic lock (Fig. (5)).
Fig. (5).
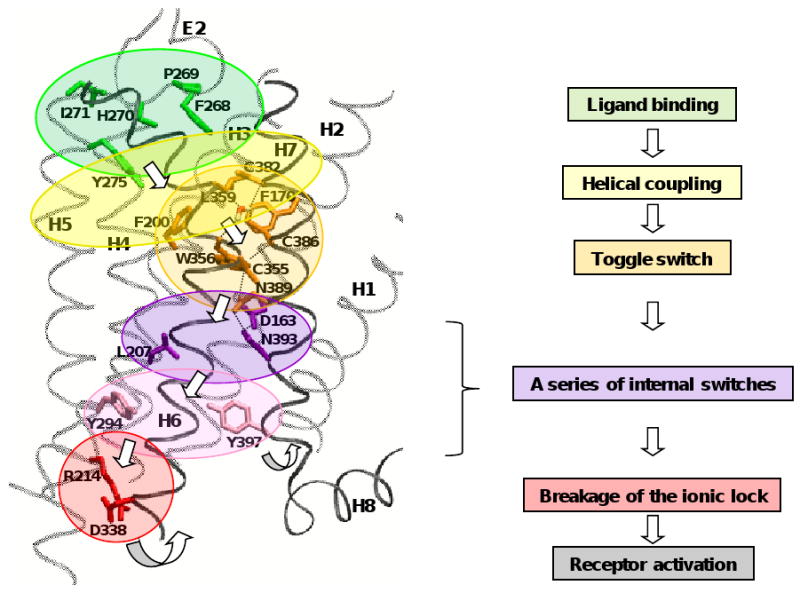
The proposed molecular mechanism of the CB1 receptor activation by CP55940. The CB1 receptor model of the inactive state [136] is used. Propagation of the molecular signal from the extracellular side to the intracellular side of the receptor is depicted by a series of the sequential steps: ligand binding (in green) -> E2 coupling to H5/H7 (in yellow) -> the W3566.48 rotameric trigger and the breakage of the H-bond between W3566.48 and N3897.45 (in orange) -> a series of internal switches, including the interaction between L2073.43 and N3937.49 (in purple) and the interaction between Y2945.58 and Y3977.52 (in pink) -> the breakage of the ionic lock between R2143.50 and D3386.30 (in red). The rigid-body movements in H6 and H7 [50,103] are represented by arrows.
Ligand-Specific CB1 Receptor Activation
Supported by similar results from other GPCR systems [169], several studies on the CB1 receptor [170,171] suggest that different CB1 receptor ligand classes evoke selective G-protein signaling. Considering the concept that one receptor can couple to different signaling pathways by different ligands [10], the binding of structurally distinct CP55940 and WIN55212-2 contributes to the ligand-specific conformational change in the receptor [131]. In this regard, it is insightful to see that among the residues of the CB1 receptor involved in the ligand-induced receptor activation (i.e., Bd residues), F268E2-I271E2 [96] and C3556.47 [91] are unique to CP55940, while G1953.31 [118], F2003.36 [95,120], W2795.43 [95] and V2825.46 [108] are unique to WIN55212-2 (Table 1). Thus, these unique residues play important roles in inducing ligand-specific receptor activation. Considering the suggested important role of aromatic stacking in receptor activation [95,113,157,172], it is likely that F268E2 [96] and F2003.36 [95,120] are important for receptor activation by CP55940 and WIN55212-2, respectively. Thus, employing the known type Bd residues, albeit limited in number, it is possible to describe the ligand-specific interference with the CWxP motif as the key step of the CB1 receptor activation mechanism [112,165]. Here, the ligand-specific CB1 receptor activation is illustrated by comparing CP55940 with WIN55212-2.
For CP55940, the Bd residues include F268E2/P269E2/H270E2/I271E2 [96], Y2755.39 [107] and C3556.47 [91]. It has been suggested that the E2 C-terminal F268E2 is crucial for CP55940 binding but not for that of SR141716A [96]. It has also been suggested that Y2755.39 is crucial for receptor activation by CP55940 [107]. Thus, upon CP55940 binding, the aromatic stacking interaction between the ligand aromatic A-ring and the receptor residues, including F268E2 [96] and Y2755.39 [107], composes the key initial contacts, which allows the hydrophobic C3 side chain of CP55940, the steric trigger for receptor activation [173], to position deep in the core. It is likely that the interaction between the C3 side chain of the ligand and L3596.51 on the solvent-accessible surface of the binding site crevice of CP55940 [100] contributes to the breakage of the water-mediated H-bonds (Fig. (4B)), which, in turn, interferes with the extracellular side of H7 and with the CWxP motif (Fig. (5)). As a result, the molecular constraints on W3566.48 are released and W3566.48 becomes free to rotate for receptor activation. It is possible that the coupling movements of H5/H7 [8,155,156] by the ligand binding to E2 [96] initially and then to the extracellular side of the H5 through H7 region [95,144] assist of the rigid-body rotation of H6 [52,174-179]. As shown in Fig. (3B), C3556.47 [91], whose side chain is pointing toward H7 and located off the receptor core region, is less likely to be accessible to CP55940 in the receptor inactive state, but becomes available for ligand binding with the rigid-body rotation of H6 upon receptor activation. The findings that C3556.47 [91] and S3837.39 [114] of the CB1 receptor were important for CP55940 but not for WIN55212-2 suggest that these residues play a role in CP55940-specific receptor activation. Overall, as shown in Fig. (5), the molecular signal by CP55940 binding from the extracellular side passes through the receptor in the following order: E2 -> H5/H7 -> H6 (the rotamer toggle switch) -> H7 (the internal switch) -> H6 (the ionic lock).
For WIN55212-2, the Bd residues include G1953.31 [118], F2003.36 [95,120], Y2755.39 [107], W2795.43 [95,120] and V2825.46 [108]. Upon binding, WIN55212-2 interacts directly with Y2755.39 [107] and W2795.43 [95,120] within the proposed aromatic microdomain [95] which allows the aromatic naphthyl ring, the steric trigger of WIN55212-2 for receptor activation (Shim and Howlett, 2006), to bind to F2003.36 of the proposed rotamer toggle switch of the CB1 receptor [112,165]. It is likely that the interaction between the naphthyl ring of the ligand and F2003.36 [95,120] contributes to the disruption of aromatic stacking between W3566.48 and F2003.36, interfering with the extracellular side of H3 and with the CWxP motif. As a result of the rotameric change in F2003.36, the molecular constraints on W3566.48 are released and W3566.48 becomes free to rotate for receptor activation. Because the water-mediated H-bonds above W3566.48 are in close proximity to F2003.36 (Fig. (4B)), it is possible that the water-mediated H-bonds are broken by WIN55212-2 binding to F2003.36. The findings that W2795.43 [95,120] and V2825.46 [108] of the CB1 receptor were important for WIN55212-2 but not for CP55940 suggest that these residues play a role in WIN55212-2-specific receptor activation. Overall, the molecular signal by WIN55212-2 binding passes through the receptor in the following order: H5 -> H5/H3 -> H6 (the rotamer toggle switch) -> H7 (the internal switch) -> H6 (the ionic lock).
Interestingly, in order to disrupt the CWxP motif, CP55940 interferes with H7 and WIN55212-2 interferes with H3, through unique interactions with H5. Both paths of interference appear to be effective in disrupting the CWxP motif according to the global toggle switch model [50] where upon activation, W3566.48 of the CWxP motif was displaced with the outward movement of the intracellular segment of H6. The observed ligand-specific G-protein signaling is likely to be attributed different ways to disrupt W3566.48, through H5/H7 in CP55940 or through H5/H3 in WIN55212-2. It has been proposed that H5 plays an important role in agonist-specific conformational change [180]. Thus, it is tempting to speculate that different paths of disrupting the CWxP motif contribute to the ligand-specific receptor activation.
CB1 Receptor Homology Models Suitable for Drug Design
Even before the X-ray structures of GPCRs became available, in their early pioneering works, Reggio and her colleagues [181] successfully determined the length and orientation of the membrane spanning 7 TM helices and the presence of H8 in the CB1 receptor. After the X-ray structure of rhodopsin [27] became available, this X-ray structure was utilized as the template to construct cannabinoid receptor homology models [95,124,128-130,132,182-184]. Recent CB1 receptor homology models [96,136] were constructed using the X-ray structure of β2AR [40] as templates. In spite of the similarity in overall structural topology, homology receptor models using these X-ray structures as templates may result in structures which are locally quite different. Although the CB1 receptor shows a low sequence homology to β2AR and rhodopsin (43 % and 42 % identical to human β2AR and bovine rhodopsin), homology models of the CB1 receptor using these GPCRs as templates have been justified by reliable sequence alignment with these GPCRs according to the highly conserved amino acid residues and functional motifs within the TM region (Fig. (1)), which are conserved in greater than 90 % of all GPCRs [158]. Yuzlenko and Kieć-Kononowicz [185] tested the feasibility of the homology models of adenosine receptors using the X-ray structures of rhodopsin and β2AR as templates and concluded that the β2AR-based homology models were better than the rhodopsin-based homology models judging from the stability of the ligands inside the binding pockets. The feasibility of the homology models of the CB1 receptor, using either rhodopsin or β2AR as the template, remains to be seen. The CB1 receptor is 44 %, 45 %, 43 %, 42 %, and 45 % identical to AA2R (human), β1AR (turkey), β2AR (human), rhodopsin (bovine), and rhodopsin (squid), respectively. This suggests that the X-ray structures of AA2R [42], β1AR [38] and squid rhodopsin [186] would be better templates for constructing homology models of the CB1 receptor.
Some of recent homology models of the CB1 receptor [136,184,187] have taken advantage of increasing computational resources and were determined in a fully hydrated lipid bilayer to mimic the physiological environment. Such homology models of the CB1 receptor provide a detailed understanding of its interaction with the lipid bilayer, including hydrophobic core and hydrophilic interfaces, and with water [188]. Some studies [95,132,184] also included CB1 receptor activated state models by modifying their receptor homology models in the inactive state on the basis of the receptor activation information obtained from biophysical studies [52,174-179]. On one hand, the active state of a GPCR is the state that perfectly fits to an agonist; a homology model of the CB1 receptor in its active state would better serve to understand the molecular interactions with an agonist. On the other hand, it is still very challenging to construct a computational model of a fully activated GPCR due to the reasons described earlier even with the large amount of accumulated biophysical data on the GPCR active state.
Structure-Based Drug Design in the CB1 Receptor Models
The X-ray structures of GPCRs and high quality homology models have been used for identifying potential lead compounds by the popular GPCR drug-design approach of structure-based high throughput screening (HTS) of a large collection of commercially available compounds [189-196] (for recent reviews, see [54,197]). Similarly, HTS has been applied to the cannabinoid receptor area [129,198,199]. A new drug-design approach, fragment-based approach (FBA) [200-202] has been successfully applied to screen relatively few compounds (approximately 1,000 compounds) using NMR or X-ray crystallography and to identify lead-like fragments with relatively low binding affinity (in the μM range) positioned non-redundantly at the binding pocket that can be connected to derive novel compounds with relatively high binding affinity (in the nM range). Compared with HTS, which needs an exhaustive search of the chemistry space allowed for the library of larger molecules, the chemistry space is efficiently probed by screening collections of small ligand fragments [202]. FBA using NMR or X-ray crystallography cannot be applied widely to GPCRs owing to the difficulty in obtaining their X-ray structures, particularly for small fragments within the active site [56]. To overcome this problem, a similar approach to FBA, multiple copy simultaneous search (MCSS) [203] can be applied to homology modeled GPCRs. MCSS generates several thousand replicas of a given functional group and orients the functional group in a favorable way within the binding-site region of the receptor [203-205]. A recent study [206], where MCSS approach was applied against the homology models of the ARs constructed by using the X-ray structure of rhodopsin as a template, successfully identified a few potent antagonists selective to the α1dAR.
It appears that if any homology model is constructed using a template of the X-ray GPCR in its apo state or inverse agonist bound type, the binding region needs be modified to accommodate various types of ligands [207-208]. For the purpose of rational drug design based upon a homology model of the CB1 receptor, it appears that the early intermediate stages of the active state of the receptor bound to an agonist, resembling the inactive state of the receptor, are sufficient as alternative structures to the fully activated structure for the receptor-based agonist design [209]. In this case, it is crucial to identify the receptor residues involved in kinetically distinct steps in the receptor activation. For β2AR, it has been proposed that the agonist binding of β2AR occurs through at least three distinct steps [11,157]: 1) the first step involves an ionic/H-bond interaction between the protonated amine of the agonist and D1133.32 and N3127.39 of the receptor and aromatic stacking between the catechol ring of the ligand and F2906.52 of the receptor. A combination of these interactions composes the key initial ligand contacts to a minimal low-affinity receptor binding site; 2) the second step involves H-bonding interactions between the catechol hydroxyl groups of the ligand and S2035.42, S2045.43, and S2075.46 of the receptor [98]. These interactions, which properly position the catechol ring of the ligand and H5, are required for the receptor to achieve the fully active state [210]; and 3) the third step involves a rotamer toggle switch of W2866.48/F2906.52 [113], which modulates H6 movement about the P6.50 kink, and H-bond interaction between the side chain hydroxyl group of the ligand and N2936.55 of the receptor. Accordingly, de Graaf and Rognan [209] modified the rotameric states of S2045.43 and S2075.46 of the X-ray of β2AR and successfully screened selective full and partial agonists.
Thus, similar steps of agonist binding can be considered for the CB1 receptor. Several issues need to be addressed: First, with a variety of ligands with distinct structures, the cannabinoid ligand binding pocket has not been well established. It is generally accepted that structurally diverse cannabinoid agonists bind to the binding sites uniquely defined for the individual classes of the ligand but with partial overlap [142] (see Fig. (3)). Second, in contrast to β2AR agonists, classical and non-classical cannabinoid agonists do not contain the protonated amine, while WIN55212-2 contains the morpholino N that can be protonated. On the other hand, similar to β2AR agonists, most cannabinoid agonists do contain at least one aromatic moiety. Thus, aromatic residues [95,96,107,120] in the extracellular side of the CB1 receptor would serve as the key initial ligand contacts to a minimal low-affinity receptor binding site. Third, it is conceivable that several aromatic residues on H5 of the CB1 receptor, including Y2755.39 [107] for CP55940 and Y2755.39 [107] and W2795.43 [95] for WIN55212-2, perform a similar role as the agonist-contact residues S2035.42, S2045.43 and S2075.46 [98] on H5 of β2AR. These residues need to be repositioned with a rotameric change for a full activation of the receptor [210]. Fourth, the toggle switch W3566.48/F2003.36 [165] and the VxxI helical groove [127] just proximal to the CWxP sequence of H6 can be the key interaction sites for the agonist binding contacts that induce the agonist-initiated receptor micro-conformational change [131,173].
Based upon the results from CB1 receptor mutagenesis, pharmacological and computational studies, it is possible to generate a CB1 receptor model in an early active state by modifying the rotameric states of the binding site residues. Candidate modifications include C3556.47 for CP55940 [91] and F2003.36 [95,120]. These residues are known to be involved in receptor activation as well as ligand binding. Such early active state CB1 receptor models are useful in screening CB1 receptor selective full and partial agonists, as similarly demonstrated for β2AR [209]. As one example of this strategy, Salo et al. [129] modified a homology model of the CB2 receptor and docked HU210 in the binding pocket prior to database screening to identify novel agonist candidates.
Summary
The CB1 receptor is a GPCR with conformational flexibility as indicated by its high basal activity. The CB1 receptor functional residues reported from mutational studies are utilized to describe the molecular events of the ligand-specific CB1 receptor activation from ligand binding to G protein signaling. For the CB1 receptor, as shown in many GPCRs, the integrity of the extracellular loops in ligand binding and coupling to the TM helical domain, the interference of the CWxP motif and the breakage of the ionic lock are important for receptor activation. Yet, little is known about the molecular structure of the CB1 receptor in the active state. But, it is possible to construct homology models of the CB1 receptor corresponding to the early stage of its activation that are suitable for identifying new therapeutic agents by applying the validated structure-based approaches, such as virtual HTS and FBA.
Acknowledgments
Support from NIH NIDA 5K01DA020663 and NCSA under MCB080037N is acknowledged. Drs. L. Padgett and A. Howlett are greatly acknowledged for their careful reading of the manuscript. Drs. L. Pedersen and L. Perera are acknowledged for their insightful discussion and suggestions.
References
- 1.Ji TH, Grossmann M, Ji I. G Protein-Coupled Receptors. J Biol Chem. 1998;273:17299–17302. doi: 10.1074/jbc.273.28.17299. [DOI] [PubMed] [Google Scholar]
- 2.Gether U. Uncovering molecular mechanisms involved in activation of G protein-coupled receptors. Endocr Rev. 2000;21:90–113. doi: 10.1210/edrv.21.1.0390. [DOI] [PubMed] [Google Scholar]
- 3.Drews J. Drug Discovery: A Historical Prospective. Science. 2002;287:1960–1964. doi: 10.1126/science.287.5460.1960. [DOI] [PubMed] [Google Scholar]
- 4.Jacoby E, Bouhelal R, Gerspacher M, Seuwen K. The 7 TM G-protein-coupled receptor target family. ChemMedChem. 2006;1:761–782. doi: 10.1002/cmdc.200600134. [DOI] [PubMed] [Google Scholar]
- 5.De Amici M, Dallanoce C, Holzgrabe U, Tränkle C, Mohr K. Allosteric ligands for G protein-coupled receptors: A novel strategy with attractive therapeutic opportunities. Med Res Rev. 2009 Jun 25; doi: 10.1002/med.20166. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 6.Heilker R, Wolff M, Tautermann CS, Bieler M. G-protein-coupled receptor-focused drug discovery using a target class platform approach. Drug Discov Today. 2009;14:231–240. doi: 10.1016/j.drudis.2008.11.011. [DOI] [PubMed] [Google Scholar]
- 7.Lagerström MC, Schiöth HB. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat Rev Drug Discov. 2008;7:339–357. doi: 10.1038/nrd2518. [DOI] [PubMed] [Google Scholar]
- 8.Jäger D, Schmalenbach C, Prilla S, Schrobang J, Kebig A, Sennwitz M, Heller E, Tränkle C, Holzgrabe U, Höltje HD, Mohr K. Allosteric small molecules unveil a role of an extracellular E2/transmembrane helix 7 junction for G protein-coupled receptor activation. J Biol Chem. 2007;282:34968–34976. doi: 10.1074/jbc.M705563200. [DOI] [PubMed] [Google Scholar]
- 9.Williams C, Hill SJ. GPCR signaling: understanding the pathway to successful drug discovery. Methods Mol Biol. 2009;552:39–50. doi: 10.1007/978-1-60327-317-6_3. [DOI] [PubMed] [Google Scholar]
- 10.Kenakin T. Ligand-selective receptor conformations revisited: the promise and the problem. Trends Pharmacol Sci. 2003;24:346–354. doi: 10.1016/S0165-6147(03)00167-6. [DOI] [PubMed] [Google Scholar]
- 11.Kobilka BK, Deupi X. Conformational complexity of G-protein-coupled receptors. Trends Pharmacol Sci. 2007;28:397–406. doi: 10.1016/j.tips.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 12.Lefkowitz RJ, Cotecchia S, Samama P, Costa T. Constitutive activity of receptors coupled to guanine nucleotide regulatory proteins. Trends Pharmacol Sci. 1993;14:303–307. doi: 10.1016/0165-6147(93)90048-O. [DOI] [PubMed] [Google Scholar]
- 13.Parnot C, Miserey-Lenkei S, Bardin S, Corvol P, Clauser E. Lessons from constitutively active mutants of G protein-coupled receptors. Trends Endocrinol Metab. 2002;13:336–343. doi: 10.1016/s1043-2760(02)00628-8. [DOI] [PubMed] [Google Scholar]
- 14.Smit MJ, Vischer HF, Bakker RA, Jongejan A, Timmerman H, Pardo L, Leurs R. Pharmacogenomic and structural analysis of constitutive g protein-coupled receptor activity. Annu Rev Pharmacol Toxicol. 2007;47:53–87. doi: 10.1146/annurev.pharmtox.47.120505.105126. [DOI] [PubMed] [Google Scholar]
- 15.Kenakin T, Onaran O. The Ligand Paradox between Affinity and Efficacy: Can You Be There and Not Make a Difference? Trends Pharmacol Sci. 2002;23:275–280. doi: 10.1016/s0165-6147(02)02036-9. [DOI] [PubMed] [Google Scholar]
- 16.Banères JL, Mesnier D, Martin A, Joubert L, Dumuis A, Bockaert J. Molecular characterization of a purified 5-HT4 receptor: a structural basis for drug efficacy. J Biol Chem. 2005;280:20253–20260. doi: 10.1074/jbc.M412009200. [DOI] [PubMed] [Google Scholar]
- 17.Alves ID, Salamon Z, Varga E, Yamamura HI, Tollin G, Hruby VJ. Direct observation of G-protein binding to the human delta-opioid receptor using plasmon-waveguide resonance spectroscopy. J Biol Chem. 2003;278:48890–48897. doi: 10.1074/jbc.M306866200. [DOI] [PubMed] [Google Scholar]
- 18.Li JH, Hamdan FF, Kim SK, Jacobson KA, Zhang X, Han SJ, Wess J. Ligand-specific changes in M3 muscarinic acetylcholine receptor structure detected by a disulfide scanning strategy. Biochemistry. 2008;47:2776–2788. doi: 10.1021/bi7019113. [DOI] [PubMed] [Google Scholar]
- 19.Ruan KH, Cervantes V, Wu J. Ligand-specific conformation determines agonist activation and antagonist blockade in purified human thromboxane A2 receptor. Biochemistry. 2009;48:3157–3165. doi: 10.1021/bi801443g. [DOI] [PubMed] [Google Scholar]
- 20.Perez DM, Karnik SS. Multiple signaling states of G-protein-coupled receptors. Pharmacol Rev. 2005;57:147–161. doi: 10.1124/pr.57.2.2. [DOI] [PubMed] [Google Scholar]
- 21.Vauquelin G, Van Liefde I. G protein-coupled receptors: a count of 1001 conformations. Fundam Clin Pharmacol. 2005;19:45–56. doi: 10.1111/j.1472-8206.2005.00319.x. [DOI] [PubMed] [Google Scholar]
- 22.Okada T, Ernst OP, Palczewski K, Hofmann KP. Activation of rhodopsin: new insights from structural and biochemical studies. Trends Biochem Sci. 2001;26:318324. doi: 10.1016/s0968-0004(01)01799-6. [DOI] [PubMed] [Google Scholar]
- 23.Schertler GF. Structure of rhodopsin and the metarhodopsin I photointermediate. Curr Opin Struct Biol. 2005;15:408–415. doi: 10.1016/j.sbi.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 24.Ridge KD, Palczewski K. Visual rhodopsin sees the light: structure and mechanism of G protein signaling. J Biol Chem. 2007;282:9297–9301. doi: 10.1074/jbc.R600032200. [DOI] [PubMed] [Google Scholar]
- 25.Yao X, Parnot C, Deupi X, Ratnala VR, Swaminath G, Farrens D, Kobilka B. Coupling ligand structure to specific conformational switches in the beta2-adrenoceptor. Nat Chem Biol. 2006;2:417–4122. doi: 10.1038/nchembio801. [DOI] [PubMed] [Google Scholar]
- 26.Ghanouni P, Gryczynski Z, Steenhuis JJ, Lee TW, Farrens DL, Lakowicz JR, Kobilka BK. Functionally different agonists induce distinct conformations in the G protein coupling domain of the beta 2 adrenergic receptor. J Biol Chem. 2001;276:24433–24436. doi: 10.1074/jbc.C100162200. [DOI] [PubMed] [Google Scholar]
- 27.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 28.Li J, Edwards PC, Burghammer M, Villa C, Schertler GF. Structure of bovine rhodopsin in a trigonal crystal form. J Mol Biol. 2004;343:1409–1438. doi: 10.1016/j.jmb.2004.08.090. [DOI] [PubMed] [Google Scholar]
- 29.Teller DC, Okada T, Behnke CA, Palczewski K, Stenkamp RE. Advances in determination of a high-resolution three-dimensional structure of rhodopsin, a model of G-protein-coupled receptors (GPCRs) Biochemistry. 2001;40:7761–7772. doi: 10.1021/bi0155091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luecke H, Schobert B, Lanyi JK, Spudich EN, Spudich JL. Crystal structure of sensory rhodopsin II at 2.4 angstroms: insights into color tuning and transducer interaction. Science. 2001;293:1499–1503. doi: 10.1126/science.1062977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okada T, Fujiyoshi Y, Silow M, Navarro J, Landau EM, Shichida Y. Functional role of internal water molecules in rhodopsin revealed by X-ray crystallography. Proc Natl Acad Sci USA. 2002;99:5982–5987. doi: 10.1073/pnas.082666399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Salom D, Lodowski DT, Stenkamp RE, Le Trong I, Golczak M, Jastrzebska B, Harris T, Ballesteros JA, Palczewski K. Crystal structure of a photoactivated deprotonated intermediate of rhodopsin. Proc Natl Acad Sci USA. 2006;103:16123–16128. doi: 10.1073/pnas.0608022103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Standfuss J, Xie G, Edwards PC, Burghammer M, Oprian DD, Schertler GF. Crystal structure of a thermally stable rhodopsin mutant. J Mol Biol. 2007;372:1179–1188. doi: 10.1016/j.jmb.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murakami M, Kouyama T. Crystal structure of squid rhodopsin. Nature. 2008;453:363–367. doi: 10.1038/nature06925. [DOI] [PubMed] [Google Scholar]
- 35.Stenkamp RE. Alternative models for two crystal structures of bovine rhodopsin. Acta Crystallogr D Biol Crystallogr. 2008;D64:902–904. doi: 10.1107/S0907444908017162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park JH, Scheerer P, Hofmann KP, Choe HW, Ernst OP. Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature. 2008;454:183–187. doi: 10.1038/nature07063. [DOI] [PubMed] [Google Scholar]
- 37.Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krauss N, Choe HW, Hofmann KP, Ernst OP. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455:497–4502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- 38.Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AG, Tate CG, Schertler GF. Structure of a beta1-adrenergic G-protein-coupled receptor. Nature. 2008;454:486–491. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VR, Sanishvili R, Fischetti RF, Schertler GF, Weis WI, Kobilka BK. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 40.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hanson MA, Cherezov V, Griffith MT, Roth CB, Jaakola VP, Chien EY, Velasquez J, Kuhn P, Stevens RC. A specific cholesterol binding site is established by the 2.8 A structure of the human beta2-adrenergic receptor. Structure. 2008;16:897–905. doi: 10.1016/j.str.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, Ijzerman AP, Stevens RC. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zezula J, Freissmuth M. The A(2A)-adenosine receptor: a GPCR with unique features. Br J Pharmacol. 2008;153 1:S184–190. doi: 10.1038/sj.bjp.0707674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huber T, Sakmar TP. Rhodopsin's active state is frozen like a DEER in the headlights. Proc Natl Acad Sci USA. 2008;105:7343–7344. doi: 10.1073/pnas.0804122105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shukla AK, Sun JP, Lefkowitz RJ. Crystallizing thinking about the beta2-adrenergic receptor. Mol Pharmacol. 2008;73:1333–1338. doi: 10.1124/mol.108.045849. [DOI] [PubMed] [Google Scholar]
- 46.Okada T, Sugihara M, Bondar AN, Elstner M, Entel P, Buss V. The retinal conformation and its environment in rhodopsin in light of a new 2.2 A crystal structure. J Mol Biol. 2004;342:571–583. doi: 10.1016/j.jmb.2004.07.044. [DOI] [PubMed] [Google Scholar]
- 47.Dunham TD, Farrens DL. Conformational changes in rhodopsin. Movement of helix f detected by site-specific chemical labeling and fluorescence spectroscopy. J Biol Chem. 1999;274:1683–1690. doi: 10.1074/jbc.274.3.1683. [DOI] [PubMed] [Google Scholar]
- 48.Sheikh SP, Zvyaga TA, Lichtarge O, Sakmar TP, Bourne HR. Rhodopsin activation blocked by metal-ion-binding sites linking transmembrane helices C and F. Nature. 1996;383:347–350. doi: 10.1038/383347a0. [DOI] [PubMed] [Google Scholar]
- 49.Crocker E, Eilers M, Ahuja S, Hornak V, Hirshfeld A, Sheves M, Smith SO. Location of Trp265 in metarhodopsin II: implications for the activation mechanism of the visual receptor rhodopsin. J Mol Biol. 2006;357:163–172. doi: 10.1016/j.jmb.2005.12.046. [DOI] [PubMed] [Google Scholar]
- 50.Elling CE, Frimurer TM, Gerlach LO, Jorgensen R, Holst B, Schwartz TW. Metal ion site engineering indicates a global toggle switch model for seven-transmembrane receptor activation. J Biol Chem. 2006;281:17337–17346. doi: 10.1074/jbc.M512510200. [DOI] [PubMed] [Google Scholar]
- 51.Altenbach C, Kusnetzow AK, Ernst OP, Hofmann KP, Hubbell WL. High-resolution distance mapping in rhodopsin reveals the pattern of helix movement due to activation. Proc Natl Acad Sci USA. 2008;105:7439–7444. doi: 10.1073/pnas.0802515105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ballesteros JA, Jensen AD, Liapakis G, Rasmussen SG, Shi L, Gether U, Javitch JA. Activation of the beta 2-adrenergic receptor involves disruption of an ionic lock between the cytoplasmic ends of transmembrane segments 3 and 6. J Biol Chem. 2001;276:29171–29177. doi: 10.1074/jbc.M103747200. [DOI] [PubMed] [Google Scholar]
- 53.Ballesteros JA, Weinstein H. Integrated methods for modeling G-protein coupled receptors. In: Conn PM, Sealfon SC, editors. Methods in Neuroscience. Academic Press; San Francisco: 1995. pp. 366–428. [Google Scholar]
- 54.Bissantz C, Schalon C, Guba W, Stahl M. Focused library design in GPCR projects on the example of 5-HT(2c) agonists: comparison of structure-based virtual screening with ligand-based search methods. Proteins. 2005;61:938–952. doi: 10.1002/prot.20651. [DOI] [PubMed] [Google Scholar]
- 55.Archer E, Maigret B, Escrieut C, Pradayrol L, Fourmy D. Rhodopsin crystal: new template yielding realistic models of G-protein-coupled receptors? Trends Pharmacol Sci. 2003;24:36–40. doi: 10.1016/s0165-6147(02)00009-3. [DOI] [PubMed] [Google Scholar]
- 56.Kobilka B, Schertler GF. New G-protein-coupled receptor crystal structures: insights and limitations. Trends Pharmacol Sci. 2008;29:79–83. doi: 10.1016/j.tips.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 57.Kolb P, Rosenbaum DM, Irwin JJ, Fung JJ, Kobilka BK, Shoichet BK. Structure-based discovery of beta2-adrenergic receptor ligands. Proc Natl Acad Sci USA. 2009;106:6843–6848. doi: 10.1073/pnas.0812657106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wenzel-Seifert K, Seifert R. Molecular analysis of beta(2)-adrenoceptor coupling to G(s)-, G(i)-, and G(q)-proteins. Mol Pharmacol. 2000;58:954–966. doi: 10.1124/mol.58.5.954. [DOI] [PubMed] [Google Scholar]
- 59.Ratnala VR, Kobilka B. Understanding the ligand-receptor-G protein ternary complex for GPCR drug discovery. Methods Mol Biol. 2009;552:67–77. doi: 10.1007/978-1-60327-317-6_5. [DOI] [PubMed] [Google Scholar]
- 60.Grossfield A, Pitman MC, Feller SE, Soubias O, Gawrisch K. Internal hydration increases during activation of the G-protein-coupled receptor rhodopsin. J Mol Biol. 2008;381:478–486. doi: 10.1016/j.jmb.2008.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dror RO, Arlow DH, Borhani DW, Jensen MØ, Piana S, Shaw DE. Identification of two distinct inactive conformations of the beta2-adrenergic receptor reconciles structural and biochemical observations. Proc Natl Acad Sci USA. 2009;106:4689–4694. doi: 10.1073/pnas.0811065106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Röhrig UF, Guidoni L, Rothlisberger U. Early steps of the intramolecular signal transduction in rhodopsin explored by molecular dynamics simulations. Biochemistry. 2002;41:10799–10809. doi: 10.1021/bi026011h. [DOI] [PubMed] [Google Scholar]
- 63.Saam J, Tajkhorshid E, Hayashi S, Schulten K. Molecular dynamics investigation of primary photoinduced events in the activation of rhodopsin. Biophys J. 2002;83:3097–3112. doi: 10.1016/S0006-3495(02)75314-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lemaître V, Yeagle P, Watts A. Molecular dynamics simulations of retinal in rhodopsin: from the dark-adapted state towards lumirhodopsin. Biochemistry. 2005;44:12667–12680. doi: 10.1021/bi0506019. [DOI] [PubMed] [Google Scholar]
- 65.Kong Y, Karplus M. The signaling pathway of rhodopsin. Structure. 2007;15:611–623. doi: 10.1016/j.str.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 66.Crozier PS, Stevens MJ, Woolf TB. How a small change in retinal leads to G-protein activation: initial events suggested by molecular dynamics calculations. Proteins. 2007;66:559–574. doi: 10.1002/prot.21175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lau PW, Grossfield A, Feller SE, Pitman MC, Brown MF. Dynamic structure of retinylidene ligand of rhodopsin probed by molecular simulations. J Mol Biol. 2007;372:906–917. doi: 10.1016/j.jmb.2007.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bhattacharya S, Hall SE, Vaidehi N. Agonist-induced conformational changes in bovine rhodopsin: insight into activation of G-protein-coupled receptors. J Mol Biol. 2008;382:539–555. doi: 10.1016/j.jmb.2008.06.084. [DOI] [PubMed] [Google Scholar]
- 69.Isin B, Schulten K, Tajkhorshid E, Bahar I. Mechanism of signal propagation upon retinal isomerization: insights from molecular dynamics simulations of rhodopsin restrained by normal modes. Biophys J. 2008;95:789–803. doi: 10.1529/biophysj.107.120691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jardón-Valadez E, Bondar AN, Tobias DJ. Dynamics of the internal water molecules in squid rhodopsin. Biophys J. 2009;96:2572–2576. doi: 10.1016/j.bpj.2008.12.3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hayashi S, Tajkhorshid E, Schulten K. Photochemical reaction dynamics of the primary event of vision studied by means of a hybrid molecular simulation. Biophys J. 2009;96:403–416. doi: 10.1016/j.bpj.2008.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Devane WA, Dysarz FA, III, Johnson MR, Melvin LS, Howlett AC. determination and characterization of a cannabinoid receptor in rat brain. Mol Pharmacol. 1988;34:605–613. [PubMed] [Google Scholar]
- 73.Howlett AC, Johnson MR, Melvin LS, Milne GM. Nonclassical cannabinoid analgetics inhibit adenylate cyclase: development of a cannabinoid receptor model. Mol Pharmacol. 1988;33:297–302. [PubMed] [Google Scholar]
- 74.Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 75.Howlett AC. Pharmacology of cannabinoid receptors. Annu Rev Pharmacol Toxicol. 1995;35:607–634. doi: 10.1146/annurev.pa.35.040195.003135. [DOI] [PubMed] [Google Scholar]
- 76.Felder CC, Glass M. Cannabinoid receptors and their endogenous agonist. Annu Rev Pharmacol Toxicol. 1998;38:179–200. doi: 10.1146/annurev.pharmtox.38.1.179. [DOI] [PubMed] [Google Scholar]
- 77.Joost P, Methner A. Phylogenetic analysis of 277 human G-protein-coupled receptors as a tool for the prediction of orphan receptor ligands. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-11-research0063. RESEARCH0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fredriksson R, Lagerström MC, Lundin LG, Schiöth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63:1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- 79.Hoffmann C, Moro S, Nicholas RA, Harden TK, Jacobson KA. The role of amino acids in extracellular loops of the human P2Y1 receptor in surface expression and activation processes. J Biol Chem. 1999;274:14639–14647. doi: 10.1074/jbc.274.21.14639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mirzadegan T, Benkö G, Filipek S, Palczewski K. Sequence analyses of G-protein-coupled receptors: similarities to rhodopsin. Biochemistry. 2003;42:2759–2767. doi: 10.1021/bi027224+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Davidson FF, Loewen PC, Khorana HG. Structure and function in rhodopsin: replacement by alanine of cysteine residues 110 and 187, components of a conserved disulfide bond in rhodopsin, affects the light-activated metarhodopsin II state. Proc Natl Acad Sci USA. 1994;91:4029–4033. doi: 10.1073/pnas.91.9.4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bouaboula M, Perrachon S, Milligan L, Canat X, Rinaldi-Carmona M, Portier M, Barth F, Calandra B, Pecceu F, Lupker J, Maffrand JP, Le Fur G, Casellas P. A selective inverse agonist for central cannabinoid receptor inhibits mitogen-activated protein kinase activation stimulated by insulin or insulin-like growth factor 1. Evidence for a new model of receptor/ligand interactions. J Biol Chem. 1997;272:22330–22339. doi: 10.1074/jbc.272.35.22330. [DOI] [PubMed] [Google Scholar]
- 83.Holst B, Schwartz TW. Molecular mechanism of agonism and inverse agonism in the melanocortin receptors: Zn(2+) as a structural and functional probe. Ann N Y Acad Sci. 2003;994:1–11. doi: 10.1111/j.1749-6632.2003.tb03156.x. [DOI] [PubMed] [Google Scholar]
- 84.Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, O'Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med. 2003;348:1085–1095. doi: 10.1056/NEJMoa022050. [DOI] [PubMed] [Google Scholar]
- 85.Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, Felder CC, Herkenham M, Mackie K, Martin BR, Mechoulam R, Pertwee RG. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev. 2002;54:161–202. doi: 10.1124/pr.54.2.161. [DOI] [PubMed] [Google Scholar]
- 86.Ryberg E, Larsson N, Sjögren S, Hjorth S, Hermansson NO, Leonova J, Elebring T, Nilsson K, Drmota T, Greasley PJ. The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol. 2007;152:1092–1101. doi: 10.1038/sj.bjp.0707460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lauckner JE, Jensen JB, Chen HY, Lu HC, Hille B, Mackie K. GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc Natl Acad Sci USA. 2008;105:2699–2704. doi: 10.1073/pnas.0711278105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pan X, Ikeda SR, Lewis DL. Rat brain cannabinoid receptor modulates N-type Ca2+ channels in a neuronal expression system. Mol Pharmacol. 1996;49:707–714. [PubMed] [Google Scholar]
- 89.Meschler JP, Kraichely DM, Wilken GH, Howlett AC. Inverse agonist properties of N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2, 4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide HCl (SR141716A) and 1-(2-chlorophenyl)-4-cyano-5-(4-methoxyphenyl)-1H-pyrazole-3-carboxyl ic acid phenylamide (CP-272871) for the CB(1) cannabinoid receptor. Biochem Pharmacol. 2000;60:1315–23. doi: 10.1016/s0006-2952(00)00447-0. [DOI] [PubMed] [Google Scholar]
- 90.Mato S, Pazos A, Valdizán EM. Cannabinoid receptor antagonism and inverse agonism in response to SR141716A on cAMP production in human and rat brain. Eur J Pharmacol. 2002;443:43–46. doi: 10.1016/s0014-2999(02)01575-3. [DOI] [PubMed] [Google Scholar]
- 91.Picone RP, Khanolkar AD, Xu W, Ayotte LA, Thakur GA, Hurst DP, Abood ME, Reggio PH, Fournier DJ, Makriyannis A. (-)-7′-Isothiocyanato-11-hydroxy-1′1′-dimethylheptylhexahydrocannabinol (AM841), a high-affinity electrophilic ligand, interacts covalently with a cysteine in helix six and activates the CB1 cannabinoid receptor. Mol Pharmacol. 2005;68:1623–1635. doi: 10.1124/mol.105.014407. [DOI] [PubMed] [Google Scholar]
- 92.Beukers MW, Ijzerman AP. Techniques: how to boost GPCR mutagenesis studies using yeast. Trends Pharmacol Sci. 2005;26:533–539. doi: 10.1016/j.tips.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 93.Rader AJ, Anderson G, Isin B, Khorana HG, Bahar I, Klein-Seetharaman J. Identification of core amino acids stabilizing rhodopsin. Proc Natl Acad Sci USA. 2004;101:7246–7251. doi: 10.1073/pnas.0401429101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rhee MH. Functional role of serine residues of transmembrane dopamin VII in signal transduction of CB2 cannabinoid receptor. J Vet Sci. 2002;3:185–191. [PubMed] [Google Scholar]
- 95.McAllister SD, Rizvi G, Anavi-Goffer S, Hurst DP, Barnett-Norris J, Lynch DL, Reggio PH, Abood ME. An aromatic microdomain at the cannabinoid CB(1) receptor constitutes an agonist/inverse agonist binding region. J Med Chem. 2003;46:5139–5152. doi: 10.1021/jm0302647. [DOI] [PubMed] [Google Scholar]
- 96.Ahn KH, Bertalovitz AC, Mierke DF, Kendall DA. Dual Role of the Second Extracellular Loop of the Cannabinoid Receptor One: Ligand Binding and Receptor Localization. Mol Pharmacol. 2009;76:833–842. doi: 10.1124/mol.109.057356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wess J, Nanavati S, Vogel Z, Maggio R. Functional role of proline and tryptophan residues highly conserved among G protein-coupled receptors studied by mutational analysis of the m3 muscarinic receptor. EMBO J. 1993;12:331–338. doi: 10.1002/j.1460-2075.1993.tb05661.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ambrosio C, Molinari P, Cotecchia S, Costa T. Catechol-binding serines of beta(2)-adrenergic receptors control the equilibrium between active and inactive receptor states. Mol Pharmacol. 2000;57:198–210. [PubMed] [Google Scholar]
- 99.Rovati GE, Capra V, Neubig RR. The highly conserved DRY motif of class A G protein-coupled receptors: beyond the ground state. Mol Pharmacol. 2007;71:959–964. doi: 10.1124/mol.106.029470. [DOI] [PubMed] [Google Scholar]
- 100.Nebane NM, Hurst DP, Carrasquer CA, Qiao Z, Reggio PH, Song ZH. Residues accessible in the binding-site crevice of transmembrane helix 6 of the CB2 cannabinoid receptor. Biochemistry. 2008;47:13811–13821. doi: 10.1021/bi8007802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Song ZH, Feng W. Absence of a conserved proline and presence of a conserved tyrosine in the CB2 cannabinoid receptor are crucial for its function. FEBS Lett. 2002;531:290–294. doi: 10.1016/s0014-5793(02)03537-8. [DOI] [PubMed] [Google Scholar]
- 102.Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 103.Ahuja S, Smith SO. Multiple switches in G protein-coupled receptor activation. Trends Pharmacol Sci. 2009;30:494–502. doi: 10.1016/j.tips.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 104.Feng W, Song ZH. Effects of D3.49A, R3.50A, and A6.34E mutations on ligand binding and activation of the cannabinoid-2 (CB2) receptor. Biochem Pharmacol. 2003;65:1077–1085. doi: 10.1016/s0006-2952(03)00005-4. [DOI] [PubMed] [Google Scholar]
- 105.D'Antona AM, Ahn KH, Kendall DA. Mutations of CB1 T210 produce active and inactive receptor forms: correlations with ligand affinity, receptor stability, and cellular localization. Biochemistry. 2006;45:5606–5617. doi: 10.1021/bi060067k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.D'Antona AM, Ahn KH, Wang L, Mierke DF, Lucas-Lenard J, Kendall DA. A cannabinoid receptor 1 mutation proximal to the DRY motif results in constitutive activity and reveals intramolecular interactions involved in receptor activation. Brain Res. 2006;1108:1–11. doi: 10.1016/j.brainres.2006.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.McAllister SD, Tao Q, Barnett-Norris J, Buehner K, Hurst DP, Guarnieri F, Reggio PH, Nowell Harmon KW, Cabral GA, Abood ME. A critical role for a tyrosine residue in the cannabinoid receptors for ligand recognition. Biochem Pharmacol. 2002;63:2121–2136. doi: 10.1016/s0006-2952(02)01031-6. [DOI] [PubMed] [Google Scholar]
- 108.Song ZH, Slowey CA, Hurst DP, Reggio PH. The difference between the CB(1) and CB(2) cannabinoid receptors at position 5.46 is crucial for the selectivity of WIN55212-2 for CB(2) Mol Pharmacol. 1999;56:834–840. [PubMed] [Google Scholar]
- 109.Tao Q, McAllister SD, Andreassi J, Nowell KW, Cabral GA, Hurst DP, Bachtel K, Ekman MC, Reggio PH, Abood ME. Role of a conserved lysine residue in the peripheral cannabinoid receptor (CB2): evidence for subtype specificity. Mol Pharmacol. 1999;55:605–613. [PubMed] [Google Scholar]
- 110.Shire D, Calandra B, Delpech M, Dumont X, Kaghad M, Le Fur G, Caput D, Ferrara P. Structural features of the central cannabinoid CB1 receptor involved in the binding of the specific CB1 antagonist SR 141716A. J BiolChem. 1996;271:6941–6946. doi: 10.1074/jbc.271.12.6941. [DOI] [PubMed] [Google Scholar]
- 111.Murphy JW, Kendall DA. Integrity of extracellular loop 1 of the human cannabinoid receptor 1 is critical for high-affinity binding of the ligand CP 55,940 but not SR 141716A. Biochem Pharmacol. 2003;65:1623–1631. doi: 10.1016/s0006-2952(03)00155-2. [DOI] [PubMed] [Google Scholar]
- 112.McAllister SD, Hurst DP, Barnett-Norris J, Lynch D, Reggio PH, Abood ME. Structural mimicry in class A GPCR rotamer toggle switches: The importance of the F3.36(201)/W6.48(357) interaction in cannabinoid CB1 receptor activation. J Biol Chem. 2004;279:48024–48037. doi: 10.1074/jbc.M406648200. [DOI] [PubMed] [Google Scholar]
- 113.Shi L, Liapakis G, Xu R, Guarnieri F, Ballesteros JA, Javitch JA. Beta2 adrenergic receptor activation. Modulation of the proline kink in transmembrane 6 by a rotamer toggle switch. J Biol Chem. 2002;277:40989–40996. doi: 10.1074/jbc.M206801200. [DOI] [PubMed] [Google Scholar]
- 114.Kapur A, Hurst DP, Fleischer D, Whitnell R, Thakur GA, Makriyannis A, Reggio PH, Abood ME. Mutation studies of Ser7.39 and Ser2.60 in the human CB1 cannabinoid receptor: evidence for a serine-induced bend in CB1 transmembrane helix 7. Mol Pharmacol. 2007;71:1512–1524. doi: 10.1124/mol.107.034645. [DOI] [PubMed] [Google Scholar]
- 115.Dryja TP, Berson EL, Rao VR, Oprian DD. Heterozygous missense mutation in the rhodopsin gene as a cause of congenital stationary night blindness. Nat Genet. 1993;4:280–283. doi: 10.1038/ng0793-280. [DOI] [PubMed] [Google Scholar]
- 116.Tao Q, Abood ME. Mutation of a highly conserved aspartate residue in the second transmembrane domain of the cannabinoid receptors, CB1 and CB2, disrupts G-protein coupling. J Pharmacol Exp Ther. 1998;285:651–658. [PubMed] [Google Scholar]
- 117.Roche JP, Bounds S, Brown S, Mackie K. A mutation in the second transmembrane region of the CB1 receptor selectively disrupts G protein signaling and prevents receptor internalization. Mol Pharmacol. 1999;56:611–618. doi: 10.1124/mol.56.3.611. [DOI] [PubMed] [Google Scholar]
- 118.Chin CN, Murphy JW, Huffman JW, Kendall DA. The third transmembrane helix of the cannabinoid receptor plays a role in the selectivity of aminoalkylindoles for CB2, peripheral cannabinoid receptor. J Pharmacol ExpTher. 1999;291:837–844. [PubMed] [Google Scholar]
- 119.Reggio PH, Basu-Dutt S, Barnett-Norris J, Castro MT, Hurst DP, Seltzman HH, Roche MJ, Gilliam AF, Thomas BF, Stevenson LA, Pertwee RG, Abood ME. The bioactive conformation of aminoalkylindoles at the cannabinoid CB1 and CB2 receptors: insights gained from (E)- and (Z)-naphthylidene indenes. J Med Chem. 1998;41:5177–5187. doi: 10.1021/jm9801197. [DOI] [PubMed] [Google Scholar]
- 120.Shen CP, Xiao JC, Armstrong H, Hagmann W, Fong TM. F200A substitution in the third transmembrane helix of human cannabinoid CB1 receptor converts AM2233 from receptor agonist to inverse agonist. Eur J Pharmacol. 2006;531:41–46. doi: 10.1016/j.ejphar.2005.12.026. [DOI] [PubMed] [Google Scholar]
- 121.Fay JF, Dunham TD, Farrens DL. Cysteine residues in the human cannabinoid receptor: only C257 and C264 are required for a functional receptor, and steric bulk at C386 impairs antagonist SR141716A binding. Biochemistry. 2005;44:8757–8769. doi: 10.1021/bi0472651. [DOI] [PubMed] [Google Scholar]
- 122.Song ZH, Bonner TI. A lysine residue of the cannabinoid receptor is critical for receptor recognition by several agonists but not WIN55212-2. Mol Pharmacol. 1996;49:891–896. [PubMed] [Google Scholar]
- 123.Chin CN, Lucas-Lenard J, Abadji V, Kendall DA. Ligand binding and modulation of cyclic AMP levels depend on the chemical nature of residue 192 of the human cannabinoid receptor 1. J Neurochem. 1998;70:366–373. doi: 10.1046/j.1471-4159.1998.70010366.x. [DOI] [PubMed] [Google Scholar]
- 124.Hurst DP, Lynch DL, Barnett-Norris J, Hyatt SM, Seltzman HH, Zhong M, Song ZH, Nie J, Lewis D, Reggio PH. N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (SR141716A) interaction with LYS 3.28(192) is crucial for its inverse agonism at the cannabinoid CB1 receptor. Mol Pharmacol. 2002;62:1274–1287. doi: 10.1124/mol.62.6.1274. [DOI] [PubMed] [Google Scholar]
- 125.Hurst D, Umejiego U, Lynch D, Seltzman H, Hyatt S, Roche M, McAllister S, Fleischer D, Kapur A, Abood M, Shi S, Jones J, Lewis D, Reggio P. Biarylpyrazole inverse agonists at the cannabinoid CB1 receptor: importance of the C-3 carboxamide oxygen/lysine3.28(192) interaction. J Med Chem. 2006;49:5969–5987. doi: 10.1021/jm060446b. [DOI] [PubMed] [Google Scholar]
- 126.Semus SF, Martin BR. A computergraphic investigation into the pharmacological role of the THC-cannabinoid phenolic moiety. Life Sci. 1990;46:1781–1785. doi: 10.1016/0024-3205(90)90142-e. [DOI] [PubMed] [Google Scholar]
- 127.Barnett-Norris J, Hurst DP, Lynch DL, Guarnieri F, Makriyannis A, Reggio PH. Conformational Memories and the Endocannabinoid Binding Site at the Cannabinoid CB1 Receptor. J Med Chem. 2002;45:3649–3659. doi: 10.1021/jm0200761. [DOI] [PubMed] [Google Scholar]
- 128.Shim JY, Welsh WJ, Howlett AC. Homology model of the CB1 cannabinoid receptor: sites critical for nonclassical cannabinoid agonist interaction. Biopolymers. 2003;71:169–189. doi: 10.1002/bip.10424. [DOI] [PubMed] [Google Scholar]
- 129.Salo OM, Raitio KH, Savinainen JR, Nevalainen T, Lahtela-Kakkonen M, Laitinen JT, Järvinen T, Poso A. Virtual screening of novel CB2 ligands using a comparative model of the human cannabinoid CB2 receptor. J Med Chem. 2005;48:7166–7171. doi: 10.1021/jm050565b. [DOI] [PubMed] [Google Scholar]
- 130.Montero C, Campillo NE, Goya P, Páez JA. Homology models of the cannabinoid CB1 and CB2 receptors. A docking analysis study. Eur J Med Chem. 2005;40:75–83. doi: 10.1016/j.ejmech.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 131.Shim JY, Howlett AC. Aminoalkylindole (R)-[2,3-Dihydro-5-methyl-3-[(4-morpholin-yl)methyl] pyrrolo[1,2,3-de]-1,4-benzoxa-zin-6-yl](1-naphthalenyl)methanone (WIN55212-2) Docking to the CB1 Cannabinoid Receptor and a Mechanism for Conformational Induction. J Chem Inf Model. 2006;46:1286–1300. doi: 10.1021/ci0504824. [DOI] [PubMed] [Google Scholar]
- 132.Tuccinardi T, Ferrarini PL, Manera C, Ortore G, Saccomanni G, Martinelli A. Cannabinoid CB2/CB1 selectivity. Receptor modeling and automated docking analysis. J Med Chem. 2006;49:984–994. doi: 10.1021/jm050875u. [DOI] [PubMed] [Google Scholar]
- 133.Padgett LW, Howlett AC, Shim JY. Binding mode prediction of conformationally restricted anandamide analogs within the CB1 receptor. J Mol Signal. 2008;3:5 doi: 10.1186/1750-2187-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Melvin LS, Milne GM, Johnson MR, Subramaniam B, Wilken GH, Howlett AC. Structure-activity relationships for cannabinoid receptor-binding and analgesic activity: studies of bicyclic cannabinoid analogs. Mol Pharmacol. 1993;44:1008–1015. [PubMed] [Google Scholar]
- 135.Fersht AR. Relationships between apparent binding energies measured in site-directed mutagenesis experiments and energetics of binding and catalysis. Biochemistry. 1988;27:1577–1580. doi: 10.1021/bi00405a027. [DOI] [PubMed] [Google Scholar]
- 136.Shim JY. Transmembrane helical domain of the cannabinoid CB1 receptor. Biophys J. 2009;96:3251–3262. doi: 10.1016/j.bpj.2008.12.3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Segrest JP, De Loof H, Dohlman JG, Brouillette CG, Anantharamaiah GM. Amphipathic helix motif: classes and properties. Proteins. 1990;8:103–117. doi: 10.1002/prot.340080202. [DOI] [PubMed] [Google Scholar]
- 138.Strandberg E, Killian JA. Snorkeling of lysine side chains in transmembrane helices: how easy can it get? FEBS Lett. 2003;544:69–73. doi: 10.1016/s0014-5793(03)00475-7. [DOI] [PubMed] [Google Scholar]
- 139.Deol SS, Bond PJ, Domene C, Sansom MS. Lipid-protein interactions of integral membrane proteins: a comparative simulation study. Biophys J. 2004;87:3737–3749. doi: 10.1529/biophysj.104.048397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Aliste MP, MacCallum JL, Tieleman DP. Molecular dynamics simulations of pentapeptides at interfaces: salt bridge and cation-pi interactions. Biochemistry. 2003;42:8976–8987. doi: 10.1021/bi027001j. [DOI] [PubMed] [Google Scholar]
- 141.Anderson MA, Ogbay B, Arimoto R, Sha W, Kisselev OG, Cistola DP, Marshall GR. Relative strength of cation-pi vs salt-bridge interactions: the Gtalpha(340-350) peptide/rhodopsin system. J Am Chem Soc. 2006;128:7531–7541. doi: 10.1021/ja058513z. [DOI] [PubMed] [Google Scholar]
- 142.Shim JY, Collantes ER, Welsh WJ, Subramaniam B, Howlett AC, Eissenstat MA, Ward SJ. A 3D-QSAR Study of the Cannabimimetic Aminoalkylindoles using Comparative Molecular Field Analysis (CoMFA) J Med Chem. 1998;41:4521–4532. doi: 10.1021/jm980305c. [DOI] [PubMed] [Google Scholar]
- 143.Karlin A, Akabas MH. Substituted-cysteine accessibility method. Methods Enzymol. 1998;293:123–45. doi: 10.1016/s0076-6879(98)93011-7. [DOI] [PubMed] [Google Scholar]
- 144.Reggio PH. Ligand-ligand and ligand-receptor approaches to modeling the cannabinoid CB1 and CB2 receptors: achievements and challenges. Curr Med Chem. 1999;6:665–683. [PubMed] [Google Scholar]
- 145.Shire D, Calandra B, Bouaboula M, Barth F, Rinaldi-Carmona M, Casellas P, Ferrara P. Cannabinoid receptor interactions with the antagonists SR 141716A and SR 144528. Life Sci. 1999;65:627–635. doi: 10.1016/s0024-3205(99)00285-4. [DOI] [PubMed] [Google Scholar]
- 146.Shi L, Javitch JA. The second extracellular loop of the dopamine D2 receptor lines the binding-site crevice. Proc Natl Acad Sci USA. 2004;101:440–445. doi: 10.1073/pnas.2237265100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Klco JM, Wiegand CB, Narzinski K, Baranski TJ. Essential role for the second extracellular loop in C5a receptor activation. Nat Struct Mol Biol. 2005;12:320–326. doi: 10.1038/nsmb913. [DOI] [PubMed] [Google Scholar]
- 148.Massotte D, Kieffer BL. The second extracellular loop: a damper for G protein-coupled receptors? Nat Struct Mol Biol. 2005;12:287–288. doi: 10.1038/nsmb0405-287. [DOI] [PubMed] [Google Scholar]
- 149.Gao ZG, Chen A, Barak D, Kim SK, Müller CE, Jacobson KA. Identification by site-directed mutagenesis of residues involved in ligand recognition and activation of the human A3 adenosine receptor. J Biol Chem. 2002;277:19056–19063. doi: 10.1074/jbc.M110960200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Khasawneh FT, Huang JS, Turek JW, Le Breton GC. Differential mapping of the amino acids mediating agonist and antagonist coordination with the human thromboxane A2 receptor protein. J Biol Chem. 2006;281:26951–26965. doi: 10.1074/jbc.M507469200. [DOI] [PubMed] [Google Scholar]
- 151.Gouldson P, Calandra B, Legoux P, Kernéis A, Rinaldi-Carmona M, Barth F, Le Fur G, Ferrara P, Shire D. Mutational analysis and molecular modelling of the antagonist SR 144528 binding site on the human cannabinoid CB(2) receptor. Eur J Pharmacol. 2000;401:17–25. doi: 10.1016/s0014-2999(00)00439-8. [DOI] [PubMed] [Google Scholar]
- 152.Lu R, Hubbard JR, Martin BR, Kalimi MY. Roles of sulfhydryl and disulfide groups in the binding of CP-55,940 to rat brain cannabinoid receptor. Mol Cell Biochem. 1993;121:119–126. doi: 10.1007/BF00925970. [DOI] [PubMed] [Google Scholar]
- 153.Conner M, Hawtin SR, Simms J, Wootten D, Lawson Z, Conner AC, Parslow RA, Wheatley M. Systematic analysis of the entire second extracellular loop of the V(1a) vasopressin receptor: key residues, conserved throughout a G-protein-coupled receptor family, identified. J Biol Chem. 2007;282:17405–17412. doi: 10.1074/jbc.M702151200. [DOI] [PubMed] [Google Scholar]
- 154.Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, Kobilka BK. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 155.Goodwin JA, Hulme EC, Langmead CJ, Tehan BG. Roof and floor of the muscarinic binding pocket: variations in the binding modes of orthosteric ligands. Mol Pharmacol. 2007;72:1484–1496. doi: 10.1124/mol.107.038265. [DOI] [PubMed] [Google Scholar]
- 156.Ahuja S, Hornak V, Yan EC, Syrett N, Goncalves JA, Hirshfeld A, Ziliox M, Sakmar TP, Sheves M, Reeves PJ, Smith SO, Eilers M. Helix movement is coupled to displacement of the second extracellular loop in rhodopsin activation. Nat Struct Mol Biol. 2009a;16:168–75. doi: 10.1038/nsmb.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Swaminath G, Deupi X, Lee TW, Zhu W, Thian FS, Kobilka TS, Kobilka B. Probing the beta2 adrenoceptor binding site with catechol reveals differences in binding and activation by agonists and partial agonists. J Biol Chem. 2005;280:22165–22171. doi: 10.1074/jbc.M502352200. [DOI] [PubMed] [Google Scholar]
- 158.Baldwin JM, Schertler GF, Unger VM. An alpha-carbon template for the transmembrane helices in the rhodopsin family of G-protein-coupled receptors. J Mol Biol. 1997;272:144–164. doi: 10.1006/jmbi.1997.1240. [DOI] [PubMed] [Google Scholar]
- 159.Abadji V, Lucas-Lenard JM, Chin C, Kendall DA. Involvement of the carboxyl terminus of the third intracellular loop of the cannabinoid CB1 receptor in constitutive activation of Gs. J Neurochem. 1999;72:2032–2038. doi: 10.1046/j.1471-4159.1999.0722032.x. [DOI] [PubMed] [Google Scholar]
- 160.Nie J, Lewis DL. Structural domains of the CB1 cannabinoid receptor that contribute to constitutive activity and G-protein sequestration. J Neurosci. 2001;21:8758–8764. doi: 10.1523/JNEUROSCI.21-22-08758.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Palczewski K. G protein-coupled receptor rhodopsin. Annu Rev Biochem. 2006;75:743–767. doi: 10.1146/annurev.biochem.75.103004.142743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Horn F, Weare J, Beukers MW, Hörsch S, Bairoch A, Chen W, Edvardsen O, Campagne F, Vriend G. GPCRDB: an information system for G protein-coupled receptors. Nucleic Acids Res. 1998;26:275–279. doi: 10.1093/nar/26.1.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Fritze O, Filipek S, Kuksa V, Palczewski K, Hofmann KP, Ernst OP. Role of the conserved NPxxY(x)5,6F motif in the rhodopsin ground state and during activation. Proc Natl Acad Sci USA. 2003;100:2290–2295. doi: 10.1073/pnas.0435715100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ahuja S, Crocker E, Eilers M, Hornak V, Hirshfeld A, Ziliox M, Syrett N, Reeves PJ, Khorana HG, Sheves M, Smith SO. Location of the retinal chromophore in the activated state of rhodopsin. J Biol Chem. 2009;284:10190–10201. doi: 10.1074/jbc.M805725200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Singh R, Hurst DP, Barnett-Norris J, Lynch DL, Reggio PH, Guarnieri F. Activation of the cannabinoid CB1 receptor may involve a W6 48/F3 36 rotamer toggle switch. J Pept Res. 2002;60:357–370. doi: 10.1034/j.1399-3011.2002.21065.x. [DOI] [PubMed] [Google Scholar]
- 166.Sealfon SC, Chi L, Ebersole BJ, Rodic V, Zhang D, Ballesteros JA, Weinstein H. Related contribution of specific helix 2 and 7 residues to conformational activation of the serotonin 5-HT2A receptor. J Biol Chem. 1995;270:16683–16688. doi: 10.1074/jbc.270.28.16683. [DOI] [PubMed] [Google Scholar]
- 167.Perlman JH, Colson AO, Wang W, Bence K, Osman R, Gershengorn MC. Interactions between conserved residues in transmembrane helices 1, 2, and 7 of the thyrotropin-releasing hormone receptor. J Biol Chem. 1997;272:11937–11942. doi: 10.1074/jbc.272.18.11937. [DOI] [PubMed] [Google Scholar]
- 168.Urizar E, Claeysen S, Deupi X, Govaerts C, Costagliola S, Vassart G, Pardo L. An activation switch in the rhodopsin family of G protein-coupled receptors: the thyrotropin receptor. J Biol Chem. 2005;280:17135–17141. doi: 10.1074/jbc.M414678200. [DOI] [PubMed] [Google Scholar]
- 169.Gazi L, Nickolls SA, Strange PG. Functional coupling of the human dopamine D2 receptor with G alpha i1, G alpha i2, G alpha i3 and G alpha o G proteins: evidence for agonist regulation of G protein selectivity. Br J Pharmacol. 2003;138:775–786. doi: 10.1038/sj.bjp.0705116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Houston DB, Howlett AC. Differential receptor-G-protein coupling evoked by dissimilar cannabinoid receptor agonists. Cell Signal. 1998;10:667–674. doi: 10.1016/s0898-6568(98)00013-8. [DOI] [PubMed] [Google Scholar]
- 171.Glass M, Northup JK. Agonist selective regulation of G proteins by cannabinoid CB1 and CB2 receptors. Mol Pharmacol. 1999;56:1362–1369. doi: 10.1124/mol.56.6.1362. [DOI] [PubMed] [Google Scholar]
- 172.Tikhonova IG, Sum CS, Neumann S, Engel S, Raaka BM, Costanzi S, Gershengorn MC. Discovery of novel agonists and antagonists of the free fatty acid receptor 1 (FFAR1) using virtual screening. J Med Chem. 2008;51:625–633. doi: 10.1021/jm7012425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Shim JY, Howlett AC. Steric Trigger As a Mechanism for CB1 Cannabinoid Receptor Activation. J Chem Inf Comput Sci. 2004;44:1466–1476. doi: 10.1021/ci040040c. [DOI] [PubMed] [Google Scholar]
- 174.Farrens DL, Altenbach C, Yang K, Hubbell WL, Khorana HG. Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science. 1996;274:768–770. doi: 10.1126/science.274.5288.768. [DOI] [PubMed] [Google Scholar]
- 175.Javitch JA, Fu D, Liapakis G, Chen J. Constitutive activation of the beta2 adrenergic receptor alters the orientation of its sixth membrane-spanning segment. J Biol Chem. 1997;272:18546–18549. doi: 10.1074/jbc.272.30.18546. [DOI] [PubMed] [Google Scholar]
- 176.Jensen AD, Guarnieri F, Rasmussen SG, Asmar F, Ballesteros JA, Gether U. Agonist-induced conformational changes at the cytoplasmic side of transmembrane segment 6 in the beta 2 adrenergic receptor mapped by site-selective fluorescent labeling. J Biol Chem. 2001;276:9279–9290. doi: 10.1074/jbc.M004871200. [DOI] [PubMed] [Google Scholar]
- 177.Ghanouni P, Steenhuis JJ, Farrens DL, Kobilka BK. Agonist-induced conformational changes in the G-protein-coupling domain of the beta 2 adrenergic receptor. Proc Natl Acad Sci USA. 2001;98:5997–6002. doi: 10.1073/pnas.101126198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Greasley PJ, Fanelli F, Rossier O, Abuin L, Cotecchia S. Mutagenesis and modelling of the alpha(1b)-adrenergic receptor highlight the role of the helix 3/helix 6 interface in receptor activation. Mol Pharmacol. 2002;61:1025–1032. doi: 10.1124/mol.61.5.1025. [DOI] [PubMed] [Google Scholar]
- 179.Ward SD, Hamdan FF, Bloodworth LM, Siddiqui NA, Li JH, Wess J. Use of an in situ disulfide cross-linking strategy to study the dynamic properties of the cytoplasmic end of transmembrane domain VI of the M3 muscarinic acetylcholine receptor. Biochemistry. 2006;45:676–685. doi: 10.1021/bi051503q. [DOI] [PubMed] [Google Scholar]
- 180.Katritch V, Reynolds KA, Cherezov V, Hanson MA, Roth CB, Yeager M, Abagyan R. Analysis of full and partial agonists binding to beta2-adrenergic receptor suggests a role of transmembrane helix V in agonist-specific conformational changes. J Mol Recognit. 2009;22:307–318. doi: 10.1002/jmr.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Bramblett RD, Panu AM, Ballesteros JA, Reggio PH. Construction of a 3D model of the cannabinoid CB1 receptor: determination of helix ends and helix orientation. Life Sci. 1995;56:1971–1982. doi: 10.1016/0024-3205(95)00178-9. [DOI] [PubMed] [Google Scholar]
- 182.Xie XQ, Chen JZ, Billings EM. 3D structural model of the G-protein-coupled cannabinoid CB2 receptor. Proteins. 2003;53:307–319. doi: 10.1002/prot.10511. [DOI] [PubMed] [Google Scholar]
- 183.Salo OM, Lahtela-Kakkonen M, Gynther J, Järvinen T, Poso A. Development of a 3D model for the human cannabinoid CB1 receptor. J Med Chem. 2004;47:3048–3057. doi: 10.1021/jm031052c. [DOI] [PubMed] [Google Scholar]
- 184.Gonzalez A, Duran LS, Araya-Secchi R, Garate JA, Pessoa-Mahana CD, Lagos CF, Perez-Acle T. Computational modeling study of functional microdomains in cannabinoid receptor type 1. Bioorg Med Chem. 2008;16:4378–4389. doi: 10.1016/j.bmc.2008.02.070. [DOI] [PubMed] [Google Scholar]
- 185.Yuzlenko O, Kieć-Kononowicz K. Molecular modeling of A1 and A2A adenosine receptors: comparison of rhodopsin- and beta2-adrenergic-based homology models through the docking studies. J Comput Chem. 2009;30:14–32. doi: 10.1002/jcc.21001. [DOI] [PubMed] [Google Scholar]
- 186.Shimamura T, Hiraki K, Takahashi N, Hori T, Ago H, Masuda K, Takio K, Ishiguro M, Miyano M. Crystal structure of squid rhodopsin with intracellularly extended cytoplasmic region. J Biol Chem. 2008;283:17753–17756. doi: 10.1074/jbc.C800040200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Anavi-Goffer S, Fleischer D, Hurst DP, Lynch DL, Barnett-Norris J, Shi S, Lewis DL, Mukhopadhyay S, Howlett AC, Reggio PH, Abood ME. Helix 8 Leu in the CB1 cannabinoid receptor contributes to selective signal transduction mechanisms. J Biol Chem. 2007;282:25100–25113. doi: 10.1074/jbc.M703388200. [DOI] [PubMed] [Google Scholar]
- 188.White SH, Wimley WC. Membrane protein folding and stability: physical principles. Annu Rev Biophys Biomol Struct. 1999;28:319–365. doi: 10.1146/annurev.biophys.28.1.319. [DOI] [PubMed] [Google Scholar]
- 189.Klabunde T, Giegerich C, Evers A. Sequence-derived three-dimensional pharmacophore models for G-protein-coupled receptors and their application in virtual screening. J Med Chem. 2009;52:2923–2932. doi: 10.1021/jm9001346. [DOI] [PubMed] [Google Scholar]
- 190.Reynolds KA, Katritch V, Abagyan R. Identifying conformational changes of the beta(2) adrenoceptor that enable accurate prediction of ligand/receptor interactions and screening for GPCR modulators. J Comput Aided Mol Des. 2009;23:273–288. doi: 10.1007/s10822-008-9257-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Kellenberger E, Springael JY, Parmentier M, Hachet-Haas M, Galzi JL, Rognan D. Identification of nonpeptide CCR5 receptor agonists by structure-based virtual screening. J Med Chem. 2007;50:1294–1303. doi: 10.1021/jm061389p. [DOI] [PubMed] [Google Scholar]
- 192.Kiss R, Kiss B, Könczöl A, Szalai F, Jelinek I, László V, Noszál B, Falus A, Keseru GM. Discovery of novel human histamine H4 receptor ligands by large-scale structure-based virtual screening. J Med Chem. 2008;51:3145–3153. doi: 10.1021/jm7014777. [DOI] [PubMed] [Google Scholar]
- 193.Radestock S, Weil T, Renner S. Homology model-based virtual screening for GPCR ligands using docking and target-biased scoring. JChem Inf Model. 2008;48:1104–1117. doi: 10.1021/ci8000265. [DOI] [PubMed] [Google Scholar]
- 194.Engel S, Skoumbourdis AP, Childress J, Neumann S, Deschamps JR, Thomas CJ, Colson AO, Costanzi S, Gershengorn MC. A virtual screen for diverse ligands: discovery of selective G protein-coupled receptor antagonists. J Am Chem Soc. 2008;130:5115–5123. doi: 10.1021/ja077620l. [DOI] [PubMed] [Google Scholar]
- 195.Cavasotto CN, Orry AJ, Murgolo NJ, Czarniecki MF, Kocsi SA, Hawes BE, O'Neill KA, Hine H, Burton MS, Voigt JH, Abagyan RA, Bayne ML, Monsma FJ. Jr. Discovery of novel chemotypes to a G-protein-coupled receptor through ligand-steered homology modeling and structure-based virtual screening. J Med Chem. 2008;51:581–588. doi: 10.1021/jm070759m. [DOI] [PubMed] [Google Scholar]
- 196.Evers A, Klabunde T. Structure-based drug discovery using GPCR homology modeling: successful virtual screening for antagonists of the alpha1A adrenergic receptor. J Med Chem. 2005;48:1088–1097. doi: 10.1021/jm0491804. [DOI] [PubMed] [Google Scholar]
- 197.Jacob L, Hoffmann B, Stoven V, Vert JP. Virtual screening of GPCRs: an in silico chemogenomics approach. BMC Bioinformatics. 2008;9:363. doi: 10.1186/1471-2105-9-363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Chen JZ, Wang J, Xie XQ. GPCR structure-based virtual screening approach for CB2 antagonist search. J Chem Inf Model. 2007;47:1626–1637. doi: 10.1021/ci7000814. [DOI] [PubMed] [Google Scholar]
- 199.Wang H, Duffy RA, Boykow GC, Chackalamannil S, Madison VS. Identification of novel cannabinoid CB1 receptor antagonists by using virtual screening with a pharmacophore model. J Med Chem. 2008;51:2439–2446. doi: 10.1021/jm701519h. [DOI] [PubMed] [Google Scholar]
- 200.Blundell TL, Sibanda BL, Montalvão RW, Brewerton S, Chelliah V, Worth CL, Harmer NJ, Davies O, Burke D. Structural biology and bioinformatics in drug design: opportunities and challenges for target identification and lead discovery. Philos Trans R Soc Lond B Biol Sci. 2006;361:413–423. doi: 10.1098/rstb.2005.1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Hajduk PJ, Greer J. A decade of fragment-based drug design: strategic advances and lessons learned. Nat Rev Drug Discov. 2007;6:211–219. doi: 10.1038/nrd2220. [DOI] [PubMed] [Google Scholar]
- 202.Congreve M, Chessari G, Tisi D, Woodhead AJ. Recent developments in fragment-based drug discovery. J Med Chem. 2008;51:3661–3680. doi: 10.1021/jm8000373. [DOI] [PubMed] [Google Scholar]
- 203.Miranker A, Karplus M. Functionality Maps of Binding Sites: a Multiple Copy Simultaneous Search Method. Proteins: Struct Funct Genet. 1991;11:29–34. doi: 10.1002/prot.340110104. [DOI] [PubMed] [Google Scholar]
- 204.Stultz CM, Karplus M. In: Fragment-based approaches in drug discovery. Jahnke W, Erlanson DA, editors. Wiley-VCH; Weinheim: 2006. [Google Scholar]
- 205.Schubert CR, Stultz CM. The multi-copy simultaneous search methodology: a fundamental tool for structure-based drug design. J Comput Aided Mol Des. 2009 Jun 9; doi: 10.1007/s10822-009-9287-y. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 206.Leonardi A, Barlocco D, Montesano F, Cignarella G, Motta G, Testa R, Poggesi E, Seeber M, De Benedetti PG, Fanelli F. Synthesis, screening, and molecular modeling of new potent and selective antagonists at the alpha 1d adrenergic receptor. J Med Chem. 2004;47:1900–1918. doi: 10.1021/jm030944+. [DOI] [PubMed] [Google Scholar]
- 207.Bissantz C, Bernard P, Hibert M, Rognan D. Protein-based virtual screening of chemical databases. II. Are homology models of G-Protein Coupled Receptors suitable targets? Proteins. 2003;50:5–25. doi: 10.1002/prot.10237. [DOI] [PubMed] [Google Scholar]
- 208.Tanrikulu Y, Proschak E, Werner T, Geppert T, Todoroff N, Klenner A, Kottke T, Sander K, Schneider E, Seifert R, Stark H, Clark T, Schneider G. Homology model adjustment and ligand screening with a pseudoreceptor of the human histamine H4 receptor. ChemMedChem. 2009;4:820–827. doi: 10.1002/cmdc.200800443. [DOI] [PubMed] [Google Scholar]
- 209.de Graaf C, Rognan D. Selective structure-based virtual screening for full and partial agonists of the beta2 adrenergic receptor. J Med Chem. 2008;51:4978–4985. doi: 10.1021/jm800710x. [DOI] [PubMed] [Google Scholar]
- 210.Liapakis G, Chan WC, Papadokostaki M, Javitch JA. Synergistic contributions of the functional groups of epinephrine to its affinity and efficacy at the beta2 adrenergic receptor. Mol Pharmacol. 2004;65:1181–1190. doi: 10.1124/mol.65.5.1181. [DOI] [PubMed] [Google Scholar]
- 211.Notredame C, Higgins DG, Heringa J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 212.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. ClustalW and ClustalX version 2. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]