

Abstract
Chronic gastric infection by the gram-negative bacterium H. pylori is strongly associated with the development of distal gastric carcinoma and gastric mucosal lymphoma in humans. Eradication of H. pylori with combination antibiotic therapy cures most cases of gastric lymphoma and slows progression to gastric adenocarcinoma. H. pylori promotes gastric neoplasia, principally via the induction of an intense gastric inflammatory response that lasts over decades. This persistent inflammatory state produces chronic oxidative stress and adaptive changes in gastric epithelial and immune cell pathobiology that in a minority of infected subjects eventually proceeds to frank neoplastic transformation.
Keywords: Helicobacter pylori infections, gastric cancer, lymphoma, mechanisms, inflammation, carcinogenesis
1. Introduction
The two common malignant neoplasms that arise in the stomach are adenocarcinoma and lymphoma of gastric mucosal associated lymphoid tissue (MALT). While the incidence of gastric carcinoma has declined in many developing countries, it is still exceedingly prevalent in most of the developing world, and is the second leading cause of cancer-related death worldwide [1]. Most gastric cancers are still detected at an advanced stage. Consequently, the prognosis of this disease remains very poor, even after extensive surgery and adjuvant therapy [2]. Gastric MALT lymphoma is considerably less common than gastric carcinoma, accounting for 3% of all gastric tumors [3].
Both gastric carcinomas and MALT lymphomas have long been recognized to occur on a background of chronic gastric inflammation. For the past two decades it has been evident that the usual cause of this gastritis is persistent infection by the gram-negative micro-aerophilic bacterium Helicobacter pylori (H. pylori). Approximately 70% of all gastric cancer cases worldwide are directly attributable to prior H. pylori infection [4], as are the majority of gastric MALT lymphomas [3].
Currently, about half of the world’s population is infected by H. pylori, with rates in the developed world in the order of 70% [5]. Gastric colonization by H. pylori is usually asymptomatic and although about 20% of the infected population progress to some extent down the “Correa” pathway of pre-neoplastic changes over several decades, gastric neoplasms develop in fewer than 2% [6]. Gastric lymphoma is an even rarer consequence of H. pylori infection, occurring in fewer than 1% of those who are infected. However, based upon the available epidemiological evidence, the World Health Organization’s International Agency for Research on Cancer classified H. pylori as a group I or definite carcinogen (the only bacterium to be thus classified) in 1994 [7]. Since that time evidence linking H. pylori to gastric cancer has continued to accumulate and strengthen.
Numerous epidemiological, animal and experimental studies support a positive association between chronic H. pylori infection and the development of distal gastric cancer and gastric MALT lymphoma. However, the molecular cellular events responsible for the promotion of these gastric malignancies by H. pylori remain poorly defined. Current evidence suggests that the bacterium itself has carcinogenic effects and that the inflammatory response to H. pylori, which is highly variable, can contribute to lowering the threshold for gastric cancer development.
Among the molecular mechanisms that are thought to be important in H. pylori-induced gastric carcinogenesis are the induction of oxidative and nitrosative stress with consequent cellular and DNA damage followed by cycles of repair. Ultimately, as antioxidant defenses and damage- repair responses are overwhelmed and depleted, genetic errors that arise under the pressure of accelerated gastric epithelial turnover may accumulate to the point at which neoplastic transformation is inevitable. Many of these events occurring in the chronically inflamed gastric mucosa are common to other inflammation-associated malignancies, while some are unique to H. pylori infection [8]. In this review we shall discuss Helicobacter pylori as an agent in gastric carcinogenesis and consider the mechanisms responsible for its pathogenesis.
2. Helicobacter pylori
2.1 Biology, heterogeneity and niche
Helicobacter pylori (H. pylori) is a gram-negative spiral-shaped bacterium. Usually acquired in infancy, this bacterium induces chronic gastric inflammation persisting for the life of its host [5]. Spontaneous loss of H. pylori from the stomach is rare unless the gastric mucosa has become hostile to continued colonization, as may happen during extensive intestinal metaplasia of the gastric epithelium. Because H. pylori does not adhere well to intestinal mucosal cells, the evidence linking H. pylori infection with intestinal-type gastric cancer may not be apparent when intestinal metaplasia dominates gastric topography. For the same reasons, serum antibody levels against H. pylori antigens decline during progression to gastric cancer. Thus, the role of H. pylori in gastric carcinogenesis was initially underappreciated since early studies of gastric cancer relied on the detection of H. pylori exposure by histological analysis of resection specimens or simultaneous serology.
H. pylori adheres to surface epithelial cells of the stomach; however, it may also colonize the proximal duodenum (or rarely the esophagus) when there is gastric metaplasia in those sites. Rare colonization of the gastric metaplasia of a Meckel’s diverticulum or in the rectum has also been described [9]. Though H. pylori is predominantly extracellular, H. pylori has been occasionally described in an intracellular location within gastric epithelial cells, particularly in cancers [10]. It has been postulated that the intracellular location of H. pylori may facilitate persistence and the acquisition of resistance to antibiotics [11].
H. pylori possesses several mechanisms to survive and persist in the gastric lumen. For example, it utilizes its highly active urease enzyme to buffer a gastric environment of pH1-2 [12] and survival is facilitated by its helical morphology and unipolar flagella enabling movement within the gastric mucous layer overlaying gastric epithelial cells [13]. Coccoidal forms of H. pylori morphology have also been observed in vivo after antibiotic treatment and in vitro as bacterial cultures age, but it is not clear whether these coccoidal forms are viable and therefore retain pathogenic potential.
2.2 H. pylori genomics
Worldwide variations in the H. pylori genome are strongly associated with migration patterns of human populations, suggesting that the first humans were already infected by Helicobacter pylori as they moved away from East Africa about 60,000 years ago [14]. The first H. pylori genome sequence was published in 1997 [15]; there are now at least 7 full genome sequences available in the public domain. [16--21]. In general, the H. pylori genome comprises about 1.6 megabases, encoding approximately 1,500 predicted open reading frames. About 20–30% of the genome is variable between different strains. This is a relatively high percentage among bacterial species and is thought to result from a high spontaneous mutation rate and a relatively high recombination frequency [22].
Several highly variable regions within the H. pylori genome have been identified including the so-called “plasticity zone” and “cytotoxin associated gene” (cag) pathogenicity island. The cag island encodes several structural proteins important in assembling a type four secretion system capable of translocating H. pylori products (including the immunodominant 120–145 kDa CagA protein) directly inside host gastric epithelial cells [23]. The H. pylori genome also encodes several adhesins that are important for ensuring tight contact between H. pylori and gastric epithelial cells. These include blood group antigen binding adhesin (BabA) and the sialic acid binding adhesin, SabA. H. pylori’s vacA gene encodes a multimeric vacuolating exotoxin (VacA), an 88 kDa secretory protein which has the potential of forming intracellular vacuoles in gastric and other epithelial cells [24]. Presence of vacA is conserved among all H. pylori strains, but the gene exhibits a high level of genetic diversity within regions encoding the signal sequence, intermediate element, and the middle portion of the VacA protein[25].
Recent analysis of the transcriptome of H.pylori strain 26695, has revealed the presence of both sense and antisense transcription from common RNA sequences [26], introducing another level of complexity to the genome. Multiple RNA forms were also reported, including non-coding RNAs facilitating regulation of specific patterns of gene transcription under acidic stress, or as H. pylori receives signals from underlying gastric epithelial cells.
3. Gastric Adenocarcinoma
Gastric adenocarcinomas usually arise in the distal stomach (antrum or body) and are strongly associated with H. pylori. Cancers of the proximal stomach (cardia and gastroesophageal junction cancers) have different epidemiological and pathobiological characteristics, and are not commonly found in parts of the world with high H. pylori infection rates. The two main histological subtypes of gastric cancer that are highly associated with prior H. pylori infection are the “intestinal” subtype and the “diffuse” subtype [6]. The intestinal subtype is the dominant form throughout most of the developing world and is characterized by a well described preneoplastic sequence manifesting from chronic superficial gastritis through atrophic gastritis, intestinal metaplasia, and dysplasia to cancer as described originally by Correa et al [27]. Progression through this histological sequence is accompanied by the accumulation of mutations typical of a multistep process of carcinogenesis. The diffuse type of gastric cancer is a more undifferentiated type of cancer in which tumor cells are dispersed and not organized into glandular structures. Diffuse gastric cancer is preceded by a less well-defined histological progression without recognizable histological precursor lesions other than chronic gastritis.
3.1 Evidence for the carcinogenicity of H. pylori from human epidemiological studies
Although it is well established that gastric cancers normally arise in a stomach that is chronically inflamed, it was initially difficult to demonstrate the association of H. pylori with gastric cancer from histological studies. This is likely due to the diminished mucosal adherence of H. pylori as gastric cells become metaplastic, and with decreased acid secretion consequent to loss of specialized acid secreting cells (parietal cells) from gastric gland atrophy, a gastric luminal environment increasingly hostile to the survival of this acidophilic bacterium.
However, by the early 1990s, several prospective observational studies had been completed that were so conclusive of a positive association between H. pylori infection and non-cardia gastric cancer that the World Health Organization declared H. pylori a gastric carcinogen in 1994 [7]. Subsequent studies using more sensitive assays to document H. pylori (such as detecting the long-lived anti-CagA antibodies, or gastric biopsy PCR) have confirmed that H. pylori infection is common in non-cardia gastric cancers. Overall, the matched odds ratio for H. pylori infection in non-cardia gastric cancer ranges from as low of 2 to as high as 20 with no substantial differences between males or females or between gastric cancers of the intestinal subtype or of the diffuse histological subtype.
In contrast to the consistent positive results for an association between H. pylori and non-cardia gastric cancer, no significant associations have been reported between H. pylori and cancers of the gastric cardia. Overall, the matched odds ratio in meta-analysis of H. pylori infection and gastric cardia cancer is around 1.0 [28], with some studies reporting an inverse relationship, and thus, a possible “protective effect” of H. pylori against cardia gastric cancer [29].
The strong association of H. pylori with gastric cancer has spurred a large number of randomized controlled trials to investigate the effects of H. pylori eradication on gastric cancer occurrence. Most have been underpowered, and were thus unable to demonstrate a statistically significant effect of H. pylori eradication on gastric cancer as an endpoint. Nevertheless, most have shown reductions in gastric cancer development by about a third- a reduction that has been limited mainly to patients who received H. pylori eradication at a relatively early stage in their disease progression, before the development of intestinal metaplasia. In meta-analysis, the relative risk of gastric cancer following H. pylori eradication was calculated to be 0.65 overall (95% confidence interval 0.43–0.98) [30]. Given ethical concerns regarding entering patients at high risk of gastric cancer into the placebo arms of such prospective experiments, the requirement for very large, lengthy, and expensive studies to identify relatively rare endpoints, it is unlikely randomized controlled studies will ever be able to define the cancer-preventing benefits of H. pylori eradication with greater precision.
The concept that H. pylori eradication can reduce the risk for gastric cancer is strongly supported by a large study of patients who were at risk for a second gastric malignancy following endoscopic mucosal resection of their first gastric cancers. Following removal of their “early” gastric cancer” (lesion confined to the mucosa) a Japanese multicenter group randomized such patients to either eradication therapy or standard care [31]. Patients who had been successfully eradicated of H. pylori following their initial gastric cancer resection had a highly significantly reduced risk of developing a second gastric cancer (hazard ratio 0.35 at 3 years follow up).
Taken together, the epidemiological evidence to support the case for H. pylori eradication in patients at risk for gastric cancer has been sufficient to convince an Asian-Pacific consensus panel to recommend serological screening for H. pylori to start in early adulthood in this region where gastric cancer is particularly prevalent [32].
3.2 Variations in H. pylori strains and gastric cancer risk
Because the development of gastric cancer following H. pylori infection is relatively infrequent and H. pylori is a highly heterogeneous bacterium, there has been considerable effort spent investigating the relationship between the presence of specific H. pylori factors and the occurrence of gastric cancer, particularly regarding CagA, a putative bacterial oncoprotein.
i. Cag pathogenicity island
Most strains of H. pylori carry the cag pathogenicity island [33]. Strains lacking this region of the genome (cag-negative strains) are less likely to be associated with gastric cancer. In a meta-analysis of retrospective and prospective studies [34], H. pylori infection was associated with an odds ratio for non-cardia gastric cancer of 2.7, with a further additional risk of 2.0 for infections with CagA-positive compared with CagA-negative strains. The proportion of strains that are cag-positive varies among geographical regions, but in general, almost all strains are cag-positive where gastric cancer prevalence is highest (such as Southeast Asia). Within such high gastric cancer incidence regions, differences in gastric cancer susceptibility are partly related to sequence variation within the CagA protein’s carboxyl terminal EPIYA pentapeptide repeat motif. This sequence is a target of phosphorylation following translocation of the CagA protein into gastric epithelial cells [35]. Variations in the number of repeats of the EPIYA sequence correlates with different levels of intracellular CagA phosphorylation by Src kinases. High levels of phosphorylation corresponds to more cytoskeletal reorganization following CagA transfection in vitro, thus providing a mechanistic correlate underpinning the epidemiological association of specific EPIYA sequence with clinical outcomes[25, 35, 14, 36].
The importance of the cag pathogenicity island to the development of gastric cancer is supported by results in the Mongolian gerbil model, where infection with a cag-negative isogenic mutant strain was unable to produce gastric cancer, unlike its wild-type counterpart [37]. Similar results have been seen in mice that are prone to gastric cancer by virtue of genetic overexpression of the human gastrin gene [38]. In these hypergastrinemic mice, H. pylori with functional disruption of the cag secretion system due to isogenic deletion of the cagE gene were less likely to promote gastric cancer when compared with wild type H. pylori.
The consequences of translocating CagA into gastric epithelial cells by H. pylori’s type four secretory system have been studied extensively [39]. Assembly of a functional type four secretory system is dependent upon many genes within the cag locus. H. pylori’s cagL protein docks with the α5β1 integrin on gastric epithelial cell surface to initiate an intimate interaction between H. pylori’s cag pilus and the host cell plasma membrane. This then facilitates translocation of H. pylori products, including CagA. Once inside gastric epithelial cells, CagA becomes phosphorylated at EPIYA sites by gastric epithelial Src kinases. Phospho-CagA activates the SHP2 oncoprotein and reacts with and inhibits the PAR1/MARK kinase that is normally important in maintaining gastric epithelial cell polarity and junctional integrity [40]. The net effects of CagA phosphorylation include reorganization of the actin cytoskeleton, increased cell proliferation, motility and activation of inflammatory cells, and mitogenic gene transcription. The morphological changes induced by CagA in the AGS gastric epithelial cell line have been termed the “hummingbird” phenotype and are reminiscent of the responses of many epithelial cell types to hepatocyte growth factor stimulation [41]. This phenotype may reflect an epithelial to mesenchymal transition towards a more invasive state, though this has not been formally proven. Numerous other cellular effects consequent to CagA translocation have been described, but not all require CagA phosphorylation [39]. For example, interaction of non-phosphorylated CagA with occludin, zonulin 1, junctional adhesion molecules, and other elements of tight and adherens junctions between adjacent gastric epithelial cells have been described in cell culture models. Disruption of such intercellular junctions may facilitate gastric epithelial cell damage and mucosal ulceration. It may also allow H. pylori antigens access to mucosal immune cells.
While most of the effects of the cag pathogenicity island in vitro have been attributed to translocation of CagA and its consequent effects inside gastric epithelial cells, the intact type four secretion system also has the potential to translocate other H. pylori products. Muramyldipeptides, components of H. pylori’s peptidoglycan cell wall, may also be translocated inside gastric epithelial cells where they interact with NOD1, an intracellular pathogen recognition protein that functions in the activation of innate immune responses [42]. NOD1 signaling then stimulates type 1 interferon secretion, boosting innate immune defenses against H. pylori and potentially other mucosal infections [43].
Despite extensive dissection of the downstream pathways activated by intracellular CagA translocation in vitro, direct evidence of the oncogenicity of CagA has been fairly elusive.
Ohnishi et al have demonstrated that the transgenic expression of CagA in mice can promote gastric polyps and, very rarely, cancer [44]. However, despite targeting transgenic cagA expression to gastric epithelial cells, both gastric and small intestinal cancers were observed, as well as some leukemias. While this model does not convincingly recapitulate the effects of CagA as a gastric-specific oncoprotein with high penetrance, it did emphasize the importance of CagA phosphorylation because the phenotype was not reproduced by transgenic overexpression of a non-phosphorylable form of CagA. Interestingly, in this model tumors developed without preceding inflammation or gastric precursor histological lesions (such as intestinal metaplasia). In clinical studies, carriage of the cag pathogenicity island has been consistently linked to more severe gastric inflammation, but this may not necessarily be a causal association. Given the importance of the inflammatory response to H. pylori in the etiology of gastric cancer, the relatively weak oncogenic activity of transgenically expressed CagA may be explained by a lack of the associated intense inflammatory response that typically follows H. pylori infection.
ii. Other H. pylori factors possibly associated with increased gastric cancer risk
In cell culture, VacA produces endosomal vacuoles in epithelial cells, induces apoptosis, and inhibits the proliferation of T lymphocytes, which may be important in promoting gastric mucosal damage and in immune evasion. Variations in the structure of H. pylori’s VacA protein (and thus its in vitro activity) have been studied with respect to clinical outcomes. However, despite a fairly consistent association of specific VacA subtypes with peptic ulcer disease, sequence variation within the VacA signal or middle regions has not been consistently associated with heightened gastric cancer risk. Recently, another variable region within the vacA gene (the i or intermediate region) was identified. Initial studies have suggested that i region polymorphisms are associated with altered gastric cancer risk in Iran and Italy [25,45], though one study from East Asia has not confirmed this finding[36]. Interestingly, isogenic deletion of vacA did not alter the carcinogenic potential of H. pylori in the gerbil model [37], raising questions once again regarding the relevance of VacA to gastric carcinogenesis.
In an attempt to identify H. pylori markers of enhanced carcinogenic potential in humans, Gao and colleagues have screened the serum of patients with H. pylori-induced gastric cancer compared with non-cancer controls [46]. In addition to confirming the presence of serum antibodies against CagA as gastric cancer risk markers, their multiplex serology approach led to the identification of another H. pylori protein of potential relevance to gastric cancer: GroEL. The presence of serum antibodies against GroEL, a chaperonin important for bacterial protein trafficking, was highly associated with the presence of gastric cancer, suggesting that examination of GroEL function is worthy of further detailed investigation.
3.3 Lessons from animal models of H. pylori-induced gastric cancer
Several animal models of H. pylori-induced gastric cancer have been developed. The first convincing report of gastric cancer induction by H. pylori alone (without co-carcinogens) was by Watanabe and colleagues who infected outbred Mongolian gerbils [47]. Others have attempted to recapitulate these findings with varying degrees of success. Variations in the source of these animals, the strains of Helicobacters used for infection, and the conditions under which the gerbils are housed (and thus the extra-gastric microbiome) may be responsible for differences in inter-laboratory success rates with this model. Nevertheless, some groups have been able to reproducibly demonstrate H. pylori-induced gastric cancer in gerbils and thus have provided evidence that antibiotics used to eradicate the organism could prevent gastric cancer when the organism is eradicated relatively early in the animal’s lifespan[48].
Most wild-type murine strains do not develop gastric cancer following experimental infection with mouse-adapted H. pylori strains, with the exception of hybrid B6129 mice [49]. As for the gerbil model, early eradication of H. pylori with antibiotics can greatly decreases the incidence of gastric adenocarcinoma in mouse models [50].
Because of the difficulty of recapitulating H. pylori-induced gastric cancer in wild-type mice, several investigators have infected genetically altered mice that have an increased predisposition to gastric cancer. Examples include, transgenic mice over-expressing human gastrin [38] or murine interleukin-1[51], and knockout mice deficient in transforming growth factor β [52], trefoil family factor 2 [45], or the CDNK1 gene encoding the tumor suppressor protein p27 [53]. Overall, these experiments have demonstrated synergy between the predisposed genetic abnormality and exogenous H. pylori infection [54].
Cancer has also been induced in rhesus macaque monkeys by a combination of H. pylori infection and ethyl-nitro-nitrosoguanidine administration, suggesting that dietary nitrosamines may be necessary co-factors for gastric cancer development in primates [55].
Taken together, it may be concluded that the development of gastric cancer in these animal models is dependent upon the strain of H. pylori, the strain of the host, and the length of time of infection. Furthermore, H. pylori eradication can lead to significant reduction in gastric cancer incidence, especially when given at a relatively early stage of infection. Intervention studies in rodent species indicate that the biggest reduction in cancer incidence occurs when antibiotics to eradicate H. pylori are given before the mid time point between experimental infection and gastric cancer development in the uneradicated mice. Extrapolating to humans, this may correspond to intervening with eradication therapy before the age of about 40 years – a conclusion consistent with recommendations of the Asia-Pacific consensus panel regarding prophylactic H. pylori screening and eradication programs in at-risk populations [32].
3. 4 Mechanisms of H. pylori-induced gastric carcinogenesis
The chronic inflammatory state induced by persistent H. pylori infection over decades provides a rich milieu of inflammatory cytokines, as well as reactive oxygen and nitrogen species that are capable of inducing cellular damage and mutagenesis. Increased cell turnover in this environment can lead to the emergence of cell lineages not normally found in the stomach (gastric metaplasia), and in a small subset of humans infected chronically by H. pylori, the emergence of neoplastic clones under the pressure of accelerated DNA replication [56]. In this respect, a decades-long chronic inflammatory condition preceding cancer is a feature of many common human malignancies, especially of the gastrointestinal tract. Other such examples include chronic acid reflux esophagitis (predisposing to Barrett’s metaplasia and esophageal adenocarcinomas), chronic viral hepatitis (increasing the risk for hepatocellular cancer) and the chronic colonic inflammation of the inflammatory bowel diseases that is associated with increased risk of colon cancer [8]. Whereas the underlying cause of the inflammatory response is not always well defined, it is so for gastric carcinoma. Some of the mechanisms involved in H. pylori-induced gastric inflammation that are of probable importance in gastric cancer causation are outlined below and listed in Figure 1.
Figure 1.
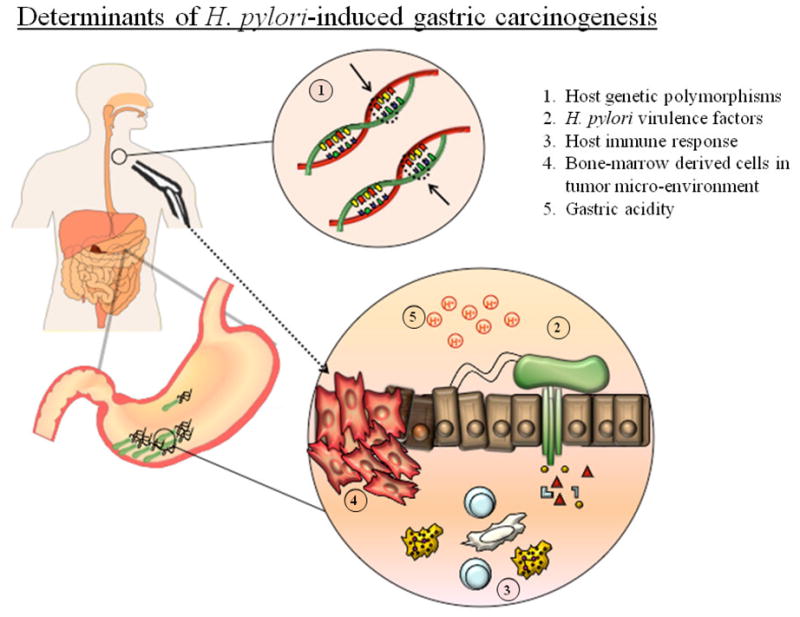
Determinants of H. pylori-induced gastric carcinogenesis. Factors contributing to increased risk of gastric cancer following infection with H. pylori are (1) certain host genetic polymorphisms, (2) H. pylori virulence factors, including those translocated inside epithelial cells via the type 4 secretory system shown in green, (3) the host immune response, (4) the recruitment of bone-marrow derived cells into the gastric mucosa and (5) high intragastric luminal pH.
i. Oxidative stress, DNA damage and cell cycle dysregulation
While nitric oxide production by mucosal macrophages may be important as a host defense mechanism against H. pylori infection[57], reactive oxygen and nitrogen species generated in the H. pylori-infected gastric mucosa also cause oxidative stress and apoptosis in gastric epithelial cells [58]. These processes may be linked through activation of apurinic/apyrimidinic endonuclease-1 [59]. Convincing evidence that H.pylori-induced inflammation can cause DNA damage in human gastric mucosa is lacking, although two groups have reported mutagenesis in experimental Helicobacter infection in mice [60, 60, 61]. The accumulation of mutations, for example in the TP53 tumor suppressor gene, may be related to H. pylori increasing the expression in gastric epithelial cells of activation-induced cytidine deaminase, a nucleic acid-editing enzyme [62].
H. pylori infection induces gastric epithelial cell apoptosis and a compensatory gastric epithelial hyperproliferative response [63]. However, in some model systems, H. pylori can inhibit the apoptotic response of gastric epithelial cells [64], a finding that has been interpreted as evidence of a strategy devised to limit host cell damage and enable bacterial persistence. A deleterious consequence of uncoupling apoptosis from proliferation is an imbalance in normal gastric mucosal homeostasis leading to dysregulated tissue growth. Impairment of normal apoptotic pathways that are commonly stimulated by chronic H. pylori infection may be mediated through down-regulation of fas-mediated signaling [65] or loss of the p27 tumor suppressor protein [66].
ii. Changes in epithelial gene expression and regulation
Numerous changes have been reported in gastric epithelial cells infected by H. pylori in vitro and in vivo, thus identifying pathways that may be of importance in promoting gastric epithelial cell transformation. While TP53 mutation is one of the common molecular hallmarks of gastric cancer, studies concerning the regulation of wild-type p53 expression by H. pylori during the preneoplastic stages have been inconsistent. However, the p53 homolog p73 has been recently identified as being highly responsive to H. pylori and might be more important than p53 in regulating apoptotic responses in gastric epithelial cells [67].
Proteomic and genomic analyses of the effects of H. pylori on gastric epithelial cells in vivo and in vitro have been utilized to explore global changes in gene expression resulting from H. pylori infection. For example, comparing the transcriptional profile of laser-capture microdissected gastric epithelial cells from the same patients before versus after H. pylori eradication led to the identification of the genes encoding gastrokine 1 and gastrokine 2 as being markedly down-regulated by chronic H. pylori infection[68]. Relatively little is known about the function of these two highly homologous proteins. However, because of their potential interactions and co-expression with members of the trefoil factor family of proteins known to be involved in gastric epithelial repair, restitution, and tumor growth, it is likely that gastrokine 1 and 2 may play an important role in the maintenance of normal gastric epithelial cell turnover. Indeed, their loss in gastric cancer, particularly in cancers of the diffuse type, has suggested that they may have tumor suppressor activity [69].
H. pylori-associated chronic gastritis and gastric carcinogenesis is accompanied by widespread epigenetic changes such as DNA methylation and histone modifications. Recent studies have demonstrated a causal role of H. pylori infection and specific alterations of DNA methylation patterns in the gastric mucosa of H. pylori infected patients and in gastric cancer cell lines in vitro. Genes specifically methylated during H. pylori infection include the E-cadherin (CDH1), gene this may be of particular importance in diffuse type gastric carcinogenesis, given that germline mutations in CDH1 are responsible for the syndrome of hereditary diffuse gastric cancer and are commonly acquired in sporadic diffuse type gastric cancer associated with H. pylori infection [70]. Methylation of the E-cadherin promoter can be reversed by eradication of H. pylori [71]. H. pylori infection also causes hypermethylation and thereby decreasing expression of the DNA repair protein O6-methylguanine DNA methyltransferase, an enzyme that normally prevents cytosine: guanine to adenine: thymine transition mutations, and thus DNA replication fidelity [72].
Eradication of H. pylori from infected patients decreased methylation patterns of a broad range of genes, though levels remained higher than in uninfected patients, suggesting there may be a persistent derangement of methylation even after successful eradication of the infection [73]. In the gerbil model of H. pylori infection inhibiting the immune response to H. pylori via immunosuppressive drugs abrogated the induction of altered DNA methylation patterns suggesting that the inflammatory response to H. pylori may have the primary role in gene methylation during H. pylori infection[73,74].
Epigenetic events other than hypermethylation have not been studied in detail; however, histone H4 acetylation of the promoter region of the TP21 tumor suppressor gene has been recently described [75].
Unique signatures of microRNA expression are being identified in many human cancers. Several groups have recently examined the microRNA signatures of H. pylori infection and their results suggest that dysregulation of miRNA expression is a plausible mechanistic link between H. pylori infection and the development of gastric disease [76]. For example,.Matsushima et al demonstrated differential expression of microRNAs between H.pylori positive and negative patients, with restoration of 14 of 30 miRNAs after H. pylori eradication [77]. Furthermore, altered expression of certain miRNAs such as let-7 family members was dependent upon the presence of the cag pathogenicity island. miR-21 and miR-155 have been reported to be upregulated during H. pylori infection; further insights into the significance of these observations in vivo is awaited [78--80].
iii. Loss of gastric acidity
Although H. pylori is the dominant bacterial species capable of survival in a pH of 2 to 3 that is typically found within the gastric lumen, bacteria that are normally resident in the oral cavity and elsewhere in the gastrointestinal tract can colonize the stomach if the gastric pH approaches 7. It has long been recognized that chronic gastric inflammation predisposes certain individuals to gastric cancer through atrophy of gastric glands, including loss of the specialized acid-secreting parietal cells [27]. In this state of hypochlorhydria, non-Helicobacter species may thrive in the gastric lumen and generate carcinogenic nitrosamines with genotoxic potential [81].
The interplay between H. pylori infection, gastric acid secretion, and clinical outcome is complex and depends to a large extent upon the region within the stomach of maximal H. pylori infection. For example, patients who develop duodenal ulcers have high acid secretory rates despite being almost universally infected by H. pylori, but they have a very low incidence of developing gastric cancer [82]. This discrepancy (high H. pylori prevalence, but low gastric cancer risk) is explicable on the basis of H. pylori infection in such patients being confined to the gastric antrum. This causes depletion of the somatostatin secreting cells residing there and subsequent exaggerated release of the acid secretory hormone gastrin from the specialized antral gastrin secreting neuroendocrine cells. Because the proximal stomach is not infected by H. pylori in duodenal ulcer patients, the high gastrin level stimulates the healthy parietal cells of the proximal stomach to hypersecrete gastric acid, thus causing damage (ulcers) in the proximal duodenum [83]. In contrast to the duodenal ulcer phenotype, patients who go on to develop gastric cancer typically have an intense and diffuse gastric inflammation when infected by H. pylori. Though their gastrin levels are also high, their parietal cells are hypofunctional in an environment of intense proximal stomach inflammation and eventually glandular atrophy consequent to H. pylori-induced inflammation. The net result is partial or even total loss of gastric acid secretion.
iv. The inflammatory response to H. pylori – necessary or sufficient?
As discussed above, gastric cancers typically develop in those patients who have the most intense inflammatory response to H. pylori. H. pylori infection is known to induce a robust cellular and humoral response composed of an infiltration of neutrophils, eosinophils, plasma cells and lymphocytes, followed by gastric epithelial cell damage [84]. Such a severe immune response leads to the secretion of pro-inflammatory cytokines, which can be both beneficial and detrimental to the host. An inflammatory environment is associated with increased cell epithelial cell turnover that can lead, stochastically, to an increased chance of mutation bearing cells arising and then becoming the precursors of a neoplastic clone [56].
The model of H. pylori promoting cancer development through increased epithelial cell turnover via inflammation has been challenged by the provocative finding of Houghton and colleagues who performed a series of bone marrow transplantation experiments in mice chronically infected with the related Helicobacter species, H. felis [85]. Surprisingly, the dysplasias and cancers that arose appeared to be the result of the malignant transformation of recruited bone marrow-derived cells (Figure 2).
Figure 2.

In the conventional model of H. pylori-associated carcinogenesis (left), the bacterium and associated inflammatory response stimulate increased cellular turnover and oxidative stress that promote oncogenic mutations, epigenetic changes and altered gene expression in epithelial cells. An alternative model (right) implicates Helicobacter species as the initiator of a chronic inflammatory response that recruits bone marrow derived cells to the gastric mucosa, contributing directly to the gastric neoplasm.
The hypothesis that bone-marrow derived lineages are critical for Helicobacter-induced gastric cancer is supported by Tu and colleagues who demonstrated that the stomach-specific overexpression of human interleukin 1β (one of the dominant pro-inflammatory cytokines secreted by the gastric mucosa of H. pylori-infected patients) induces gastric neoplasms in transgenic uninfected mice, with neoplasia being accelerated by infecting these mice with H. felis [51]. In this model, activated myeloid-derived suppressor cells recruited to the inflamed gastric mucosa were essential for cancer development - mobilization of this population and the development of cancer could be prevented through specific interleukin 1 receptor antagonism. The degree to which similar pathway may be operative in human gastric cancer remains to be determined.
v. Variations in host susceptibility to H. pylori-associated inflammation
Why only a low proportion of the population that is infected by H. pylori develops gastric cancer, even in regions of the world with high gastric cancer incidence, remained a mystery for many years. An important conceptual advance in the field was the discovery of the association of functionally important single nucleotide polymorphisms (SNP’s) in the interleukin 1 gene in association with low acid secretion and susceptibility to H. pylori-induced gastric cancer [86]. In many populations gastric cancer susceptibility following H. pylori infection is linked to increased gastric mucosal levels of interleukin-1β which are in turn dependent upon a functional SNP at position 511. SNP’s in genes encoding other cytokines, cytokine receptors and inflammatory regulators were subsequently reported, such as tumor necrosis factor-alpha, the chemokine interleukin-8, the regulatory cytokine interleukin-10, the arachidonic acid metabolizing enzyme cyclooxygenase-2, specific HLA alleles, and also toll-like receptors known to mediate signaling from microbes host in epithelial cells (summarized in [12]). This is a rapidly evolving field, and it is clear that the importance of the association of specific SNP’s with gastric cancer risk can differ when comparing different populations. Thus, despite a fairly large number of studies already published on this topic over the last 10 years, a recent systematic review of the many meta-analyses already published, has concluded that although there is likely to be interactions between individual polymorphisms, due to study heterogeneity and relatively small sample sizes, very much larger studies on genetically diverse populations are necessary to really dissect the relative importance of many of these identified polymorphisms before they can be used to help define risk on a clinical basis [87]. Evaluating both bacterial and host virulence factors simultaneously in large but well-defined populations would be optimal.
4. Gastric MALT lymphoma
Gastric MALT lymphomas are non-Hodgkins B-cell neoplasms derived from the marginal zone of lymphoid follicles [3]. Unlike the slow acceptance of the role of H. pylori in gastric adenocarcinoma development by clinicians, the evidence linking H. pylori to gastric B-cell MALT lymphomas was so strong, and the regression of the tumors following H. pylori eradication so impressive, that testing for and treating H. pylori rapidly became the standard of care for gastric MALT lymphoma management. Indeed, the observational and anecdotal data was deemed so overwhelming that no randomized clinical trial was ever done to evaluate the role of H. pylori eradication in this disease; placing patients in the placebo arm of such a study would now be considered unethical.
In the largest prospective cohort study investigating the relationship of H. pylori infection to gastric lymphoma using samples from a California serum bank, the odds ratio for the risk of low-grade gastric MALT lymphoma from H. pylori infection was 2.8 fold increased compared with H. pylori-negative subjects [88]. This study was published soon after the important observations by Hussell and colleagues, who demonstrated that the B-cell proliferation within gastric MALT lymphomas was driven by H. pylori stimulated T lymphocytes [89]. Many investigators around the world then treated patients with early stage MALT lymphoma with antibiotic treatment and observed rapid tumor regression within week to months.
Although monoclonality may persist within the residual infiltrating lymphocytes for much longer, a single short course of combination antibiotic therapy against H. pylori is generally sufficient to “cure” early stage disease - the majority of such patients have been in a sustained remission for over a decade. For MALT lymphomas of low histological grade and confined to the gastric wall or perigastric lymph nodes (stage I or stage IIe1 disease), a meta-analysis of over 30 studies has documented a remission rate of 78% overall (95% confidence interval 75–80%) [90]. Results are significantly better for stage I than stage II disease (78% versus 56%, p=0.003). From this same report, the overall rate of lymphoma recurrence rate was only 2% per year.
Many of the low-grade MALT lymphomas that are not cured by H. pylori eradication carry the AP12-MALT1 fusion gene that results from an 11:18 chromosomal translocation. MALT lymphomas of high histological grade or with more extensive tumor extension through the gastric wall have also been less responsive to H. pylori eradication. Currently, it is not known whether these high-grade MALT lymphomas and those that are AP12-MALT1 positive necessarily arise from the low-grade lesions associated with H. pylori infection [3].
The development of a MALT lymphoma in response to chronic H. pylori infection is a relatively rare occurrence. It is not known which cofactors (either bacterial, host genetic or environmental) are important in predisposing certain H. pylori-infected persons to gastric lymphoma.
5. Conclusions
Like many other cancer-inducing infections, H. pylori does not promote cancer universally. Indeed, while over 50% of the world’s population is infected with H. pylori, only 2% progress to gastric cancer, and even fewer develop a MALT lymphoma. Given this variable risk of malignancy following H. pylori infection, what are the critical factors or co-factors involved in determining which individuals infected by H. pylori will undergo H. pylori-induced gastric transformation? Some of this variability in outcome can be correlated with bacterial strain specificity, host genetic susceptibility, and the type of immune response elicited in the infected host. H. pylori contains virulence factors such as the cag pathogenicity island that induce changes in cellular morphology in vitro and alters signaling pathways and gene expression patterns; other putative virulence factors such as the BabA2 adhesin and the VacA exotoxin have not been consistently correlated with cancer susceptibility. Host polymorphisms in cytokine and cytokine receptor genes such as IL-1B, IL-1RB, TNF and IL-10 that regulate inflammatory response may explain why certain individuals within susceptible populations develop worse H. pylori-induced gastric pathology, as well as illustrate the important role that the host’s inflammatory and immune responses play in the pathobiology of H. pylori infection.
What makes H. pylori different from some other cancer causing infectious organisms, particularly certain viruses, is the fact that this extracellular bacterium can be viewed as predominantly an indirect carcinogen [91], promoting neoplastic changes through the associated chronic inflammatory response. Key elements of the carcinogenic gastric inflammatory reaction to H. pylori include oxidative stress, gastric immune cell accumulation and pro-inflammatory cytokine production leading to increased epithelial turnover and, over time, cellular transformation. The demonstration of the recruitment of bone marrow-derived cells to the tumor microenvironment during gastric carcinogenesis offers additional directions for investigating the indirect mechanisms of cancer causation by H. pylori.
Acknowledgments
Supported by grants # R01CA111533-01 and R21CA125126-01 (to SFM) and #1F31AI082948-01 (to VER) from the United States National Institutes of Health.
Footnotes
Conflict of Interest Statement
No of the authors have any potental conflicts of interest that could nnapropriately nfluence (bias) their work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 2.Quiros RM, Bui CL. Multidisciplinary approach to esophageal and gastric cancer. Surg Clin North Am. 2009;89:79–96. viii. doi: 10.1016/j.suc.2008.09.019. [DOI] [PubMed] [Google Scholar]
- 3.Du MQ, Atherton JC. Molecular subtyping of gastric MALT lymphomas: Implications for prognosis and management. Gut. 2006;55:886–893. doi: 10.1136/gut.2004.061663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parkin DM. The global health burden of infection-associated cancers in the year 2002. Int J Cancer. 2006;118:3030–3044. doi: 10.1002/ijc.21731. [DOI] [PubMed] [Google Scholar]
- 5.Suerbaum S, Michetti P. Helicobacter pylori infection. N Engl J Med. 2002;347:1175–1186. doi: 10.1056/NEJMra020542. [DOI] [PubMed] [Google Scholar]
- 6.Peek RM, Jr, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer. 2002;2:28–37. doi: 10.1038/nrc703. [DOI] [PubMed] [Google Scholar]
- 7.Schistosomes, liver flukes and Helicobacter pylori. IARC working group on the evaluation of carcinogenic risks to humans. lyon, 7–14 june 1994, IARC Monogr Eval Carcinog Risks Hum. 61 (1994) 1–241.
- 8.Moss SF, Blaser MJ. Mechanisms of disease: Inflammation and the origins of cancer. Nat Clin Pract Oncol. 2005;2:90–7. doi: 10.1038/ncponc0081. quiz 1 p following 113. [DOI] [PubMed] [Google Scholar]
- 9.Hill P, Rode J. Helicobacter pylori in ectopic gastric mucosa in Meckel’s diverticulum. Pathology. 1998;30:7–9. doi: 10.1080/00313029800169585. [DOI] [PubMed] [Google Scholar]
- 10.Necchi V, Candusso ME, Tava F, Luinetti O, Ventura U, Fiocca R, Ricci V, Solcia E. Intracellular, intercellular, and stromal invasion of gastric mucosa, preneoplastic lesions, and cancer by Helicobacter pylori. Gastroenterology. 2007;132:1009–1023. doi: 10.1053/j.gastro.2007.01.049. [DOI] [PubMed] [Google Scholar]
- 11.Dubois A, Boren T. Helicobacter pylori is invasive and it may be a facultative intracellular organism. Cell Microbiol. 2007;9:1108–16. doi: 10.1111/j.1462-5822.2007.00921.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amieva MR, El-Omar EM. Host-bacterial interactions in Helicobacter pylori infection. Gastroenterology. 2008;134:306–323. doi: 10.1053/j.gastro.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 13.Sycuro LK, Pincus Z, Gutierrez KD, Biboy J, Stern CA, Vollmer W, Salama NR. Peptidoglycan crosslinking relaxation promotes Helicobacter pylori’s helical shape and stomach colonization. Cell. 2010;141:822–833. doi: 10.1016/j.cell.2010.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Falush D, Wirth T, Linz B, et al. Traces of human migrations in Helicobacter pylori populations. Science. 2003;299:1582–5. doi: 10.1126/science.1080857. [DOI] [PubMed] [Google Scholar]
- 15.Tomb JF, White O, Kerlavage AR, et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature. 1997;388:539–47. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- 16.Tomb JF, White O, Kerlavage AR, et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature. 1997;388:539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- 17.McClain MS, Shaffer CL, Israel DA, Peek RM, Jr, Cover TL. Genome sequence analysis of Helicobacter pylori strains associated with gastric ulceration and gastric cancer. BMC Genomics. 2009;10:3. doi: 10.1186/1471-2164-10-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aim RA, Ling LS, Moir DT, et al. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature. 1999;397:176–180. doi: 10.1038/16495. [DOI] [PubMed] [Google Scholar]
- 19.Baltrus DA, Amieva MR, Covacci A, Lowe TM, Merrel DS, Ottemann KM, Stein M, Salama NR, Guillemin K. The complete genome sequence of Helicobacter pylori strain G27. J Bacteriol. 2009;191:447–448. doi: 10.1128/JB.01416-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oh JD, Kling-Backhed H, Giannakis M, et al. The complete genome sequence of a chronic atrophic gastritis Helicobacter pylori strain: Evolution during disease progression. Proc Natl Acad Sci U S A. 2006;103:9999–10004. doi: 10.1073/pnas.0603784103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thiberge JM, Boursaux-Eude C, Lehours P, et al. Array-based hybridization of Helicobacter pylori isolates to the complete genome sequence of an isolate associated with MALT lymphoma. BMC Genomics. 2010;11:368. doi: 10.1186/1471-2164-11-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Israel DA, Salama N, Krishna U, Rieger UM, Atherton JC, Falkow S, Peek RM., Jr Helicobacter pylori genetic diversity within the gastric niche of a single human host. Proc Natl Acad Sci U S A. 2001;98:14625–14630. doi: 10.1073/pnas.251551698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Higashi H, Yokoyama K, Fujii Y, Ren S, Yuasa H, Saadat I, Murata-Kamiya N, Azuma T, Hatakeyama M. EPIYA motif is a membrane-targeting signal of Helicobacter pylori virulence factor CagA in mammalian cells. J Biol Chem. 2005;280:23130–23137. doi: 10.1074/jbc.M503583200. [DOI] [PubMed] [Google Scholar]
- 24.Cover TL, Blaser MJ. Purification and characterization of the vacuolating toxin from Helicobacter pylori. J Biol Chem. 1992;267:10570–5. [PubMed] [Google Scholar]
- 25.Basso D, Zambon CF, Letley DP, et al. Clinical relevance of Helicobacter pylori cagA and vacA gene polymorphisms. Gastroenterology. 2008;135:91–9. doi: 10.1053/j.gastro.2008.03.041. [DOI] [PubMed] [Google Scholar]
- 26.Sharma CM, Hoffmann S, Darfeuille F, et al. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature. 2010;464:250–255. doi: 10.1038/nature08756. [DOI] [PubMed] [Google Scholar]
- 27.Correa P, Haenszel W, Cuello C, Tannenbaum S, Archer M. A model for gastric cancer epidemiology. Lancet. 1975;2:58–60. doi: 10.1016/s0140-6736(75)90498-5. [DOI] [PubMed] [Google Scholar]
- 28.Gastric cancer and Helicobacter pylori: A combined analysis of 12 case control studies nested within prospective cohorts. Gut. 2001;49:347–53. doi: 10.1136/gut.49.3.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Atherton JC, Blaser MJ. Coadaptation of Helicobacter pylori and humans: Ancient history, modern implications. J Clin Invest. 2009;119:2475–2487. doi: 10.1172/JCI38605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fuccio L, Zagari RM, Eusebi LH, Laterza L, Cennamo V, Ceroni L, Grilli D, Bazzoli F. Meta-analysis: Can Helicobacter pylori eradication treatment reduce the risk for gastric cancer? Ann Intern Med. 2009;151:121–128. doi: 10.7326/0003-4819-151-2-200907210-00009. [DOI] [PubMed] [Google Scholar]
- 31.Fukase K, Kato M, Kikuchi S, Inoue K, Uemura N, Okamoto S, Terao S, Amagai K, Hayashi S, Asaka M. Effect of eradication of Helicobacter pylori on incidence of metachronous gastric carcinoma after endoscopic resection of early gastric cancer: An open-label, randomised controlled trial. Lancet. 2008;372:392–7. doi: 10.1016/S0140-6736(08)61159-9. [DOI] [PubMed] [Google Scholar]
- 32.Fock KM, Katelaris P, Sugano K, et al. Second asia-pacific consensus guidelines for Helicobacter pylori infection. J Gastroenterol Hepatol. 2009;24:1587–1600. doi: 10.1111/j.1440-1746.2009.05982.x. [DOI] [PubMed] [Google Scholar]
- 33.Yamaoka Y. Helicobacter pylori typing as a tool for tracking human migration. Clin Microbiol Infect. 2009;15:829–834. doi: 10.1111/j.1469-0691.2009.02967.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang JQ, Zheng GF, Sumanac K, Irvine EJ, Hunt RH. Metaanalysis of the relationship between cagA seropositivity and gastric cancer. Gastroenterology. 2003;125:1636–44. doi: 10.1053/j.gastro.2003.08.033. [DOI] [PubMed] [Google Scholar]
- 35.Yamaoka Y, Kodama T, Kashima K, Graham DY, Sepulveda AR. Variants of the 3′ region of the cagA gene in Helicobacter pylori isolates from patients with different H. pylori-associated diseases. J Clin Microbiol. 1998;36:2258–63. doi: 10.1128/jcm.36.8.2258-2263.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ogiwara H, Graham DY, Yamaoka Y. vacA i-region subtyping. Gastroenterology. 2008;134:1267. doi: 10.1053/j.gastro.2007.11.062. author reply 1268. [DOI] [PubMed] [Google Scholar]
- 37.Franco AT, Johnston E, Krishna U, Yamaoka Y, Israel DA, Nagy TA, Wroblewski LE, Piazuelo MB, Correa P, Jr, PRM Regulation of gastric carcinogenesis by Helicobacter pylori virulence factors. Cancer Research. 2008;68:379–87. doi: 10.1158/0008-5472.CAN-07-0824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fox JG, Wang TC, Rogers AB, et al. Host and microbial constituents influence Helicobacter pylori-induced cancer in a murine model of hypergastrinemia. Gastroenterology. 2003;124:1879–90. doi: 10.1016/s0016-5085(03)00406-2. [DOI] [PubMed] [Google Scholar]
- 39.Backert S, Selbach M. Role of type IV secretion in Helicobacter pylori pathogenesis. Cell Microbiol. 2008;10:1573–81. doi: 10.1111/j.1462-5822.2008.01156.x. [DOI] [PubMed] [Google Scholar]
- 40.Saadat I, Higashi H, Obuse C, et al. Helicobacter pylori CagA targets PAR1/MARK kinase to disrupt epithelial cell polarity. Nature. 2007;447:330–333. doi: 10.1038/nature05765. [DOI] [PubMed] [Google Scholar]
- 41.Cascales E, Christie PJ. The versatile bacterial type IV secretion systems. Nat Rev Microbiol. 2003;1:137–149. doi: 10.1038/nrmicro753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Viala J, Chaput C, Boneca IG, et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol. 2004;5:1166–1174. doi: 10.1038/ni1131. [DOI] [PubMed] [Google Scholar]
- 43.Watanabe T, Asano N, Fichtner-Feigl S, Gorelick PL, Tsuji Y, Matsumoto Y, Chiba T, Fuss IJ, Kiani A, Strober W. NOD1 contributes to mouse host defense against Helicobacter pylori via induction of type I IFN and activation of the ISGF3 signaling pathway. J Clin Invest. 2010;120:1645–1662. doi: 10.1172/JCI39481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ohnishi N, Yuasa H, Tanaka S, et al. Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc Natl Acad Sci USA. 2008;105:1003–8. doi: 10.1073/pnas.0711183105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rhead JL, Letley DP, Mohammadi M, Hussein N, Mohagheghi MA, Eshagh Hosseini M, Atherton JC. A new Helicobacter pylori vacuolating cytotoxin determinant, the intermediate region, is associated with gastric cancer. Gastroenterology. 2007;133:926–36. doi: 10.1053/j.gastro.2007.06.056. [DOI] [PubMed] [Google Scholar]
- 46.Gao L, Michel A, Weck MN, Arndt V, Pawlita M, Brenner H. Helicobacter pylori infection and gastric cancer risk: Evaluation of 15 H. pylori proteins determined by novel multiplex serology. Cancer Res. 2009;69:6164–70. doi: 10.1158/0008-5472.CAN-09-0596. [DOI] [PubMed] [Google Scholar]
- 47.Watanabe T, Tada M, Nagai H, Sasaki S, Nakao M. Helicobacter pylori infection induces gastric cancer in mongolian gerbils. Gastroenterology. 1998;115:642–8. doi: 10.1016/s0016-5085(98)70143-x. [DOI] [PubMed] [Google Scholar]
- 48.Romero-Galo J, Harris EJ, Krishna U, Washington MK, Perez-Perez GI, Jr, PRM Effect of Helicobacter pylori eradication on gastric carcinogenesis. Laboratory Investigation; a Journal of Technical Methods and Pathology. 2008;88:328–36. doi: 10.1038/labinvest.3700719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rogers AB, Taylor NS, Whary MT, Stefanich ED, Wang TC, Fox JG. Helicobacter pylori but not high salt induces gastric intraepithelial neoplasia in B6129 mice. Cancer Res. 2005;65:10709–10715. doi: 10.1158/0008-5472.CAN-05-1846. [DOI] [PubMed] [Google Scholar]
- 50.Cai X, Carlson J, Stoicov C, Li H, Wang TC, Houghton J. Helicobacter felis eradication restores normal architecture and inhibits gastric cancer progression in C57BL/6 mice. Gastroenterology. 2005;128:1937–52. doi: 10.1053/j.gastro.2005.02.066. [DOI] [PubMed] [Google Scholar]
- 51.Tu S, Bhagat G, Cui G, et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell. 2008;14:408–19. doi: 10.1016/j.ccr.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hahm KB, Lee KM, Kim YB, et al. Conditional loss of TGF-beta signalling leads to increased susceptibility to gastrointestinal carcinogenesis in mice. Aliment Pharmacol Ther. 2002;16(Suppl 2):115–27. doi: 10.1046/j.1365-2036.16.s2.3.x. [DOI] [PubMed] [Google Scholar]
- 53.Kuzushita N, Rogers AB, Monti NA, Whary MT, Park MJ, Aswad BI, Shirin H, Koff A, Eguchi H, Moss SF. p27kip1 deficiency confers susceptibility to gastric carcinogenesis in Helicobacter pylori-infected mice. Gastroenterology. 2005;129:1544–156. doi: 10.1053/j.gastro.2005.07.056. [DOI] [PubMed] [Google Scholar]
- 54.Rogers AB, Houghton J. Helicobacter based mouse models of digestive system carcinogenesis. In: Kozlov SV, editor. Inflammation in Cancer, Methods in Molecular Biology. Humana Press; 2009. [DOI] [PubMed] [Google Scholar]
- 55.Liu H, Merrel DS, Semino-Mora C, Goldman M, Rahman A, Mog S, Dubois A. Diet synergistically affects Helicobacter pylori-induced gastric carcinogenesis in nonhuman primates. Gastroenterology. 2009;137:1367–79.el-6. doi: 10.1053/j.gastro.2009.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Preston-Martin S, Pike MC, Ross RK, Jones PA, Henderson BE. Increased cell division as a cause of human cancer. Cancer Research. 1990;50:7415–21. [PubMed] [Google Scholar]
- 57.Chaturvedi R, Asim M, Lewis ND, Algood HM, Cover TL, Kim PY, Wilson KT. L-arginine availability regulates inducible nitric oxide synthase-dependent host defense against Helicobacter pylori. Infect Immun. 2007;75:4305–4315. doi: 10.1128/IAI.00578-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ding SZ, Minohara Y, Fan XJ, Wang J, Reyes VE, Patel J, Dirden-Kramer B, Boldogh I, Ernst PB, Crowe SE. Helicobacter pylori infection induces oxidative stress and programmed cell death in human gastric epithelial cells. Infection and Immunity. 2007;75:4030–9. doi: 10.1128/IAI.00172-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bhattacharyya A, Chattopadhyay R, Burnette BR, Cross JV, Mitra S, Ernst PB, Bhakat KK, Crowe SE. Acetylation of apurinic/apyrimidinic endonuclease-1 regulates Helicobacter pylori-mediated gastric epithelial cell apoptosis. Gastroenterology. 2009;136:2258–2269. doi: 10.1053/j.gastro.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Touati E, Michel V, Thiberge JM, Wuscher N, Huerre M, Labigne A. Chronic Helicobacter pylori infections induce gastric mutations in mice. Gastroenterology. 2003;124:1408–19. doi: 10.1016/s0016-5085(03)00266-x. [DOI] [PubMed] [Google Scholar]
- 61.Jenks PJ, Jeremy AH, Robinson PA, Walker MM, Crabtree JE. Long-term infection with Helicobacter felis and inactivation of the tumour suppressor gene p53 cumulatively enhance the gastric mutation frequency in big blue transgenic mice. J Pathol. 2003;201:596–602. doi: 10.1002/path.1488. [DOI] [PubMed] [Google Scholar]
- 62.Matsumoto Y, Marusawa H, Kinoshita K, Endo Y, Kou T, Morisawa T, Azuma T, Okazaki IM, Honjo T, Chiba T. Helicobacter pylori infection triggers aberrant expression of activation-induced cytidine deaminase in gastric epithelium. Nat Med. 2007;13:470–476. doi: 10.1038/nm1566. [DOI] [PubMed] [Google Scholar]
- 63.Shirin H, Weinstein IB, Moss SF. Effects of H. pylori infection of gastric epithelial cells on cell cycle control. Front Biosci. 2001;6:E104–18. doi: 10.2741/shirin. [DOI] [PubMed] [Google Scholar]
- 64.Mimuro H, Suzuki T, Nagai S, et al. Helicobacter pylori dampens gut epithelial self-renewal by inhibiting apoptosis, a bacterial strategy to enhance colonization of the stomach. Cel Host Microbe. 2007;2:250–63. doi: 10.1016/j.chom.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 65.Houghton JM, Bloch LM, Goldstein M, Von Hagen S, Korah RM. In vivo disruption of the fas pathway abrogates gastric growth alterations secondary to Helicobacter infection. The Journal of Infectious Diseases. 2000;182:856–64. doi: 10.1086/315788. [DOI] [PubMed] [Google Scholar]
- 66.Shirin H, Sordillo EM, Kolevska TK, et al. Chronic Helicobacter pylori infection induces an apoptosis-resistant phenotype associated with decreased expression of p27(kipl) Infection and Immunity. 2000;68:5321–8. doi: 10.1128/iai.68.9.5321-5328.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wei J, O’Brien D, Vilgelm A, Piazuelo MB, Correa P, Washington MK, El-Rifai W, Peek RM, Zaika A. Interaction of Helicobacter pylori with gastric epithelial cells is mediated by the p53 protein family. Gastroenterology. 2008;134:1412–23. doi: 10.1053/j.gastro.2008.01.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Resnick MB, Sabo E, Meitner PA, Kim SS, Cho Y, Kim HK, Tavares R, Moss SF. Global analysis of the human gastric epithelial transcriptome altered by Helicobacter pylori eradication n vivo. Gut. 2006;55:1717–1724. doi: 10.1136/gut.2006.095646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Moss SF, Lee JW, Sabo E, et al. Decreased expression of gastrokine 1 and the trefoil factor interacting protein TFIZ1/GKN2 in gastric cancer: Influence of tumor histology and relationship to prognosis. Clin Cancer Res. 2008;14:4161–4167. doi: 10.1158/1078-0432.CCR-07-4381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hamon JP, Meltzer SJ. A review of the genomics of gastric cancer. Clin Gastroenterol Hepatol. 2006;4:416–425. doi: 10.1016/j.cgh.2006.01.019. [DOI] [PubMed] [Google Scholar]
- 71.Chan AO, Peng JZ, Lam SK, Lai KC, Yuen MF, Cheung HK, Kwong YL, Rashid A, Chan CK, Wong BC. Eradication of Helicobacter pylori infection reverses E-cadherin promoter hypermethylation. Gut. 2006;55:463–8. doi: 10.1136/gut.2005.077776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sepulveda AR, Yao Y, Yan W, Park DI, Kim JJ, Gooding W, Abudayyeh S, Graham DY. CpG methylation and reduced expression of O(6)-methylguanine DNA methylransferase is associated with Helicobacter pylori infection. Gastroenterology. 2010;138:1836–44. doi: 10.1053/j.gastro.2009.12.042. [DOI] [PubMed] [Google Scholar]
- 73.Nakajima T, Enomoto S, Yamashita S, Ando T, Nakanishi Y, Nakazawa K, Oda I, Gotoda T, Ushijima T. Persistence of a component of DNA methylation in gastric mucosae after Helicobacter pylori eradication. J Gastroenterol. 2010;45:37–44. doi: 10.1007/s00535-009-0142-7. [DOI] [PubMed] [Google Scholar]
- 74.Niwa T, Tsukamoto T, Toyoda T, Mori A, Tanaka H, Maekita T, Ichinose M, Tatematsu M, Ushijima T. Inflammatory processes triggered by Helicobacter pylori infection cause aberrant DNA methylation in gastric epithelial cells. Cancer Res. 2010;70:1430–1440. doi: 10.1158/0008-5472.CAN-09-2755. [DOI] [PubMed] [Google Scholar]
- 75.Xia G, Schneider-Stock R, Diestel A, Habold C, Krueger S, Roessner A, Naumann M, Lendeckel U. Helicobacter pylori regulates p21(WAFl) by histone H4 acetylation. Biochem Biophys Res Commun. 2008;369:26–31. doi: 10.1016/j.bbrc.2008.02.073. [DOI] [PubMed] [Google Scholar]
- 76.Belair C, Darfeuille F, Staedel C. Helicobacter pylori and gastric cancer: Possible role of mcroRNAs in this intimate relationship. Clin Microbiol Infect. 2009;15:806–812. doi: 10.1111/j.1469-0691.2009.02960.x. [DOI] [PubMed] [Google Scholar]
- 77.Matsushima K, Isomoto H, Inoue N, Nakayama T, Hayashi T, Nakayama M, Nakao K, Hirayama T, Kohno S. MicroRNA signatures in Helicobacter pylori-infected gastric mucosa. Int J Cancer. 2010 doi: 10.1002/ijc.25348. [DOI] [PubMed] [Google Scholar]
- 78.Fassi Fehri L, Koch M, Belogolova E, et al. Helicobacter pylori induces miR-15 n T cells in a cAMP-Foxp3-dependent manner. PLoS One. 2010;5:e9500. doi: 10.1371/journal.pone.0009500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang Z, Li Z, Gao C, Chen P, Chen J, Liu W, Xiao S, Lu H. mR-21 plays a pivotal role n gastric cancer pathogenesis and progression. Lab Invest. 2008;88:1358–66. doi: 10.1038/labinvest.2008.94. [DOI] [PubMed] [Google Scholar]
- 80.Xiao B, Liu Z, Li BS, et al. Induction of microRNA-155 during Helicobacter pylori infection and its negative regulatory role in the inflammatory response. J Infect Dis. 2009;200:916–25. doi: 10.1086/605443. [DOI] [PubMed] [Google Scholar]
- 81.Sanduleanu S, Jonkers D, de Bruine A, Hameeteman W, Stockbrugger RW. Changes in gastric mucosa and luminal environment during acid-suppressive therapy: A review in depth. Dig Liver Dis. 2001;33:707–19. doi: 10.1016/s1590-8658(01)80050-5. [DOI] [PubMed] [Google Scholar]
- 82.Hansson LE, Nyren O, Hsing AW, Bergstrom R, Josefsson S, Chow WH, Jr, FJF, Adami HO. The risk of stomach cancer in patients with gastric or duodenal ulcer disease. N Engl J Med. 1996;335:242–9. doi: 10.1056/NEJM199607253350404. [DOI] [PubMed] [Google Scholar]
- 83.Calam J. The somatostatin-gastrin link of Helicobacter pylori infection. Ann Med. 1995;27:69–73. doi: 10.3109/07853899509002471. [DOI] [PubMed] [Google Scholar]
- 84.Algood HM, Gallo-Romero J, Wilson KT, Peek RM, Jr, Cover TL. Host response to Helicobacter pylori infection before initiation of the adaptive immune response. FEMS Immunol Med Microbiol. 2007;51:577–586. doi: 10.1111/j.1574-695X.2007.00338.x. [DOI] [PubMed] [Google Scholar]
- 85.Houghton J, Stoicov C, Nomura S, Rogers AB, Carlson J, Li H, Cai X, Fox JG, Goldenring JR, Wang TC. Gastric cancer originating from bone marrow-derived cells. Science. 2004;306:1568–71. doi: 10.1126/science.1099513. [DOI] [PubMed] [Google Scholar]
- 86.El-Omar EM, Carrington M, Chow WH, et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature. 2000;404:398–402. doi: 10.1038/35006081. [DOI] [PubMed] [Google Scholar]
- 87.Gianfagna F, De Feo E, van Duijn CM, Ricciardi G, Boccia S. A systematic review of meta-analyses on gene polymorphisms and gastric cancer risk. Curr Genomics. 2008;9:361–374. doi: 10.2174/138920208785699544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Parsonnet J, Hansen S, Rodriguez L, Gelb AB, Warnke RA, Jellum E, Orentreich N, Vogelman JH, Friedman GD. Helicobacter pylori infection and gastric lymphoma. N Engl J Med. 1994;330:1267–1271. doi: 10.1056/NEJM199405053301803. [DOI] [PubMed] [Google Scholar]
- 89.Hussell T, Isaacson PG, Crabtree JE, Spencer J. Helicobacter pylori-specific tumour-infiltrating T cells provide contact dependent help for the growth of malignant B cells in low-grade gastric lymphoma of mucosa-associated lymphoid tissue. J Pathol. 1996;178:122–127. doi: 10.1002/(SICI)1096-9896(199602)178:2<122::AID-PATH486>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 90.Zullo A, Hassan C, Cristofari F, Andriani A, De Francesco V, Ierardi E, Tomao S, Stolte M, Morini S, Vaira D. Effects of Helicobacter pylori eradication on early stage gastric mucosa-associated lymphoid tissue lymphoma. Clin Gastroenterol Hepatol. 2010;8:105–10. doi: 10.1016/j.cgh.2009.07.017. [DOI] [PubMed] [Google Scholar]
- 91.Hausen HZ. Infections causing human cancer. 1. Wiley-VCH; Weinheim: 2006. [Google Scholar]