

Abstract
Considerable evidence points to important roles for inflammation in Alzheimer’s disease (AD) pathophysiology. Epidemiological studies have suggested that long-term NSAID therapy reduces the risk for AD; however, the mechanism remains unknown. We report that a 9 month treated of aged R1.40 mice resulted in 90% decrease in plaque burden and a similar reduction in microglial activation. Ibuprofen treatment reduced levels of lipid peroxidation, tyrosine nitration, and protein oxidation, demonstrating a dramatic effect on oxidative damage in vivo. Fibrillar Aβ stimulation has previously been demonstrated to induce the assembly and activation of the microglial NADPH oxidase leading to superoxide production through a tyrosine kinase-based signaling cascade. Ibuprofen treatment of microglia or monocytes with racemic or S-ibuprofen inhibited Aβ-stimulated Vav tyrosine phosphorylation, NADPH oxidase assembly and superoxide production. Interestingly, Aβ-stimulated Vav phosphorylation was not inhibited by COX inhibitors. These findings suggest that ibuprofen acts independently of COX inhibition to disrupt signaling cascades leading to microglial NOX2 activation, preventing oxidative damage and enhancing plaque clearance in the brain.
Keywords: NADPH oxidase, NSAIDs, Alzheimer’s disease, microglia, Vav, oxidative stress
1. Introduction
Alzheimer’s disease (AD) is characterized by the formation of focal, compact β-amyloid (Aβ deposits within the brain. These deposits are surrounded by phenotypically activated microglia, which are responsible for a locally-induced chronic inflammatory response and affect Aβ homeostasis. It has been proposed that inflammation plays an important role in AD pathogenesis as the AD brain exhibits elevated levels of inflammatory molecules or other immune mediators (Akiyama, et al., 2000,Bamberger and Landreth, 2002). Chronically activated microglia also generate reactive oxygen (ROS) and nitrogen species. Several markers of oxidative damage including lipid peroxidation (Mark, et al., 1997,Sayre, et al., 1997), nucleic acid oxidation (Nunomura, et al., 1999), and protein oxidation (Smith, et al., 1997) are increased in the AD brain (Sonnen, et al., 2009). There is compelling evidence that much of the oxidative damage observed in the AD brain is due to free radical production by microglia and precedes Aβ deposition (Pratico, 2008,Wilkinson, et al., 2006). The etiological events leading to AD remain unknown; however, our findings suggest inflammation and oxidative damage play critical roles in AD pathogenesis.
Microglia, the brain’s principal immune effector cells are a potential source of oxidative stress (Akiyama, et al., 2000,Banati, et al., 1993). We have previously demonstrated that microglia employ a multi-receptor cell surface complex, comprised of CD36, α6β1 integrin, CD47, and the class A scavenger receptor (Bamberger, et al., 2003), TLR2/4 and CD14 (Reed-Geaghan, et al., 2009) to detect and respond to Aβ fibrils. Fibrillar Aβ engagement of this receptor complex initiates a tyrosine kinase-based intracellular signaling cascade. Tyrosine phosphorylation of Vav faciliates Rac activation, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX2) assembly and superoxide production (Wilkinson, et al., 2006). The sustained microglial proinflammatory response results in overproduction of ROS, which can ultimately be neurotoxic.
A number of epidemiological studies have reported that chronic nonsteroidal anti-inflammatory drug (NSAID) therapy was associated with a dramatically reduced incidence of AD (McGeer, et al., 1996), conferring a 60-80% reduction in risk (in t’ Veld, et al., 2001,Stewart, et al.,1997, Vlad, et al., 2008). Long-term ibuprofen treatment also suppresses inflammation, reduces amyloid deposition, alters APP processing, and improves cognitive performance in murine models of AD (Jantzen, et al., 2002,Kotilinek, et al., 2008,Lim, et al., 2000,Lim, et al., 2001b,McKee, et al., 2008,Yan, et al., 2003). Together, these findings led to clinical trials of NSAIDs in AD that failed to demonstrate any benefit to patients (Aisen, et al., 2003,Arvanitakis, et al., 2008,Breitner, et al., 2009,Group, et al., 2007,Reines, et al., 2004) A recent renewed interest in this class of drugs for AD treatment stems from findings suggesting that some, but not all, NSAIDs can act independently from their classic anti-inflammatory mechanisms, which may play a role in their disease-modifying actions (Combs, et al., 2000,Eriksen, et al., 2003,Lehmann, et al., 1997,Lleo, et al., 2004,Weggen, et al., 2001,Zhou, et al., 2003). These findings raise the question of how NSAIDs might influence other pathogenic features of AD such as oxidative damage. We have investigated whether chronic ibuprofen treatment could alter AD-related oxidative damage in a mouse model, and how ibuprofen might inhibit intracellular signaling cascades responsible for NOX2 assembly and release of ROS.
2. Materials and Methods
2.1 Materials
Ibuprofen was obtained from Sigma (St. Louis, MO). This compound was formulated into standard, color-coded animal chow by Research Diets (New Brunswick, NJ) at a final concentration of 375 ppm ibuprofen.
The Aβ peptide corresponding to the human Aβ amino acids 25-35 was purchased from American Peptide Co. (Sunnyvale, CA). The method used to fibrillarize Aβ peptides has been well characterized (Burdick, et al., 1992,Lorenzo and Yankner, 1994).
2.2 Transgenic mice and ibuprofen treatment
The B6-R1.40 transgenic mouse line contains hAPP with the Swedish (K670M/N671L) FAD mutation as previously described (Lamb, et al., 1999,Lehman, et al., 2003). Fifteen-month-old male and female B6-R1.40 mice were fed drug-supplemented or control chow ad libitum for 9 months. The amount of animal chow consumed was approximately 5 g/day/animal, resulting in a final dosage of 62.5 mg/kg/day as previously described (Yan, et al., 2003). Mice were observed on a weekly basis and exhibited no overt signs of distress. Mice were sacrificed at 24 months of age. All animal studies were approved by the Case Western Reserve University School of Medicine Institutional Animal Care and Use Committee.
2.3 Histology and Immunohistochemistry
Mice were anesthetized with Avertin (0.02 cc/mg body weight) and perfused transcardially with 0.1 M sodium phosphate buffer followed by 4% paraformaldehyde. Brains were dissected, post-fixed, cryoprotected and sagittally sectioned (10 μm). Tissue sections were incubated overnight at 4°C with either 6E10 (Signet Laboratories, USA; 1:1000), CD45 (Serotec, USA; 1:300), or Iba1 (Wako, Japan; 1:500) antibodies. Sections treated with anti-Aβ (6E10) and anti-CD45 antibodies were then incubated with the appropriate biotinylated secondary antibodies, and detected through the avidin-biotin-peroxidase complex (Vector, USA). Peroxidase activity was visualized by diaminobenzidine (Vector, USA). For immunofluorescent staining, Iba1 was detected with an Alexa Fluor 488 antibody and 6E10 was detected with Alexa Fluor 546 antibody (Molecular Probes, USA; 1:1000).
Thioflavin-S staining was performed to visualize dense core plaques. Sections were rehydrated and then stained with 1% Thioflavin S (Sigma). Nuclei were visualized with a propidium iodide (0.15 μM) counterstain.
2.4 Tissue homogenization and Western blotting
Animals were sacrificed by cervical dislocation and brain tissue was immediately removed. Brains were bisected sagittally along the midline. Hemibrains, excluding the cerebellum, were homogenized in ice-cold tris-buffered saline with protease inhibitors (0.5 mM PMSF, 0.2 mM Na3VO4, protease inhibitor cocktail (Sigma, 1:100), 1 mM EDTA) using a glass-on-glass homogenizer. The homogenate was centrifuged at 5,000 rcf for 10 min at 4°C. Protein concentration was determined by the Bradford method (Bradford, 1976).
Lysates from brain homogenates were resolved by SDS PAGE on a 4-12% Bis Tris gel (Invitrogen, USA) and transferred to polyvinylidene difluoride (PVDF) membranes. Blots were incubated overnight with either anti-3-nitrotyrosine (Alpha Diagnostics, USA; 1:1000), anti-4-HNE (Chemicon, USA; 1:2000), or anti-dinitrophenylhydrazine (DNPH) (Chemicon, USA; 1:150) antibodies at 4°C. Proteins were detected by chemiluminescence (Pierce, USA). Blots were stripped and reprobed with anti-GAPDH (Trevigen, USA; 1:5000) as a protein loading control. Band intensities were quantified using NIH Image 1.62 software (Bethesda, MD).
2.5 Tissue Culture
Human THP-1 monocytes (American Type Culture Collection, USA) were grown in RPMI 1640 medium (Whittaker Bioproducts, USA) containing 10% heat-inactivated fetal bovine serum (Hyclone, USA), 5 × 10−5 M 2-mercaptoethanol, 5 mM HEPES, and 15 μg/ml gentamycin in 5% CO2. THP-1 monocytes are used in these assays as they do not attach to the tissue culture substrate through integrin-based adhesive mechanisms, allowing dissection of Aβ fibril-dependent signaling mechanisms in the absence of high basal levels of tyrosine kinase-based integrin signaling. THP-1 monocytes response to fAβ peptides faithfully replicates the response observed in primary microglia (Bamberger, et al., 2003,Combs, et al., 2001,Combs, et al., 1999,Combs, et al., 2000,Koenigsknecht and Landreth, 2004). Primary microglia were obtained from postnatal day 1-3 mouse brains as described previously (Combs, et al., 2001,Combs, et al., 1999,Combs, et al., 2000).
2.6 Cell Stimulation and Immunoprecipitations
THP-1 monocytes were collected and resuspended in Hank’s Balanced Salt Solution (HBSS) and preincubated with racemic ibuprofen, the S- or-R-enantiomers of ibuprofen or cycloxygenase inhibitors for 1 h at 37°C. Vav immunoprecipitations were performed as previously described (Wilkinson, et al., 2006). For western blotting, samples were resolved on 9 or 12% SDS-PAGE gels and transferred as mentioned above. Blots were probed with either anti-phospho-Tyr (4G10; Upstate, USA; 1:1000), -Vav (Santa Cruz, USA; 1:1000) -phospho-p38 (Cell Signaling; USA 1:1000), or -p38 (Santa Cruz, USA; 1:1000) antibodies overnight at 4°C. The proteins were detected by as mentioned above and reprobed with primary antibody for load controls.
2.7 Cellular Fractionation
THP-1 cells were fractionated as previously described (Wilkinson, et al. 2006). THP-1 cells (6 × 106 cells) were collected and resuspended in HBSS for 30 min at 37°C followed by preincubation with S-ibuprofen for 1 hr. Cells were then stimulated for 10 min with fAβ25-35 (60 μM) and lysed in relaxation buffer (100 mM KCl, 3 mM NaCl, 3.5 mM MgCl2, 1.25 mM EGTA, and 10 mM PIPES, pH 7.3) on ice for 15 min followed by 10 s of sonication. Cells were cleared by centrifugation at 5000 x g for 5 min at 4°C. The supernatant was then centrifuged for 1 h at 110,000 x g at 4 °C in a Beckman Coulter SW50.1 rotor. The resulting supernatant was removed and saved as the “cytosolic” fraction, and the membrane pellet was resuspended in relaxation buffer with 1% Igepal (NP40). Lysates were resolved on a 4-12% Bis-Tris gel, transferred and blocked. The blots were probed overnight with an anti-Rac antibody (1:1000) to determine the relative amount of Rac in each fraction and an anti-flotillin (1:1000) antibody as a membrane marker to assess the efficacy of the fractionation procedure.
2.8 Measurement of superoxide production
Intracellular superoxide radical generation was assayed by nitroblue tetrazolium (NBT, Roche, Basel, Switzerland) reduction as previously described (Wilkinson, et al., 2006). For these experiments, primary microglia from C57BL/6 mice were plated overnight in serum-free DMEM-F12. The microglia were pretreated for 1hr at 37°C with S-ibuprofen. NBT (1mg/ml) with or without fAβ25–35 (60 μM) in serum-free DMEM-F12 was then added to the wells for 30 min. Phorbol 12-myristate 13-acetate (PMA; 390 nM)) was used as a positive control (Bamberger, et al., 2003,Bianca, et al., 1999,McDonald, et al., 1997). Three random fields of cells (>100 cells) were counted on an inverted microscope.
2.9 Statistical Analysis
All experiments were performed a minimum of three times. Data from each experiment are expressed as mean ±standard deviation. Two-tailed Student’s t-test was performed between +IBU and −IBU samples. Values statistically different from controls were calculated using a one-way ANOVA, and the Tukey-Kramer multiple-comparisons test was used to determine p-values. Significance was considered at a probability (p) value equal or less than 0.05.
3. Results
3.1 Chronic ibuprofen treatment significantly reduces amyloid deposition in aged B6-R1.40 mice
The R1.40 mouse develops extracellular Aβ deposits and associated neuropathology that closely resembles alterations observed in human AD (Kulnane and Lamb, 2001,Lamb, et al., 1999,Lehman, et al., 2003). Few extracellular Aβ plaques are evident in the B6-R1.40 mouse between 14-15 months of age. We began chronic ibuprofen treatment at 15 months of age and sacrificed the animals at 24 months of age. We observed an approximate 90% reduction in Aβ plaque deposition (number of plaques/section) in the parenchyma of aged, 24-month-old ibuprofen-treated B6-R1.40 animals (n=5) when compared to age-matched non-treated B6-R1.40 animals (n=5) as measured by quantitative 6E10 immunohistochemistry (p< 0.001; Figure 1A-B). We also observed a corresponding reduction in Thioflavin-S positive, compact Aβ plaques in the parenchyma of the ibuprofen-treated animals (Figure 1C-D). Notably, Thioflavin-S positive amyloid deposition is present in the cerebral vessel walls of both the ibuprofen-treated and the control animals. These results in the B6-R1.40 mice are similar to effects observed in other ibuprofen-treated AD animal models (Lim, et al., 2000,Lim, et al., 2001b,Yan, et al., 2003), but differ from those reported by Jantzen et al. who found a reduction in overall plaque burden with no changes in Congo red stained plaques (Jantzen, et al., 2002). Ibuprofen treatment has previously been shown to suppress the phenotypic activation of microglia in AD animal models (Lim, et al., 2000,Yan, et al., 2003). We observed microglia with an amoeboid, “activated” morphology clustered around amyloid plaques in the control B6-R1.40 mice. However, microglia had a more ramified or “resting” morphology in the ibuprofen-treated B6-R1.40 mice (Figure 2A). Chronic ibuprofen treatment in the B6-R1.40 mice resulted in little, if any, amyloid deposition, and we found very few microglia adjacent to the 6E10-positive amyloid plaques remaining in these mice.
Figure 1. Chronic ibuprofen treatment reduces AD-related plaque pathology in B6-R1.40 mice.
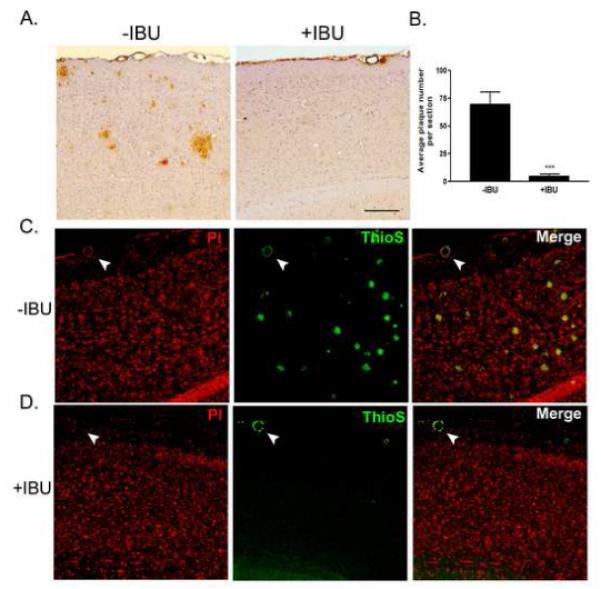
(A) Sagittal sections from age-matched non-treated (−IBU) and ibuprofen-treated (+IBU) 24-month-old B6-R1.40 mice were immunstained with anti-human Aβ monoclonal antibody 6E10. (B) Average plaque number/section in the parenchyma was reduced by 90% (n=5/group, ***p<0.001) in +IBU animals. Dense core plaques were identified in (C) −IBU and (D) +IBU mice by Thioflavin-S positive staining (green). Nuclei were visualized with propidium iodide (red). White arrows indicate blood vessels with amyloid deposition.
Figure 2. Chronic ibuprofen treatment reduces microglial activation in B6-R1.40 mice.
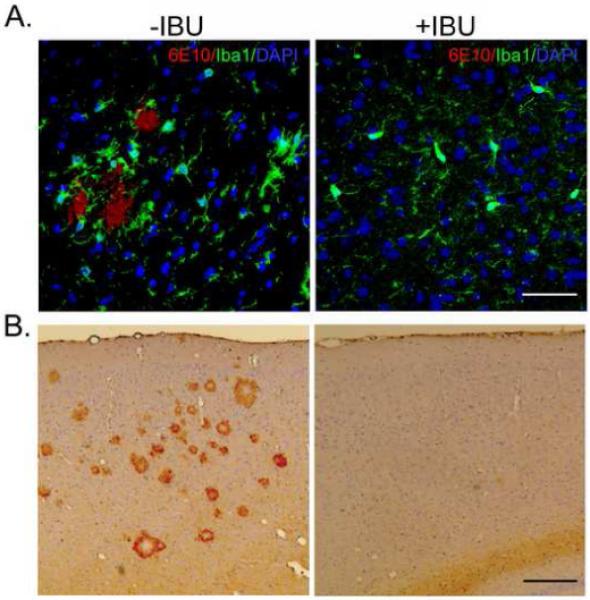
(A) Representative photomicrograph depicting phenotypically activated microglia stained with Iba1 (green) adjacent to a 6E10+ plaque (red) in the non-treated (−IBU) but not the ibuprofen-treated (+IBU) B6-R1.40 mouse. Nuclei are stained with DAPI (blue); scale bar=50 μm. (B) Sagittal sections from −IBU and +IBU-treated B6-R1.40 mice were stained for anti-CD45; scale bar=200μm.
Microglia activation was also assessed by CD45-immunoreactivity. CD45 is a tyrosine phosphatase that is important for immune cell signaling, and this molecule has been shown to be elevated in activated microglia in both the human AD brain (Masliah, et al., 1991) and AD animal models (Wilcock, et al., 2001). Following chronic ibuprofen treatment in the B6-R1.40 mouse, we observed a profound reduction in CD45-positive microglia staining compared to control B6-1.40 mice (Figure 2B), consistent with previous reports (Lim, et al., 2000,Yan, et al., 2003).
3.2 AD-related oxidative damage is attenuated by chronic ibuprofen treatment in aged B6-R1.40 mice
Activated of microglia are a significant source of reactive oxygen species (ROS) production and oxidative damage in a variety of neurodegenerative diseases including AD (Block, et al., 2007). The reduction in microglial activation following chronic ibuprofen treatment in the B6-R1.40 mice led us to examine potential alterations in oxidative damage. We first examined whether the B6-R1.40 mice had increased basal levels of oxidative stress, a phenomena observed in other APP mutant mice (Mohmmad Abdul, et al., 2006). We measured the levels of 4-hydroxynonenal (4HNE) protein adducts in brain lysates from 24 month-old age-matched C57BL/6 (wildtype) mice and B6-R1.40 mice. 4HNE is a product of lipid peroxidation and has been shown to exert a host of adverse biological side-effects (Uchida and Stadtman, 1992). A robust elevation of 4HNE is evident in the brains of the B6-R1.40 mice (Figure 3A). We next examined whether chronic ibuprofen treatment could ameliorate the substantial oxidative damage found in aged B6-R1.40 mice. Indeed, ibuprofen-treated B6-R1.40 mice show a 70% reduction in the accumulation of 4HNE protein adducts when compared to control animals (p < 0.01; Figure 3B) indicating lipid peroxidation is attenuated by ibuprofen treatment.
Figure 3. Ibuprofen treatment reduces AD-related oxidative damage.
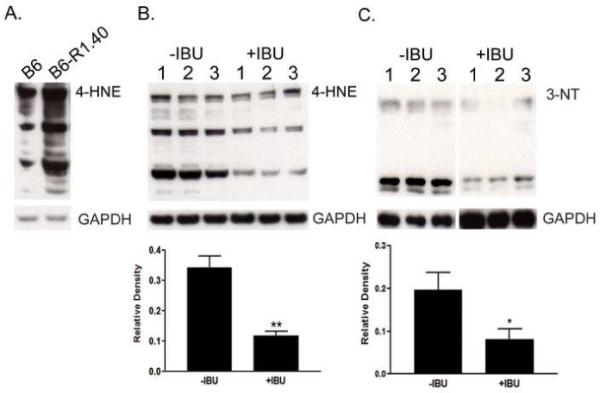
(A) Effect of the hAPP transgene on oxidative damage as measured by 4-HNE levels in brain homogenates. Samples from 24-month-old age-matched C57BL/6 (B6) and B6-R1.40 mice are shown. (B) Representative immunoblot from individual non-treated (−IBU) and ibuprofen-treated (+IBU) brain homogenates analyzed for lipid peroxidation measured by 4-HNE protein adduct levels (n=5, **p<0.01). (C) Representative immunoblot from −IBU and +IBU-treated animals analyzed for 3-nitrotyrosine (3-NT) levels (n=5, *p<0.05). Blots were stripped and reprobed with GAPDH as a protein loading control.
The increased production of nitrotyrosine has also been described in AD, and has shown a high correlation with disease state. The nitration of tyrosine residues is the result of the highly reactive peroxynitrite radical, which is produced by a reaction of nitric oxide with the superoxide anion. The nitration of tyrosine residues in proteins compromises their action in cellular signaling and alters protein structure. We examined the presence of nitrotyrosine-containing proteins in brain lysates from ibuprofen-treated and control B6-R1.40 mice. The presence of nitrotyrosine was 60% lower in the ibuprofen-treated animals than in the control animals (p < 0.05; Figure 3C).
We also evaluated the addition of carbonyl groups to protein side chains; a sensitive method for the detection and quantification of protein oxidation (Stadtman and Levine, 2000). The carbonylation of brain proteins was 4-fold greater in the control 24 month-old B6-R1.40 mice than in the ibuprofen-treated mice (p < 0.05; Figure 4). Taken together, these data indicate that chronic ibuprofen treatment suppresses oxidative damage in the B6-R1.40 AD mouse model.
Figure 4. Chronic ibuprofen treatment reduced protein oxidation in aged B6-R1.40 mice.
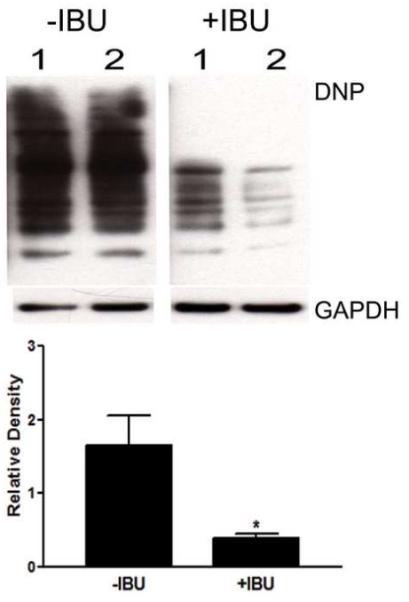
Protein oxidation was measured using an Oxyblot kit. Representative blot from non-treated (−IBU) and ibuprofen-treated (+IBU) brain homogenates analyzed by immunoblot analysis with an anti-DNP antibody (n=5, *p< 0.05). Blots were stripped and reprobed with GAPDH as a protein loading control.
3.3 Ibuprofen treatment inhibits fAβ-stimulated ROS production in primary microglia
Several potential sources of ROS exist within microglia including the NADPH oxidase, mitochondria respiratory chain, xanthine oxidase, microsomal enzymes, cycloxygenase and lipoxygenase. In response to fAβ; however, it has been postulated that the primary source of ROS and the source of widespread oxidative damage found in the AD brain is the microglial NADPH oxidase (NOX2) (Bianca, et al., 1999,Markesbery, 1997,McDonald, et al., 1997,Qin, et al., 2005,Shimohama, et al., 2000,Wilkinson, et al., 2006). The reduction in microglia activation and oxidative damage in the ibuprofen-treated R1.40 mice led us to examine whether ibuprofen could inhibit NOX2-derived ROS production in fAβ-stimulated microglia. To examine the effect of ibuprofen treatment on the fAβ-stimulated respiratory burst, we utilized primary microglia obtained from C57BL/6 mice. Analysis of intracellular superoxide production was monitored by the reduction of NBT, and PMA was used as a positive control. We observed that pretreatment with racemic ibuprofen attenuated fAβ-stimulated superoxide production in primary microglia by 30% when compared to non-treated controls (Figure 5A). This data suggests ibuprofen acts to inhibit a NOX2-derived respiratory burst in primary microglia.
Figure 5. Fibrillar Aβ-stimulated Vav phosphorylation is inhibited by ibuprofen pretreatment.
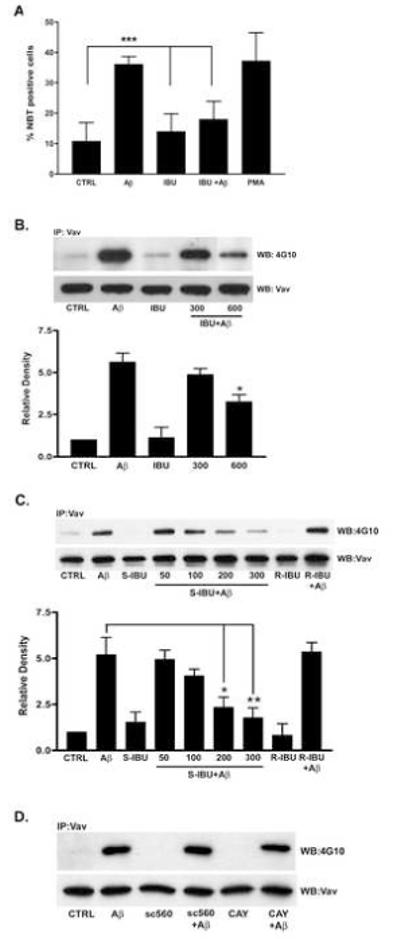
(A) C57Bl/6 microglia were pretreated with ibuprofen (600 μM) for 1hr followed by incubation in serum-free DMEM-F12 containing NBT +/− fAβ25–35 (60 μM) for 30 min. PMA (390 nM) was a positive control. Superoxide generation was monitored by the presence of insoluble formazan and visualized on a Leica DMIRB inverted microscope. Three random fields of cells (>100 cells) were counted (n=6, ***p< 0.001). (B) THP-1 cells (5 × 106 cells) were pretreated 1 hr +/− ibuprofen and then stimulated with fAβ25-35 for 3 min. Vav immunoprecipitates were analyzed by Western blot analysis using a phospho-Tyr antibody (4G10). Blots were stripped and reprobed with Vav as a protein-loading control. Band intensity was analyzed as the level of phophorylated Vav normalized to total Vav protein levels and expressed as relative density (n=3, *p<0.05). (C) Dose response of S-ibuprofen pretreatment on Vav protein-Tyr phosphorylation in THP-1 cells treated with fAβ25-35 (60 μM) for 3 min. (*p<0.05 at 200 μM and **p<0.01 at 300 μM). R-ibuprofen (200 μM) pretreatment had no effect (n=4). (D) THP-1 cells pretreated 1 hr with either a COX1 (sc560) or a COX2 (CAY10404) inhibitor were stimulated with fAβ25-35 for 3 min. Vav immunoprecipitates were analyzed by Western blot using a phospho-Tyr antibody (4G10). Blots were stripped and reprobed with Vav as a protein-loading control.
3.4 Ibuprofen inhibits fAβ-stimulated Vav phosphorylation in THP-1 cells
We reported a mechanistic link between the microglia fAβ cell surface receptor complex (Bamberger, et al., 2003) and downstream signaling events leading to NOX2 complex-derived reactive oxygen production ((Reed-Geaghan, et al., 2009,Wilkinson, et al., 2006). Assembly and function of the NOX2 enzyme complex is dependent on the fAβ-stimulated phosphorylation of Vav, a guanine nucleotide exchange factor (GEF) for the Rac1 GTPase (Wilkinson, et al., 2006). We examined whether ibuprofen could inhibit fAβ-stimulated Vav phosphorylation. THP-1 cells were pretreated with racemic ibuprofen for 1 h followed by exposure to fAβ25–35. Indeed, ibuprofen pretreatment suppressed the Tyr-phosphorylation of Vav (p < 0.05; Figure 5B).
We evaluated the ability of the individual enantiomers of ibuprofen to inhibit fAβ-stimulated Vav phosphorylation. The S-enantiomer of ibuprofen is the active enantiomer with respect to COX inhibition. Pretreatment with the S-enantiomer was able to inhibit Vav phosphorylation in a dose-dependent manner; however, pretreatment with the R-enantiomer had no effect (p < 0.05; Figure 5C). These data argue that the inhibition of COX activity may play a role in suppressing fAβ-induced reactive oxygen production. We next wanted to determine whether COX-1 or COX-2 was responsible for inhibiting Vav phosphorylation. THP-1 cells were pretreated for 1 h with sc-560, a COX-1 specific inhibitor, or CAY10404, a highly specific COX-2 inhibitor, followed by stimulation with fAβ Neither inhibitors was able to prevent an increase in Vav Tyr-phosphorylation in response to the fAβ peptide (Figure 5D). These data suggest that the action of the S-enantiomer of ibuprofen on reducing Vav phosphorylation is mediated through a COX-independent mechanism.
3.5 S-ibuprofen disrupts downstream signaling to NOX2 complex assembly
We next established whether additional signaling events leading to NOX2 complex assembly were disrupted by ibuprofen treatment. The downstream target of Vav GEF activity is the small GTPase, Rac1, which is an integral component of the NOX2 enzyme complex. Vav facilitates Rac GDP/GTP exchange converting Rac into its active conformation. Rac then translocates to the plasma membrane and interacts with other NOX2 enzyme components to assemble the catalytically active oxidase. Previously, we have demonstrated that fAβ stimulation increased Rac GTP-loading and promoted redistribution of Rac from the cytosol to the plasma membrane (Wilkinson, et al., 2006). Here, we report that S-ibuprofen pretreatment of THP-1 cells leads to a dose-dependent inhibition of fAβ-mediated Rac translocation to the membrane (Figure 6A). These data demonstrate that treatment with S-ibuprofen impairs NOX2 complex assembly.
Figure 6. S-ibuprofen disrupts NOX2 complex assembly.
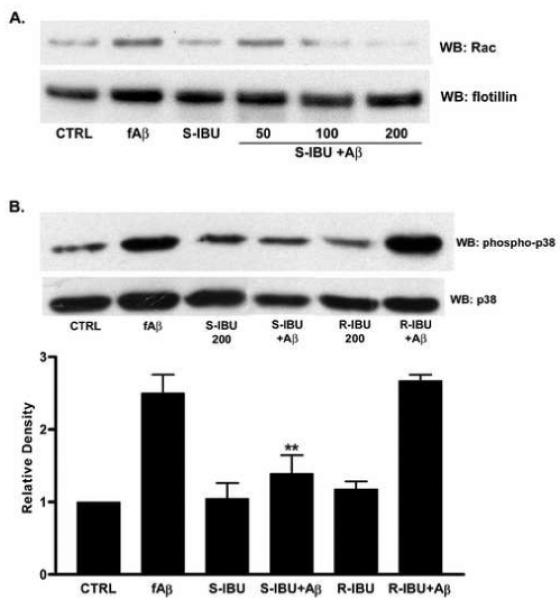
(A) Dose response for THP-1 cells pretreated with S-ibuprofen for 1 hr followed by stimulation with fAβ25-35 (60 μM) for 10 min. Lysates were subjected to differential centrifugation and membrane fractions were immunoblotted for Rac. Cell fractions were also immunoblotted with a flotillin antibody to assess the efficacy of the fractionation procedure. (B) THP-1 cells pretreated with either S- or R-ibuprofen for 1 hr were stimulated with fAβ25-35. Lysates were immunoblotted for phosphorylated p38. Blots were stripped and reprobed with p38 as a protein loading control. Band intensity was analyzed as the level of phophorylated p38 normalized to total p38 protein levels and expressed as relative density (n=4, **p<0.01).
Since S-ibuprofen treatment inhibits signaling cascades leading to a defect in oxidase assembly, we next determined whether parallel signaling pathways responsible for activation of p47phox were also impaired. p47phox, a cytosolic component of the NOX2 enzyme complex, must be phosphorylated on serine residues to initiate translocation to the membrane where it interacts with the membrane-bound cytochrome b558. It is well established that upstream p38 MAPK activity is critical for both superoxide production and p47phox phosphorylation in phagocytes (Detmers, et al., 1998,Yamamori, et al., 2000). Important for our studies, we have previously demonstrated that p38 phosphorylation is upregulated following exposure to Aβ fibrils in THP-1 cells (McDonald, et al., 1998). To determine the effect of S-ibuprofen on p38 activity, THP-1 cells were pretreated with S-ibuprofen followed by exposure to fAβ; cellular lysates were then analyzed for levels of p38 phosphorylation. We report that S-ibuprofen pretreatment diminished Aβ-induced p38 activity and R-ibuprofen had no effect (p < 0.001; Figure 6B). These findings suggest that S-ibuprofen acts to disrupt parallel signaling cascades leading to improper assembly of the NOX2 enzyme complex. It is also possible that p38 activation is a result of ROS production.
3.6 S-ibuprofen inhibits fAβ-stimulated ROS production in primary microglia
In light of our previous findings that several critical signaling molecules are regulated by ibuprofen treatment, we hypothesized that S-ibuprofen might inhibit the generation of ROS in primary murine microglia stimulated with fAβ peptides. Following pretreatment with S-ibuprofen, Aβ-stimulated intracellular superoxide production was monitored in primary microglia. We observed that fAβ-induced ROS production was dramatically reduced by pretreatment with S-ibuprofen when compared to either fAβ or PMA, a positive control for respiratory burst (p < 0.001; Figure 7). These results indicate that the S-ibuprofen impairs NADPH oxidase function and ROS production in response to Aβ fibrils.
Figure 7. S-ibuprofen inhibits the generation of NOX2-derived radicals in microglia stimulated with fAβ.
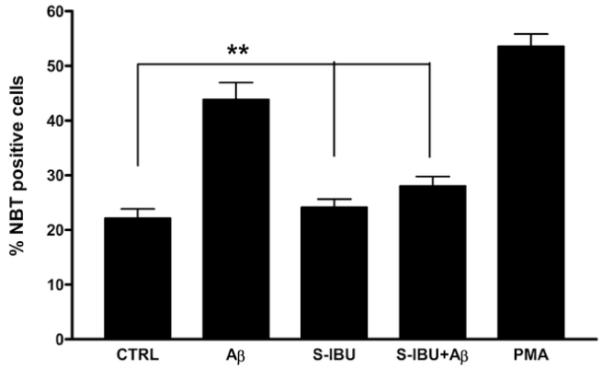
Primary C57Bl/6 microglia were preincubated with S-ibuprofen (200 μM) for 1 hr in serum-free DMEM-F12. NBT was added to the media and microglia were stimulated with fAβ25-35 (60 μM) or PMA (390 nM) for 30 min. Superoxide anion generation was monitored by the presence of insoluble formazan. Three random fields of cells (>100 cells) were counted (n=3, **p < 0.01).
Discussion
NSAIDs have received considerable attention as a potential therapy for AD owing to numerous epidemiological studies that provide evidence for an association between the chronic intake of NSAIDs and a decreased risk for AD (in t’ Veld, et al., 2001,Stewart, et al., 1997,Vlad, et al., 2008,Zandi, et al., 2002). However, several recent studies found no benefit from NSAID intake on incidence of AD (Aisen, et al., 2003,Arvanitakis, et al., 2008,Breitner, et al., 2009,Group, et al., 2007,Reines, et al., 2004). Thus, there remains considerable confusion about the effects of NSAIDS in altering AD risk and pathogenesis. In the epidemiological study, Vlad et al. reported that ibuprofen exhibited a strong protective effect, and this protection increases with prolonged usage (Vlad, et al., 2008). Additionally, in vivo studies in murine models of AD have demonstrated that preventative long-term ibuprofen treatment inhibited brain inflammation, reduced amyloid pathology, decreased Aβ levels, and improved cognition (Jantzen, et al., 2002,Lim, et al., 2001a,Lim, et al., 2000,McKee, et al., 2008,Yan, et al., 2003). In the present study, we report that R1.40 mice treated with ibuprofen had a 90% reduction in Aβ plaque number within the parenchyma. These results obtained in R1.40 mice recapitulate previously reported findings in the Tg2576 model (Jantzen, et al., 2002,Lim, et al., 2000,Yan, et al., 2003), but are notable for the near complete absence of amyloid deposits in the brains of these aged mice. In addition, we observe a significant reduction in microglial activation and association with Aβ plaques in the R1.40 mice treated with ibuprofen, a phenomenon previously reported in ibuprofen-treated Tg2576 mice (Lim, et al., 2000,Yan, et al., 2003).
We further report that ibuprofen acts to suppress oxidative damage in the AD brain through its capacity to inhibit NOX2-derived free radical production. The brain’s high metabolic rate and reduced capacity for cellular regeneration makes the brain susceptible to oxidative damage. Damage from oxidative stress has been postulated to be an antecedent event in AD pathogenesis (Pratico and Sung, 2004,Pratico, et al., 2001). Markers of oxidative damage can be detected prior to Aβ deposition in both brains of humans(Markesbery, 1997) and in Tg2576 mice (Park, et al., 2005,Pratico, et al., 2001). Interestingly, it has been reported that some NSAIDs exhibit antioxidant activity (Asanuma, et al., 2001,Hamburger and McCay, 1990). We found that chronic ibuprofen treatment of R1.40 mice results in significant reductions of three independent measures of oxidative damage--lipid peroxidation, tyrosine nitration and protein oxidation, in contrast to Lim and colleagues who reported no change in the level of protein oxidation between ibuprofen and control-treated Tg2576 mice (Lim, et al., 2001b). It is significant to note that Lim et al. used a different AD animal model (Tg2576), a shorter treatment paradigm (6 months), and only analyzed a single marker for oxidative damage. Importantly, our current findings provide the first in vivo evidence that chronic ibuprofen treatment can alleviate AD-related oxidative damage.
The microglia phagocytic NADPH oxidase (NOX2) complex has been hypothesized to be a major source of ROS in AD (Bianca, et al., 1999,McDonald, et al., 1997,Qin, et al., 2006). Microglia and monocytes generate a NOX2-derived respiratory burst in response to fibrillar Aβ peptides (Bianca, et al., 1999,McDonald, et al., 1997) that is dependent on Aβ fibril engagement of a microglial cell surface receptor complex (Bamberger, et al., 2003,Wilkinson, et al., 2006). The NOX2 enzyme complex localizes to both intracellular and plasma membranes catalyzing the production of superoxide from oxygen. NOX2 is comprised of several different subunits including two integral membrane-bound proteins, p22phox and gp91phox, which form the catalytic subunit (cytochrome b558) of the complex. In resting cells, the NOX2 enzyme complex has cytosolic components (p47phox, p67phox, p40phox, and Rac1) that are distributed throughout the cytoplasm. Upon stimulation, these cytosolic components are activated by parallel signaling pathways, initiating translocation to the membrane where they interact with the membrane-bound subunits forming the active complex (Bedard and Krause, 2007,Lambeth, 2004).
Recently, we have identified a mechanistic linkage between fAβ engagement of the microglial Aβ receptor complex and the initiation of intracellular signaling events regulating oxidase assembly and activation. Tyrosine phosphorylation of Vav, a guanine nucleotide exchange factor (GEF) for Rac1, and Vav’s association with Src-kinases were identified as proximal signaling events critical for ROS production in fAβ-stimulated microglia (Wilkinson, et al., 2006). Here, we demonstrate that racemic ibuprofen and S-ibuprofen act to inhibit Aβ fibril-stimulated ROS production through disruption of the intracellular signaling pathways leading to NOX2 complex assembly and activation. Treatment with these NSAIDs abrogated Vav tyrosine phosphorylation resulting in the inability of Rac to become activated and translocate to the membrane. We confirmed that NOX2 enzyme activity and ROS production was indeed reduced in primary microglia following pretreatment with S-ibuprofen. Pretreatment with R-ibuprofen was without effect. These data argue for the involvement of a COX-dependent signaling mechanism. Surprisingly, the COX specific inhibitors, sc-560 (COX-1) or CAY10404 (COX-2) failed to attenuate Vav tyrosine phosphorylation. A parallel effect of S-ibuprofen pretreatment was observed for the inhibition of fAβ-stimulated p38 phosphorylation. Together, these findings indicate an S-enantiomer-specific, COX-independent action of this drug. Several COX-independent actions of NSAIDs have been documented; however, COX-independent actions of S-ibuprofen have not, to our knowledge, been reported. While the exact upstream signaling targets modulated by S-ibuprofen remain to be identified, our data suggests that S-ibuprofen may act through disruption of the action of upstream Src kinases, as global inhibition of Src-family tyrosine kinases or inhibition of phosphatidylinositol-3 kinase has been previously shown to attenuate ROS production (Bianca, et al., 1999,Wilkinson, et al., 2006).
The consequence of inhibiting NOX2-derived radicals in AD models has recently been examined. The contribution of NOX2-derived ROS was validated using a NOX2-deficient (gp91phox null) macrophage cell line, which failed to kill APP-expressing neuroblastoma cells. Interestingly, Tg2576 mice in which NOX2 was genetically inactivated do not exhibit alterations in AD plaque pathology at the age of initial deposition, suggesting that ROS generated by the NOX2 complex do not contribute to processes affecting initial Aβ deposition. As expected, these animals did not develop oxidative stress, cerebrovascular dysfunction, or behavioral deficits that normally would occur in the Tg2576 mice (Park, et al., 2008).
A central finding in this current study is the dramatic reduction in murine plaque burden with ibuprofen treatment, a finding consistent with previous reports (Jantzen, et al., 2002,Lim, et al., 2001a,Lim, et al., 2000,McKee, et al., 2008,Yan, et al., 2003). The mechanistic basis of this effect remains unclear. A recent renewed interest in this class of drugs for AD treatment stems from findings suggesting that some, but not all, NSAIDs can act independently from their classic anti-inflammatory mechanisms, which may play a role in their disease-modifying actions (Combs, et al., 2000,Eriksen, et al., 2003,Lehmann, et al., 1997,Lleo, et al., 2004,Weggen, et al., 2001,Zhou, et al., 2003). Koenigsknecht et al. reported that the microglial phagocytic function was suppressed in presence of inflammatory cytokines found in the AD brain and that this function could be restored upon treatment of ibuprofen or COX2 inhibitors in vitro (Koenigsknecht-Talboo and Landreth, 2005), a finding consistent with those reported by Liang et al. in vivo(Liang, et al., 2005). It remains possible that the reduction in activated microglia and oxidative damage in the brain is secondary to reduction in plaque burden in these animals. The role of microglia in plaque formation and remodeling has been questioned. In a recent paper by Grathwohl et al., the authors demonstrate a loss of microglia had no effect on plaque number and size over a period of 2-4 weeks, they suggest endogenous microglia play no role in formation and maintenance of Aβ plaques. However, the authors also report a 3-4 fold increase in soluble Aβ levels in the microglia-deficient animals (Grathwohl, et al., 2009), consistent with a role for microglia in clearance of Aβ.
In summary, we provide evidence that ibuprofen acts in an enantiomer-specific manner to inhibit NADPH oxidase activation and ROS production. This is associated with a dramatic reduction in oxidative damage and amyloid deposition in a murine model of AD. The existing data from mouse models suggest that ibuprofen acts through multiple independent pathways to affect AD-related pathology. It is important to note that the outcomes of experiments in animal models have not been predictive of the effects of NSAIDs in humans and it remains unclear how these drugs affect AD risk and pathogenesis.
Acknowledgements
This work was supported by grants from the National Institutes of Health (AG16740, G.E.L.; AG023012, B.T.L.; AG024494, K.H.), the Blanchette Hooker Rockefeller Foundation, and the American Health Assistance Foundation (G.E.L.). B.L.W. and E.R.G.were supported in part through Ruth L. Kirschstein National Research Service Awards from the National Institutes of Health (F32 AG24031 and F31-NS057867). Thanks to Natalie Cherosky for technical help.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aisen PS, Schafer KA, Grundman M, Pfeiffer E, Sano M, Davis KL, Farlow MR, Jin S, Thomas RG, Thal LJ. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. Jama. 2003;289(21):2819–26. doi: 10.1001/jama.289.21.2819. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O’Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21(3):383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvanitakis Z, Grodstein F, Bienias JL, Schneider JA, Wilson RS, Kelly JF, Evans DA, Bennett DA. Relation of NSAIDs to incident AD, change in cognitive function, and AD pathology. Neurology. 2008;70(23):2219–25. doi: 10.1212/01.wnl.0000313813.48505.86. [DOI] [PubMed] [Google Scholar]
- Asanuma M, Nishibayashi-Asanuma S, Miyazaki I, Kohno M, Ogawa N. Neuroprotective effects of non-steroidal anti-inflammatory drugs by direct scavenging of nitric oxide radicals. J Neurochem. 2001;76(6):1895–904. doi: 10.1046/j.1471-4159.2001.00205.x. [DOI] [PubMed] [Google Scholar]
- Bamberger ME, Harris ME, McDonald DR, Husemann J, Landreth GE. A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J Neurosci. 2003;23(7):2665–74. doi: 10.1523/JNEUROSCI.23-07-02665.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamberger ME, Landreth GE. Inflammation, apoptosis, and Alzheimer’s disease. Neuroscientist. 2002;8(3):276–83. doi: 10.1177/1073858402008003013. [DOI] [PubMed] [Google Scholar]
- Banati RB, Gehrmann J, Schubert P, Kreutzberg GW. Cytotoxicity of microglia. Glia. 1993;7(1):111–8. doi: 10.1002/glia.440070117. [DOI] [PubMed] [Google Scholar]
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87(1):245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Bianca VD, Dusi S, Bianchini E, Dal Pra I, Rossi F. beta-amyloid activates the O-2 forming NADPH oxidase in microglia, monocytes, and neutrophils. A possible inflammatory mechanism of neuronal damage in Alzheimer’s disease. J Biol Chem. 1999;274(22):15493–9. doi: 10.1074/jbc.274.22.15493. [DOI] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8(1):57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Breitner JC, Haneuse SJ, Walker R, Dublin S, Crane PK, Gray SL, Larson EB. Risk of dementia and AD with prior exposure to NSAIDs in an elderly community-based cohort. Neurology. 2009;72(22):1899–905. doi: 10.1212/WNL.0b013e3181a18691. doi:WNL.0b013e3181a18691 [pii] 10.1212/WNL.0b013e3181a18691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdick D, Soreghan B, Kwon M, Kosmoski J, Knauer M, Henschen A, Yates J, Cotman C, Glabe C. Assembly and aggregation properties of synthetic Alzheimer’s A4/beta amyloid peptide analogs. J Biol Chem. 1992;267(1):546–54. [PubMed] [Google Scholar]
- Combs C, Bates P, Karlo J, Landreth G. Regulation of beta-amyloid stimulated proinflammatory responses by peroxisome proliferator-activated receptor alpha. Neurochem Intl. 2001 doi: 10.1016/s0197-0186(01)00052-3. [DOI] [PubMed] [Google Scholar]
- Combs CK, Johnson DE, Cannady SB, Lehman TM, Landreth GE. Identification of microglial signal transduction pathways mediating a neurotoxic response to amyloidogenic fragments of beta-amyloid and prion proteins. J Neurosci. 1999;19(3):928–39. doi: 10.1523/JNEUROSCI.19-03-00928.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs CK, Johnson DE, Karlo JC, Cannady SB, Landreth GE. Inflammatory mechanisms in Alzheimer’s disease: inhibition of beta-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARgamma agonists. J Neurosci. 2000;20(2):558–67. doi: 10.1523/JNEUROSCI.20-02-00558.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detmers PA, Zhou D, Polizzi E, Thieringer R, Hanlon WA, Vaidya S, Bansal V. Role of stress-activated mitogen-activated protein kinase (p38) in beta 2-integrin-dependent neutrophil adhesion and the adhesion-dependent oxidative burst. J Immunol. 1998;161(4):1921–9. [PubMed] [Google Scholar]
- Eriksen JL, Sagi SA, Smith TE, Weggen S, Das P, McLendon DC, Ozols VV, Jessing KW, Zavitz KH, Koo EH, Golde TE. NSAIDs and enantiomers of flurbiprofen target gamma-secretase and lower Abeta 42 in vivo. J Clin Invest. 2003;112(3):440–9. doi: 10.1172/JCI18162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grathwohl SA, Kalin RE, Bolmont T, Prokop S, Winkelmann G, Kaeser SA, Odenthal J, Radde R, Eldh T, Gandy S, Aguzzi A, Staufenbiel M, Mathews PM, Wolburg H, Heppner FL, Jucker M. Formation and maintenance of Alzheimer’s disease beta-amyloid plaques in the absence of microglia. Nat Neurosci. 2009;12(11):1361–3. doi: 10.1038/nn.2432. doi:nn.2432 [pii] 10.1038/nn.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Group AR, Lyketsos CG, Breitner JC, Green RC, Martin BK, Meinert C, Piantadosi S, Sabbagh M. Naproxen and celecoxib do not prevent AD in early results from a randomized controlled trial. Neurology. 2007;68(21):1800–8. doi: 10.1212/01.wnl.0000260269.93245.d2. [DOI] [PubMed] [Google Scholar]
- Hamburger SA, McCay PB. Spin trapping of ibuprofen radicals: evidence that ibuprofen is a hydroxyl radical scavenger. Free Radic Res Commun. 1990;9(3-6):337–42. doi: 10.3109/10715769009145692. [DOI] [PubMed] [Google Scholar]
- in t’ Veld BA, Ruitenberg A, Hofman A, Launer LJ, van Duijn CM, Stijnen T, Breteler MM, Stricker BH. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer’s disease. N Engl J Med. 2001;345(21):1515–21. doi: 10.1056/NEJMoa010178. [DOI] [PubMed] [Google Scholar]
- Jantzen PT, Connor KE, DiCarlo G, Wenk GL, Wallace JL, Rojiani AM, Coppola D, Morgan D, Gordon MN. Microglial activation and beta -amyloid deposit reduction caused by a nitric oxide-releasing nonsteroidal anti-inflammatory drug in amyloid precursor protein plus presenilin-1 transgenic mice. J Neurosci. 2002;22(6):2246–54. doi: 10.1523/JNEUROSCI.22-06-02246.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenigsknecht-Talboo J, Landreth GE. Microglial phagocytosis induced by fibrillar beta-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J Neurosci. 2005;25(36):8240–9. doi: 10.1523/JNEUROSCI.1808-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenigsknecht J, Landreth G. Microglial phagocytosis of fibrillar beta-amyloid through a beta1 integrin-dependent mechanism. J Neurosci. 2004;24(44):9838–46. doi: 10.1523/JNEUROSCI.2557-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotilinek LA, Westerman MA, Wang Q, Panizzon K, Lim GP, Simonyi A, Lesne S, Falinska A, Younkin LH, Younkin SG, Rowan M, Cleary J, Wallis RA, Sun GY, Cole G, Frautschy S, Anwyl R, Ashe KH. Cyclooxygenase-2 inhibition improves amyloid-beta-mediated suppression of memory and synaptic plasticity. Brain. 2008;131(Pt 3):651–64. doi: 10.1093/brain/awn008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulnane LS, Lamb BT. Neuropathological characterization of mutant amyloid precursor protein yeast artificial chromosome transgenic mice. Neurobiol Dis. 2001;8(6):982–92. doi: 10.1006/nbdi.2001.0446. [DOI] [PubMed] [Google Scholar]
- Lamb BT, Bardel KA, Kulnane LS, Anderson JJ, Holtz G, Wagner SL, Sisodia SS, Hoeger EJ. Amyloid production and deposition in mutant amyloid precursor protein and presenilin-1 yeast artificial chromosome transgenic mice. Nat Neurosci. 1999;2(8):695–7. doi: 10.1038/11154. [DOI] [PubMed] [Google Scholar]
- Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4(3):181–9. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- Lehman EJ, Kulnane LS, Gao Y, Petriello MC, Pimpis KM, Younkin L, Dolios G, Wang R, Younkin SG, Lamb BT. Genetic background regulates beta-amyloid precursor protein processing and beta-amyloid deposition in the mouse. Hum Mol Genet. 2003;12(22):2949–56. doi: 10.1093/hmg/ddg322. [DOI] [PubMed] [Google Scholar]
- Lehmann JM, Lenhard JM, Oliver BB, Ringold GM, Kliewer SA. Peroxisome proliferator-activated receptors alpha and gamma are activated by indomethacin and other non-steroidal anti-inflammatory drugs. J Biol Chem. 1997;272(6):3406–10. doi: 10.1074/jbc.272.6.3406. [DOI] [PubMed] [Google Scholar]
- Liang X, Wang Q, Hand T, Wu L, Breyer RM, Montine TJ, Andreasson K. Deletion of the prostaglandin E2 EP2 receptor reduces oxidative damage and amyloid burden in a model of Alzheimer’s disease. J Neurosci. 2005;25(44):10180–7. doi: 10.1523/JNEUROSCI.3591-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim GP, Chu T, Yang F, Beech W, Frautschy SA, Cole GM. The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J Neurosci. 2001a;21(21):8370–7. doi: 10.1523/JNEUROSCI.21-21-08370.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B, Tran T, Ubeda O, Ashe KH, Frautschy SA, Cole GM. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer’s disease. J Neurosci. 2000;20(15):5709–14. doi: 10.1523/JNEUROSCI.20-15-05709.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim GP, Yang F, Chu T, Gahtan E, Ubeda O, Beech W, Overmier JB, Hsiao-Ashec K, Frautschy SA, Cole GM. Ibuprofen effects on Alzheimer pathology and open field activity in APPsw transgenic mice. Neurobiol Aging. 2001b;22(6):983–91. doi: 10.1016/s0197-4580(01)00299-8. [DOI] [PubMed] [Google Scholar]
- Lleo A, Berezovska O, Herl L, Raju S, Deng A, Bacskai BJ, Frosch MP, Irizarry M, Hyman BT. Nonsteroidal anti-inflammatory drugs lower Abeta42 and change presenilin 1 conformation. Nat Med. 2004;10(10):1065–6. doi: 10.1038/nm1112. [DOI] [PubMed] [Google Scholar]
- Lorenzo A, Yankner BA. Beta-amyloid neurotoxicity requires fibril formation and is inhibited by congo red. Proc Natl Acad Sci U S A. 1994;91(25):12243–7. doi: 10.1073/pnas.91.25.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark RJ, Lovell MA, Markesbery WR, Uchida K, Mattson MP. A role for 4-hydroxynonenal, an aldehydic product of lipid peroxidation, in disruption of ion homeostasis and neuronal death induced by amyloid beta-peptide. J Neurochem. 1997;68(1):255–64. doi: 10.1046/j.1471-4159.1997.68010255.x. [DOI] [PubMed] [Google Scholar]
- Markesbery WR. Oxidative stress hypothesis in Alzheimer’s disease. Free Radic Biol Med. 1997;23(1):134–47. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Hansen L, Alford M, Albright T, Terry R, Shapiro P, Sundsmo M, Saitoh T. Immunoreactivity of CD45, a protein phosphotyrosine phosphatase, in Alzheimer’s disease. Acta Neuropathol. 1991;83(1):12–20. doi: 10.1007/BF00294425. [DOI] [PubMed] [Google Scholar]
- McDonald DR, Bamberger ME, Combs CK, Landreth GE. beta-Amyloid fibrils activate parallel mitogen-activated protein kinase pathways in microglia and THP1 monocytes. J Neurosci. 1998;18(12):4451–60. doi: 10.1523/JNEUROSCI.18-12-04451.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald DR, Brunden KR, Landreth GE. Amyloid fibrils activate tyrosine kinase-dependent signaling and superoxide production in microglia. J Neurosci. 1997;17(7):2284–94. doi: 10.1523/JNEUROSCI.17-07-02284.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee AC, Carreras I, Hossain L, Ryu H, Klein WL, Oddo S, LaFerla FM, Jenkins BG, Kowall NW, Dedeoglu A. Ibuprofen reduces Abeta, hyperphosphorylated tau and memory deficits in Alzheimer mice. Brain Res. 2008;1207:225–36. doi: 10.1016/j.brainres.2008.01.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohmmad Abdul H, Sultana R, Keller JN, St Clair DK, Markesbery WR, Butterfield DA. Mutations in amyloid precursor protein and presenilin-1 genes increase the basal oxidative stress in murine neuronal cells and lead to increased sensitivity to oxidative stress mediated by amyloid beta-peptide (1-42), HO and kainic acid: implications for Alzheimer’s disease. J Neurochem. 2006;96(5):1322–35. doi: 10.1111/j.1471-4159.2005.03647.x. [DOI] [PubMed] [Google Scholar]
- Nunomura A, Perry G, Pappolla MA, Wade R, Hirai K, Chiba S, Smith MA. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer’s disease. J Neurosci. 1999;19(6):1959–64. doi: 10.1523/JNEUROSCI.19-06-01959.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park L, Anrather J, Zhou P, Frys K, Pitstick R, Younkin S, Carlson GA, Iadecola C. NADPH-oxidase-derived reactive oxygen species mediate the cerebrovascular dysfunction induced by the amyloid beta peptide. J Neurosci. 2005;25(7):1769–77. doi: 10.1523/JNEUROSCI.5207-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park L, Zhou P, Pitstick R, Capone C, Anrather J, Norris EH, Younkin L, Younkin S, Carlson G, McEwen BS, Iadecola C. Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc Natl Acad Sci U S A. 2008;105(4):1347–52. doi: 10.1073/pnas.0711568105. doi:0711568105 [pii] 10.1073/pnas.0711568105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratico D. Evidence of oxidative stress in Alzheimer’s disease brain and antioxidant therapy: lights and shadows. Ann N Y Acad Sci. 2008;1147:70–8. doi: 10.1196/annals.1427.010. [DOI] [PubMed] [Google Scholar]
- Pratico D, Sung S. Lipid peroxidation and oxidative imbalance: early functional events in Alzheimer’s disease. J Alzheimers Dis. 2004;6(2):171–5. doi: 10.3233/jad-2004-6209. [DOI] [PubMed] [Google Scholar]
- Pratico D, Uryu K, Leight S, Trojanoswki JQ, Lee VM. Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J Neurosci. 2001;21(12):4183–7. doi: 10.1523/JNEUROSCI.21-12-04183.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin B, Cartier L, Dubois-Dauphin M, Li B, Serrander L, Krause KH. A key role for the microglial NADPH oxidase in APP-dependent killing of neurons. Neurobiol Aging. 2005 doi: 10.1016/j.neurobiolaging.2005.09.036. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Qin B, Cartier L, Dubois-Dauphin M, Li B, Serrander L, Krause KH. A key role for the microglial NADPH oxidase in APP-dependent killing of neurons. Neurobiol Aging. 2006;27(11):1577–87. doi: 10.1016/j.neurobiolaging.2005.09.036. doi:S0197-4580(05)00293-9 [pii] 10.1016/j.neurobiolaging.2005.09.036. [DOI] [PubMed] [Google Scholar]
- Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE. CD14 and toll-like receptors 2 and 4 are required for fibrillar A{beta}-stimulated microglial activation. J Neurosci. 2009;29(38):11982–92. doi: 10.1523/JNEUROSCI.3158-09.2009. doi:29/38/11982 [pii] 10.1523/JNEUROSCI.3158-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reines SA, Block GA, Morris JC, Liu G, Nessly ML, Lines CR, Norman BA, Baranak CC. Rofecoxib: no effect on Alzheimer’s disease in a 1-year, randomized, blinded, controlled study. Neurology. 2004;62(1):66–71. doi: 10.1212/wnl.62.1.66. [DOI] [PubMed] [Google Scholar]
- Sayre LM, Zelasko DA, Harris PL, Perry G, Salomon RG, Smith MA. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer’s disease. J Neurochem. 1997;68(5):2092–7. doi: 10.1046/j.1471-4159.1997.68052092.x. [DOI] [PubMed] [Google Scholar]
- Shimohama S, Tanino H, Kawakami N, Okamura N, Kodama H, Yamaguchi T, Hayakawa T, Nunomura A, Chiba S, Perry G, Smith MA, Fujimoto S. Activation of NADPH oxidase in Alzheimer’s disease brains. Biochem Biophys Res Commun. 2000;273(1):5–9. doi: 10.1006/bbrc.2000.2897. [DOI] [PubMed] [Google Scholar]
- Smith MA, Richey Harris PL, Sayre LM, Beckman JS, Perry G. Widespread peroxynitrite-mediated damage in Alzheimer’s disease. J Neurosci. 1997;17(8):2653–7. doi: 10.1523/JNEUROSCI.17-08-02653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnen JA, Larson EB, Gray SL, Wilson A, Kohama SG, Crane PK, Breitner JC, Montine TJ. Free radical damage to cerebral cortex in Alzheimer’s disease, microvascular brain injury, and smoking. Ann Neurol. 2009;65(2):226–9. doi: 10.1002/ana.21508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtman ER, Levine RL. Protein oxidation. Ann N Y Acad Sci. 2000;899:191–208. doi: 10.1111/j.1749-6632.2000.tb06187.x. [DOI] [PubMed] [Google Scholar]
- Stewart WF, Kawas C, Corrada M, Metter EJ. Risk of Alzheimer’s disease and duration of NSAID use. Neurology. 1997;48(3):626–32. doi: 10.1212/wnl.48.3.626. [DOI] [PubMed] [Google Scholar]
- Uchida K, Stadtman ER. Modification of histidine residues in proteins by reaction with 4-hydroxynonenal. Proc Natl Acad Sci U S A. 1992;89(10):4544–8. doi: 10.1073/pnas.89.10.4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlad SC, Miller DR, Kowall NW, Felson DT. Protective effects of NSAIDs on the development of Alzheimer disease. Neurology. 2008;70:1672–7. doi: 10.1212/01.wnl.0000311269.57716.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, Findlay KA, Smith TE, Murphy MP, Bulter T, Kang DE, Marquez-Sterling N, Golde TE, Koo EH. A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature. 2001;414(6860):212–6. doi: 10.1038/35102591. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, Gordon MN, Ugen KE, Gottschall PE, DiCarlo G, Dickey C, Boyett KW, Jantzen PT, Connor KE, Melachrino J, Hardy J, Morgan D. Number of Abeta inoculations in APP+PS1 transgenic mice influences antibody titers, microglial activation, and congophilic plaque levels. DNA Cell Biol. 2001;20(11):731–6. doi: 10.1089/10445490152717596. [DOI] [PubMed] [Google Scholar]
- Wilkinson B, Koenigsknecht-Talboo J, Grommes C, Lee CY, Landreth G. Fibrillar beta-amyloid-stimulated intracellular signaling cascades require Vav for induction of respiratory burst and phagocytosis in monocytes and microglia. J Biol Chem. 2006;281(30):20842–50. doi: 10.1074/jbc.M600627200. [DOI] [PubMed] [Google Scholar]
- Yamamori T, Inanami O, Nagahata H, Cui Y, Kuwabara M. Roles of p38 MAPK, PKC and PI3-K in the signaling pathways of NADPH oxidase activation and phagocytosis in bovine polymorphonuclear leukocytes. FEBS Lett. 2000;467(2-3):253–8. doi: 10.1016/s0014-5793(00)01167-4. [DOI] [PubMed] [Google Scholar]
- Yan Q, Zhang J, Liu H, Babu-Khan S, Vassar R, Biere AL, Citron M, Landreth G. Anti-inflammatory drug therapy alters beta-amyloid processing and deposition in an animal model of Alzheimer’s disease. J Neurosci. 2003;23(20):7504–9. doi: 10.1523/JNEUROSCI.23-20-07504.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandi PP, Anthony JC, Hayden KM, Mehta K, Mayer L, Breitner JC. Reduced incidence of AD with NSAID but not H2 receptor antagonists: the Cache County Study. Neurology. 2002;59(6):880–6. doi: 10.1212/wnl.59.6.880. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Su Y, Li B, Liu F, Ryder JW, Wu X, Gonzalez-DeWhitt PA, Gelfanova V, Hale JE, May PC, Paul SM, Ni B. Nonsteroidal anti-inflammatory drugs can lower amyloidogenic Abeta42 by inhibiting Rho. Science. 2003;302(5648):1215–7. doi: 10.1126/science.1090154. [DOI] [PubMed] [Google Scholar]