

Abstract
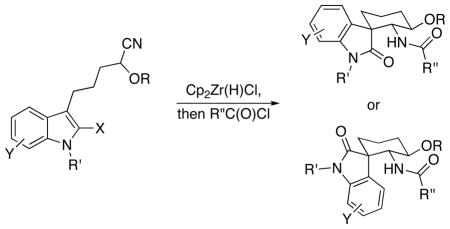
Spirooxindole amides can be prepared by the intramolecular addition of functionalized indoles into acyliminium ions that are accessed from nitriles by hydrozirconation and acylation. The stereochemical outcome at the quaternary center was controlled by the steric bulk of the substituent at the 2-position of the indole unit. The products are well-suited for diversification to prepare libraries.
The spirooxindole unit is earning the status of “privileged structure” in synthetic and medicinal chemistry, as evidenced by a number of recent reviews1 and the rapid development of new methods for their synthesis.2 The biological activity that a number of spirooxindoles exhibit has spawned library syntheses3 that led to the identification of potent MDM2 inhibitors,4 adjuvants of the actin polymerization inhibitor latrunculin B,3a and antimalarial agents.5 We have developed a new protocol for stereoselective spirooxindole synthesis in accord with our interest in preparing structurally diverse amides through nucleophilic addition reactions to acylimines. These acylimines derive from nitriles via a sequence of nitrile hydrozirconation followed by acylation.6 The oxindole preparation proceeds (Scheme 1) through the generation of indolyl acylimines (1) to form stereoisomeric spirooxindoles 2 or 3 through a Friedel-Crafts alkylation reaction, with the configuration of the quaternary stereocenter being controlled by the steric bulk of the substituent at the 2-position of the indole. The products of this sequence are structurally unique and contain multiple sites for diversification, making the route well-suited for applications in diversity-oriented synthesis.
Scheme 1.

Spirooxindole formation through acylimine addition.
Chloroindoles, shown by Horne and co-workers7 to be effective oxindole enolate surrogates, and silyloxyindoles served as the nucleophilic components for the cyclization reactions in this study. A representative route for their preparation is shown in Scheme 2. Aldehyde 4, prepared through a known8 periodate mediated cleavage of commercially-available 7-octene-1,2-diol, was converted to cyanohydrin ether 5 through a one-pot sequential acetalization reaction with BnOTMS and BiBr3 followed by TMSCN addition.9 Ozonolysis and Fischer indole synthesis with benzyl phenylhydrazine provided 6. Cyclization substrate 7 was prepared by chlorination with NCS. The corresponding silyloxyindole 8 was prepared by oxidizing 6 with DMSO and HCl through a variation on an established10 protocol to form the oxindole followed by silylation by TIPSOTf and Et3N.11
Scheme 2.

Substrate synthesis.
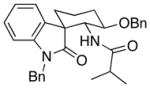
Spirooxindole formation was demonstrated (Scheme 3) by subjecting 7 to Cp2Zr(H)Cl12 followed by acylation with hydrocinnamyl chloride to form an acylimine13 that folds preferentially into a reactive conformation (9) in which the benzyloxy and acylimine groups adopt pseudoequatorial orientations and the C2-center of the indole occupies a pseudoaxial orientation. The minor conformation (10) places the indole C2 in a pseudoequatorial orientation. Addition at the 3-position of the indole followed by hydrolysis of the chloroiminium ion intermediate provided spirooxindoles 11 and 12 in 73% and 10% yields respectively. The structures of the products were assigned based on extensive analyses of their NOESY spectra. This selectivity indicates that the aryl group is sterically more demanding14 than the chlorine substituent in the transition states.
Scheme 3.

Oxindole formation through nitrile hydrozirconation, acylation, and intramolecular addition.
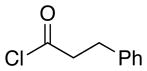
The scope of the reactions with the 2-chloroindoles is shown in Table 1. Aliphatic, branched aliphatic, α, β-unsaturated, and heteroatom-functionalized acid chlorides serve as suitable electrophiles for the transformation. N-Methoxymethylindoles are effective substrates (entries 4–9), though they are less reactive than the N-benzylindoles and in some cases require the addition of a Sc(OTf)3 to promote the cyclization (entries 6 and 7). This reactivity difference can be ascribed to the inductive attenuation of indole nucleophilicity by the methoxy group. When Sc(OTf)3 is used to promote the cyclization the stereoisomer that has a cis-relationship between the amide and benzyloxy groups forms as a minor product, presumably as a result of a competing chelation-controlled transition state. Aromatic acid chlorides can be used as electrophiles for this transformation, though product yields were <25% (not shown). Chlorination at the indole 4- and 6-positions, in anticipation of further structural manipulations of the spirooxindole products, is compatible with the reaction conditions (entries 8 and 9). These substrates reacted somewhat less efficiently than the corresponding non-halogenated substrate due to inductive deactivation by the chloro group and, in the case of 21, steric interactions in the transition state, but useful quantities of the products could be isolated.
Table 1.
Chloroindole reaction scope.a
| entry | substrateb | acid chloride | product | drc | yield(%)d |
|---|---|---|---|---|---|
| 1 | 7 | 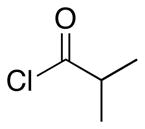 |
 13 |
3.6:1 | 87 |
| 2 | 7 | 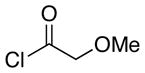 |
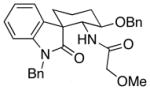 14 |
9.1:1e | 46 |
| 3 | 7 | 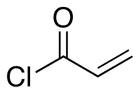 |
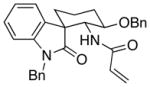 15 |
20:1 | 53 |
| 4 |
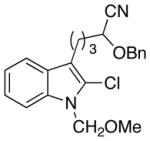 16 |
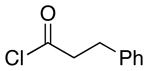 |
 17 |
15:1 | 64 |
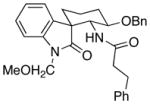
| 5 | 16 | 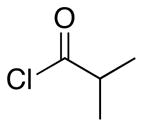 |
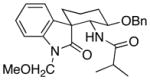 18 |
11.8:1 | 51 |
| 6f | 16 | 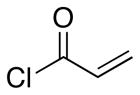 |
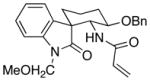 19 |
8.2:1e | 46 |
| 7f | 16 | 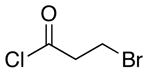 |
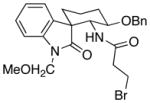 20 |
6.7:1e | 54 |
| 8 |
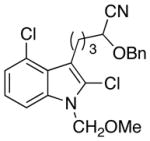 21 |
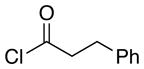 |
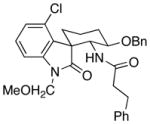 22 |
1:0 | 36 |
| 9 |
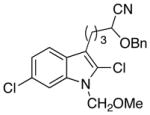 23 |
 |
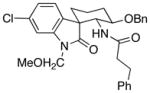 24 |
1:0 | 50 |
Representative procedure: Cp2Zr(H)Cl (1.25 equiv) was added to a 0.1 M solution of the indole nitrile in CH2Cl2 under Ar. After stirring at rt for 15 min the acid chloride was added. The mixture stirred overnight at rt.
See the Supporting Information for substrate synthesis schemes.
Ratio of isolated, purified diastereomers.
Combined yield of diastereomers.
Single isomer at the quaternary center - dr refers to the ratio of trans and cis amido ethers.
Sc(OTf)3 (0.1 equiv) was added to promote cyclization.
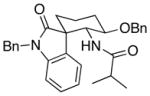
In accord with our interest15 in exploring the impact of stereochemical diversity16 in library synthesis, we sought to devise a protocol for preparing spirooxindoles in which the quaternary carbon is inverted. This can be achieved by changing the chloro substituent at the 2-position of the indole structure to a group that is more sterically demanding than the arene ring. Utilizing silyloxyindole substrate 8 in the cyclization reactions accomplished this objective (Table 2). Several issues related to these transformations are noteworthy. The overall yield of spirooxindole products from these reactions is comparable to the yields from the chloroindole substrates, indicating that the potentially labile enolsilane moiety is compatible with the reaction conditions. The reactions of 8 are slower than the reactions of the chloroindole substrates and require a full equivalent of Lewis acid despite the presence strongly electron donating silyloxy group. We attribute this effect to the steric demands in the cyclization transition states. Control of the quaternary center is good to excellent, though the trans:cis ratio between the alkoxy and amide groups is lower than what was observed for the chloroindole substrates. The trans and cis stereoisomers were readily separable by flash chromatography, with the cis-isomer reproducibly being the less polar stereoisomer. Based on the unusually downfield 1H NMR chemical shift of the C4 hydrogen in the cis-isomer, we postulate that the polarity arises from a hydrogen bond between the oxygen and the arene hydrogen. Changing the Lewis acid from Sc(OTf)3 to ZnCl2 results in the cis-isomer being the major product (entry 5) as a result of chelation between the acylimine and the benzyloxy group.
Table 2.
Cyclizations with silyloxyindole substrates.a
| entry | substrate | acid chloride | major product | drb | yield(%)c |
|---|---|---|---|---|---|
| 1 | 8 | 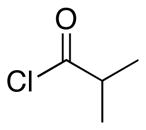 |
 25 |
4.1:1 (2.1:1) | 68 (46) |
| 2 | 8 | 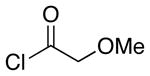 |
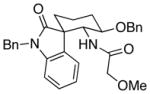 26 |
12:1 (1.7:1) | 67 (42) |
| 3 | 8 | 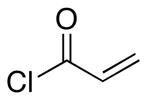 |
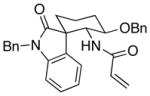 27 |
7.2:1 (3.7:1) | 38 (30) |
| 4 | 8 | 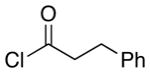 |
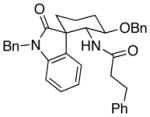 12 |
4.5:1 (2.4:1) | 66 (47) |
| 5d | 8 | 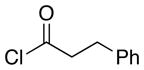 |
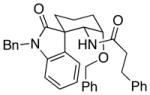 28 |
18:1 (1.8:1) | 58 (37) |
Representative procedure: Cp2Zr(H)Cl (1.25 equiv) was added to a 0.1 M solution of the indole nitrile in CH2Cl2 under Ar. After stirring at rt for 15 min the acid chloride and Sc(OTf)3 were added. The mixture stirred overnight at rt.
The ratio refers to the quaternary center. The value in parentheses refers to the ratio of the major stereoisomer to all other stereoisomers.
Combined yield of all stereoisomers. The yield of the major stereoisomer is in parentheses.
ZnCl2 was used instead of Sc(OTf)3.
The spirooxindole products have numerous points for structural diversification as a prelude to library synthesis. Several of these opportunities are shown in Scheme 4. Benzyl ether cleavage was effected through standard hydrogenolysis conditions to afford the corresponding alcohol, which can undergo subsequent acylation as seen in the conversion of 17 to 29. The acrylamide products are very versatile, undergoing Heck reaction17 to form 30, thiolate addition18 to form 31, and cross metathesis19 to form 32. Regioselective oxindole bromination can be achieved with NBS, and the resulting aryl bromide engages in a Suzuki reaction,20 as seen in the preparation of 33 from 11.
Scheme 4.

Spirooxindole functionalizations.
We have shown that indolyl cyanohydrin ethers can be converted to spirooxindole amides through a sequence of hydrozirconation, acylation, and intramolecular nucleophilic addition. Three of the four possible relative stereochemical outcomes can be prepared as major products through this process. The quaternary stereocenter can be controlled by adjusting the steric bulk of the substituent at the 2-position of the indole, while the relative configuation between the amide and benzyloxy groups can be influenced by choosing a chelating or non-chelating Lewis acid to promote the cyclization. The capacity for stereochemical diversification, the abundant opportunities for functionalizing the products, and the wide range of biological activities that spirooxindoles effect make this method a very attractive entry to library synthesis.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health, Institute of General Medicine (P50-GM067082) for financial support of this project.
Footnotes
Supporting Information Available. Experimental protocols and spectral characterization of all new products. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Zhou F, Liu YL, Zhou J. Adv Synth Catal. 2010;352:1381. [Google Scholar]; (b) Galliford CV, Scheidt KA. Angew Chem Int Ed. 2007;46:8748. doi: 10.1002/anie.200701342. [DOI] [PubMed] [Google Scholar]; (c) Marti C, Carreira EM. Eur J Org Chem. 2003:2209. [Google Scholar]
- 2.For selected recent examples, see: Zhang Y, Panek JS. Org Lett. 2009;11:3366. doi: 10.1021/ol901202t.Shintani R, Hiyashi S-y, Murakami M, Takeda M, Hiyashi T. Org Lett. 2009;11:3754. doi: 10.1021/ol901348f.Bencivenni G, Wu LY, Mazzanti A, Giannichi B, Pesciaioli F, Song MP, Bartoli G, Melchiorre P. Angew Chem Int Ed. 2009;48:7200. doi: 10.1002/anie.200903192.Chen XH, Wei Q, Luo SW, Xiao H, Gong LZ. J Am Chem Soc. 2009;131:13819. doi: 10.1021/ja905302f.Liang B, Kalidindi S, Porco JA, Jr, Stephenson CRJ. Org Lett. 2010;12:572. doi: 10.1021/ol902764k.Jiang K, Jia ZJ, Chen S, Wu L, Chen YC. Chem Eur J. 2010;16:2852. doi: 10.1002/chem.200903009.Viswambharan B, Selvakumar K, Madhavan S, Shanmugam P. Org Lett. 2010;12:2108. doi: 10.1021/ol100591r.White JD, Li Y, Ihle DC. J Org Chem. 2010;75:3569. doi: 10.1021/jo1002714.Jaegli S, Erb W, Retailleau P, Vors JP, Neuville L, Zhu J. Chem Eur J. 2010;16:5863. doi: 10.1002/chem.201000312.Chen WB, Wu ZJ, Pei QL, Cun LF, Zhang XM, Yuan WC. Org Lett. 2010;12:3132. doi: 10.1021/ol1009224.
- 3.(a) Lo MMC, Neumann CS, Nagayama S, Perlstein EO, Schreiber SL. J Am Chem Soc. 2004;126:16077. doi: 10.1021/ja045089d. [DOI] [PubMed] [Google Scholar]; (b) Chen C, Li X, Neumann CS, Lo MMC, Schreiber SL. Angew Chem Int Ed. 2005;44:2249. doi: 10.1002/anie.200462798. [DOI] [PubMed] [Google Scholar]
- 4.(a) Ding K, Lu Y, Nikolovska-Coleska Z, Qiu S, Ding Y, Gao W, Stuckey J, Krajewski K, Roller PP, Tomita Y, Parrish DA, Deschamps JR, Wang S. J Am Chem Soc. 2005;127:10130. doi: 10.1021/ja051147z. [DOI] [PubMed] [Google Scholar]; (b) Yu S, Qin D, Shangary S, Chen J, Wang G, Ding K, McEachern D, Qiu S, Nikolovska-Coleska Z, Miller R, Kang S, Yang D, Wang S. J Med Chem. 2009;52:7970. doi: 10.1021/jm901400z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rottmann M, McNamara C, Yeung BKS, Lee MCS, Zou B, Russell B, Seitz P, Plouffe DM, Dharia NV, Tan J, Cohen SB, Spencer KR, González-Páez G, Lakshminarayana SB, Goh A, Suwanarusk R, Jegla T, Schmitt EK, Beck HP, Brun R, Nosten F, Renia L, Dartois V, Keller TH, Fidock DA, Winzeler EA, Diagana TT. Science. 2010;329:1175. doi: 10.1126/science.1193225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Wan S, Green ME, Park JH, Floreancig PE. Org Lett. 2007;9:5385. doi: 10.1021/ol702184n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Xiao Q, Floreancig PE. Org Lett. 2008;10:1139. doi: 10.1021/ol8000409. [DOI] [PubMed] [Google Scholar]; (c) DeBenedetto MV, Green ME, Wan S, Park JH, Floreancig PE. Org Lett. 2009;11:835. doi: 10.1021/ol802764j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Miyake FY, Yakushijin K, Horne DA. Angew Chem Int Ed. 2004;43:5357. doi: 10.1002/anie.200460419. [DOI] [PubMed] [Google Scholar]; (b) Miyake FY, Yakushijin K, Horne DA. Org Lett. 2004;6:711. doi: 10.1021/ol030138x. [DOI] [PubMed] [Google Scholar]
- 8.Faucher AM, Bailey MD, Beaulieu PL, Brochu C, Duceppe JS, Ferland JM, Ghiro E, Gorys V, Halmos T, Kawai SH, Poirier M, Simoneau B, Tsantrizos YS, Llinàs-Brunet M. Org Lett. 2004;6:2901. doi: 10.1021/ol0489907. [DOI] [PubMed] [Google Scholar]
- 9.(a) Zhang L, Xiao Q, Ma C, Xie XQ, Floreancig PE. J Comb Chem. 2009;11:640. doi: 10.1021/cc800200h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Iwanami K, Oriyami T. Chem Lett. 2004;33:1324. [Google Scholar]
- 10.Szabó-Pusztay K, Szabó L. Synthesis. 1979:276. [Google Scholar]
- 11.Sawada T, Fuerst DE, Wood JL. Tetrahedron Lett. 2003;44:4919. [Google Scholar]
- 12.(a) Hart DW, Schwartz J. J Am Chem Soc. 1974;96:8115. [Google Scholar]; (b) Buchwald SL, LaMaire SJ, Nielsen RB, Watson BT, King SM. Tetrahedron Lett. 1987;28:3895. [Google Scholar]
- 13.Maraval A, Igau A, Donnadieu B, Majoral JP. Eur J Org Chem. 2003:385. [Google Scholar]
- 14.Overman LE, Watson DA. J Org Chem. 2006;71:2587. doi: 10.1021/jo052335a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu L, Floreancig PE. Angew Chem Int Ed. 2010;49:3069. doi: 10.1002/anie.201000033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burke MD, Schreiber SL. Angew Chem Int Ed. 2004;43:46. doi: 10.1002/anie.200300626. [DOI] [PubMed] [Google Scholar]
- 17.(a) Heck RF, Nolley JP., Jr J Org Chem. 1972;37:2320. [Google Scholar]; (b) Shen HC, Ding FX, Deng Q, Wilsie LC, Krsmanovic ML, Taggart AK, Carballo-Jane E, Ren N, Cai TQ, Wu TJ, Wu KK, Cheng K, Chen Q, Wolff MS, Tong X, Holt TG, Waters MG, Hammond ML, Tata JR, Colletti SL. J Med Chem. 2009;52:2587. doi: 10.1021/jm900151e. [DOI] [PubMed] [Google Scholar]
- 18.McGhee AM, Kizirian JC, Procter DJ. Org Biomol Chem. 2007;5:1021. doi: 10.1039/b700477j. [DOI] [PubMed] [Google Scholar]
- 19.(a) Chatterjee AK, Choi TL, Sanders DP, Grubbs RH. J Am Chem Soc. 2003;125:11360. doi: 10.1021/ja0214882. [DOI] [PubMed] [Google Scholar]; (b) Garber SB, Kingsbury JS, Gray BL, Hoveyda AH. J Am Chem Soc. 2000;122:8168. [Google Scholar]
- 20.Miyaura N, Yamada K, Suginome H, Suzuki A. J Am Chem Soc. 1985;107:972. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.