

Abstract
Purpose
To review recent developments in clinical aspects, molecular geneticsand pathogenesis of arrhythmogenic right ventricular cardiomyopathy (ARVC).
Recent findings
ARVC is a primary disease of the myocardium characterized by fibro-adipocytic replacement of myocytes, predominantly in the right ventricle.
Phenotypic expression of ARVC is variable and a significant number of patients may exhibit a subtle phenotype, particularly in the early stages of the disease. Mutations in DSP, JUP, PKP2, DSG2 and DSC2; encoding desmosomal proteins desmoplakin (DP), plakoglobin (PG), plakophilin 2 (PKP2), desmoglein 2 (DSG2), and desmocollin 2 (DSC2), respectively, cause ARVC. Thus, ARVC, at least in a subset, is a disease of desmosomes. In addition, mutations in TMEM43 and TGFB1 have been associated with ARVC. Mechanistic studies indicate that suppressed canonical Wnt signaling, imposed by nuclear PG, is the responsible mechanism for the pathogenesis of ARVC. It leads to the differentiation of a subset of second heart field cardiac progenitor cells at the epicardium to adipocytes due to enhanced expression of adipogenic factors. This mechanism explains the predominant involvement of the right ventricle in ARVC. Hence, ARVC is the first identified disease of disrupted differentiation of cardiac progenitor cells.
Summary
Advances in molecular genetics and the pathogenesis of ARVC could afford the opportunity for a genetic-based diagnosis and development of novel diagnostic markers and therapeutic targets aimed to prevent, attenuate and reverse the evolving phenotype.
Keywords: Cardiomyopathy, Genetics, Sudden death, Heart failure, Stem cells
Introduction
Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy (ARVD/ARVC) was first described in 1978 in patients who exhibited ventricular arrhythmias with a left bundle branch morphology and had fibro-fatty replacement of the right ventricle[1]. Today, ARVC is defined as an inherited primary disease of the myocardium that is characterized by fibro-adipocytic replacement of myocytes, predominantly in the right ventricle. Clinically, it is characterized by ventricular arrhythmias, principally originating from the right ventricle, sudden cardiac death (SCD), right ventricular dilatation and aneurysm and heart failure. The pathological hallmark of ARVC is the gradual replacement of cardiac myocytes by adipocytes and fibrosis (Figure 1). The left ventricle is commonly spared in early stages of the disease but is often involved in advanced stages or is the predominant chamber involved [2, 3].
Figure 1.
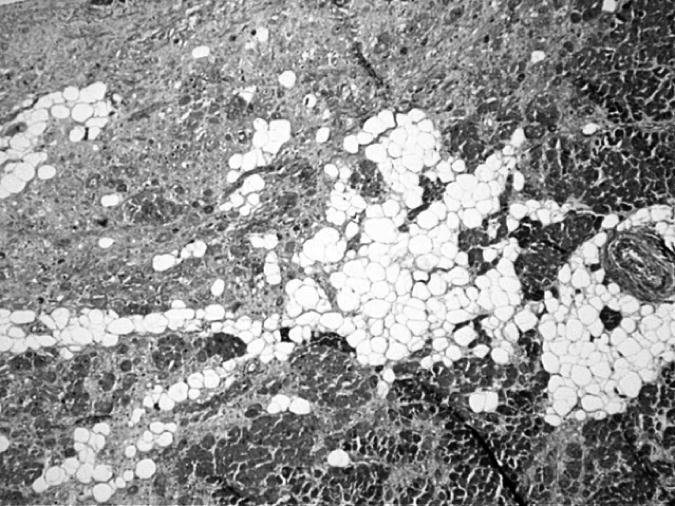
Myocardial section stained with Masson trichrome showing replacement of cardiac myocytes by adipocytes and fibrosis in a patient with ARVC
During the past few years, the molecular genetic basis has been partially elucidated. In addition, mechanistic studies have provided significant insight into the pathogenesis of this disease. Collectively, these advances have set the stage for genetic-based diagnosis and intervention to prevent the evolving phenotype and reverse the establish phenotype.
Clinical Features
ARVC is a rare and probably under-diagnosed disease. Estimation of the true prevalence of ARVC is confounded by the difficulty in its firm diagnosis because of variable and often subtle phenotypic expression. This is particularly problematic in the early stages of the disease, which typically manifest as minor ventricular arrhythmias and subtle pathological findings. Hence, it may not raise clinical suspicion. Approximately 10% of family members of probands have subtle clinical findings, which are suggestive of ARVC but per conventional criteria will not be diagnosed [4]. The disease gradually progresses to symptomatic ventricular arrhythmias, right ventricular enlargement, global or segmental hypokinesia, wall thinning and aneurismal dilatation. In advanced stages, often the left ventricle is involved and the disease many present with global cardiac failure, which may be difficult to distinguish from garden-variety forms of heart failure.
Clinical manifestations of ARVC are variable. The most common symptoms are palpitations and syncope, due to occurrences of ventricular tachycardia. SCD unfortunately may be the first manifestation of the disease, when ventricular tachycardia degenerates into ventricular fibrillation [5]. Exercise may precipitate electrical instability, presumably because of myocardial stretch following ventricular overload and sympathetic imbalance during effort. ARVC is a relatively common cause of unexpected SCD in the young, particularly in athletes [5-8]. In the US population, it accounts for 3-4% of all cases of SCD associated with physical activity in young athletes [9]. A higher prevalence has been found in autopsy studies of non-traumatic SCD [8]. In the Veneto region of Italy, ARVC was found in 6/22 (27%) of the cases of SCD in young athletes [6]. Collectively, the data suggests ARVC is an important cause of SCD in young competitive athletes.
As the name denotes, ARVC is primarily but not exclusively a disease of the right ventricle. Fibrofatty replacement of cardiac myocytes usually starts from the sub-epicardium of the right ventricle and progresses toward the endocardium. Right ventricular inflow tract, the apex and the infundibulum are the most frequently involved sites. This area is referred to as “the triangle of dysplasia”. The left ventricle is also involved in advanced stages of the disease and is occasionally the predominant site of involvement [3, 10]. The basis for predilection of the right ventricle was recently elucidated through a series of genetic-fate mapping experiments, which are discussed later [11**].
The clinical diagnosis of ARVC requires a high index of suspicion, particularly in young and middle age individuals who present with ventricular arrhythmias and/or syncope. Diagnosis in early stages is particularly difficult because of subtle nature of the findings and their non-specificity. In addition, many diagnostic tools used for the diagnosis of ARVC have limited clinical utility because of poor sensitivity and inadequate specificity. Abnormalities in standard electrocardiograms (ECGs) are frequent in patients with ARVC and correlate with the extent of the disease. A stereotypical electrocardiographic pattern does not exist. Patients with mild forms of ARVC can present with a normal ECG, therefore the presence of a normal 12-lead ECG should not preclude the diagnosis of ARVC. Conversely, patients with severe forms typically show a pathologic ECG [12]. The most common ECG abnormalities are non-specific depolarization and repolarization abnormalities commonly in the right precordial leads. The characteristic and yet uncommonelectrocardiographic feature of ARVC is the epsilon wave, which is a terminal deflection at the end of QRS. It is detected in the right precordial leads in approximately 1/3 of patients. The finding of an epsilon wave on ECG has low sensitivity but relatively high specificity for the diagnosis of ARVC. Complete or incomplete right bundle branch block is observed in about 1/3 of patients. The presence of ventricular arrhythmias with left bundle branch morphology further strengthens the diagnosis [13].
Echocardiography and magnetic resonance imaging (MRI) may show dilation and thinning of the right ventricle, localized right ventricular aneurysms, regional right ventricular hypokinesia, and fibrofatty infiltration predominantly of the right ventricle [14]. MRI has superior diagnostic utility because it allows visualization of the right ventricle in its three dimensions without restriction by acoustic windows. In addition, it has the potential to demonstrate fatty infiltration of the right ventricle [14]. However, MRI has a relatively low sensitivity in the early stages of the disease, wherein fatty infiltration may be patchy and localized to a small area and where the right ventricle is normal [15]. It is best to combine information from multiple tests to achieve the best diagnostic yield in patients in early stages of ARVC [16].
The most reliable diagnostic modality is light microscopic examination of myocardial samples, which show patchy or diffuse areas of fibrosis and adipocytic replacement of myocardium in conjunction with degenerating myocytes (apoptosis). Histological diagnosis in suspected patients requires endomyocardial biopsy, which is a relatively safe but invasive procedure. The presence of adipocytes in the myocardium alone is not sufficient to establish the diagnosis, as adipocytes are often detected in Cor adiopsum (fatty heart) and many other pathological states. An important point in the diagnosis of ARVC is the presence of a family history of ARVC, which considerably enhances the positive predictive value of any finding. Decreased plakoglobin levels at the myocardial cell–cell junctions in myocardial samples has been implicated as a marker for ARVC [17*]. Likewise, prolonged right ventricular endocardial activation time has been suggested as a marker for ARVC[18].
ARVC phenocopy
The presence of fat deposits in the myocardium can be detected in normal individuals, particularly in the elderly and is referred to as Cor adiposum [19]. It is a distinct phenotype from true ARVC as myocardial thickness is normal or increased and inflammation and myocardial atrophy are absent. Right ventricular dilatation, fibrosis, myocyte atrophy and excess adipocytes could be observed in patients with muscular dystrophies [20]. The clinical distinction, however, is seldom problematic because of involvement of skeletal muscles in muscular dystrophies.
Catecholaminergic (stress-induced) polymorphic ventricular tachycardia (CPVT) often presents with arrhythmias resembling those in ARVC. However, there are no structural or histological cardiac abnormalities in CPVT to suggest ARVC. Hence, it is considered a phenocopy and not true ARVC. Recent genetic studies have identified mutations in cardiac ryanodine receptor (RyR2) as causes of CPVT [21, 22].
Molecular genetics
ARVC is a genetic disease and familial in approximately 30 to 50% of cases. The most common mode of inheritance is autosomal dominant. Recessive forms in conjunction with palmoplantar keratoderma and woolly hair (Naxos disease) or with predominant involvement of the left ventricle (Carvajal syndrome) have also been described and referred to as “cardio-cutaneous syndromes” [23]. The genetic basis of ARVC is partially known (table 1). Several chromosomal loci have been mapped and five causal genes namely, DSP, JUP, PKP2, DSG2 and DSC2, encoding desmosomal proteins: desmoplakin (DP), plakoglobin (PG), plakophilin 2 (PKP2), desmoglein 2 (DSG2) and desmocollin 2 (DSC2), respectively, have been identified [23-31]. Collectively, the known causal genes account for approximately half of all ARVC cases. The majority of these mutations are insertion/deletion or nonsense mutations, which are expected to cause premature termination of the encoded proteins. Among the known genes, mutations in PKP2 appear to be the most common causes of ARVC, accounting for approximately 20% of cases [24, 29]. Mutations in DSG2 and DSP each account for approximately 10-15% of cases [29]. The finding of disease causing mutations in the desmosomal proteins suggests that ARVC, at least in a subset, is a disease of desmosomes.
Three genes encoding for proteins other than desmosomal proteins have also been implicated in ARVC. A point mutation in the 5′ untranslated region (UTR) of the TGFb3 gene was identified in a family mapped to 14q24.3 [32]. Whether this point mutation is a true causal mutation or simply a DNA marker remains to be established. Mutations in the RYR2 gene causes CPVT or stress-induced polymorphic ventricular tachycardia, which is a distinct phenotype from ARVC [21,22]. A missense mutation in the TMEM43 gene has been identified in the affected members of families with ARVC [33]. The gene codes for the transmembrane protein [43]. The function of this protein is largely unknown. The gene contains a response element for PPARγ, an adipogenic transcription factor, which may provide a mechanism for adipogenesis in ARVC [33].
Pathogenesis
The pathogenesis of ARVC, since its initial description, has been an enigma. Identification of mutations in desmosomal proteins provided the first set of robust clues to the pathogenesis of ARVC. Desmosomes are complex intercellular junctions responsible for cell-cell adhesion that also function as signaling molecules. Desmosomes are particularly abundant in epidermal cells and in cardiac myocytes. Three groups of proteins constitute the desmosomes: the cadherin family, the Armadillo family and the plakin family. The cadherin family is comprised of 3 desmocollins and 3 desmogleins. They are primarily responsible for anchoring the structure to the membrane. The Armadillo family is comprised of PG (also known as γ-catenin) and 3 plakophilins, which are named because they contain Armadillo repeat domains. They form the core structure and possess signaling capabilities. The plakin family is comprised of DSP, envoplakin, periplakin, plectin and pinin. The plakin family is responsible for the attachment of the desmosomes to intermediary filaments (Figure 2).
Figure 2.
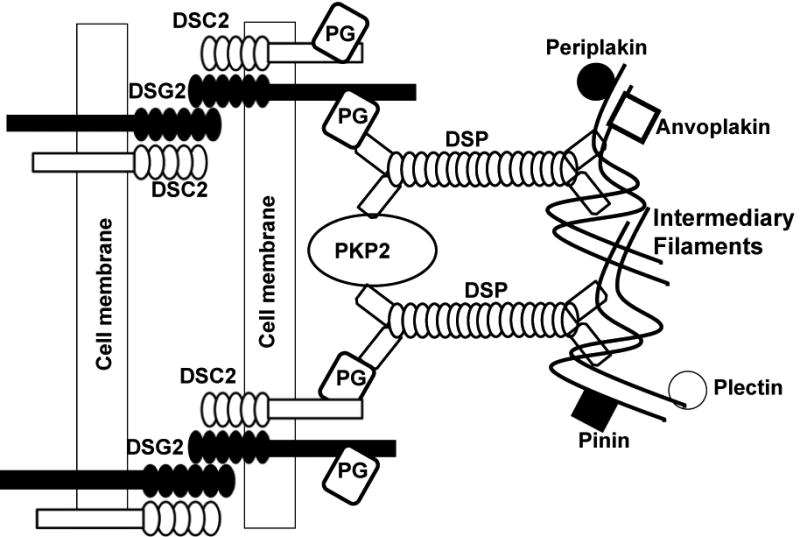
Schematic structure of a desmosome and its protein constituents.
We have focused on the signaling functions of the desmosomal proteins to elucidate the pathogenesis of ARVC. Specifically, we have focused on the transcriptional activity of Armadillo repeat family members of desmosomal protein PG. We have shown that suppression of expression of desomosomal proteins lead to partial translocation of PG into the nucleus [34]. PG and β-catenin are two closely related Armadillo repeat proteins with high degrees of sequence identity and homology [35]. Functional and phenotypic consequences of interactions between PG and β-catenin are complex and in general, antagonistic [36]. PG and β-catenin compete for nuclear localization; binding to Tcf/Lef transcription factors; degradation by ubiquitination; binding to axin and adenomatous polyposis coli; incorporation into the adherens junctions; and desmosomal assembly. Of particular interest to the pathogenesis of ARVC are the bindings of PG and β-catenin to Tcf7l2 (a.k.a. Tcf-4) transcription factor, which impart contrasting effects. PG-bound Tcf/Lef binds to DNA weaker than β-catenin-bound Tcf/Lef [37]. Thus, in the presence of nuclear PG, binding of β-catenin to Tcf7l2 is reduced [11**]. The ensuing interactions between PG and β-catenin for binding to Tcf7l2 result in suppression of canonical Wnt signaling [34]. Wnt/β-catenin signaling is an important switch regulator of myogenesis versus adipogenesis and a differentiation of cardiac progenitor cells [38-40]. Nuclear localization of PG is associated with increased expression of the non-canonical Wnt molecule Wnt5b and Bone Morphogenic protein 7 (BMP7); which are the major mediators of adipogenesis. Thus, the main tenet of our hypothesis, which is supported by strong experimental data, is based on the interaction of the nuclear PG with β-catenin and the ensuing suppression of canonical Wnt signaling (Figure 3) [11**, 34].
Figure 3.
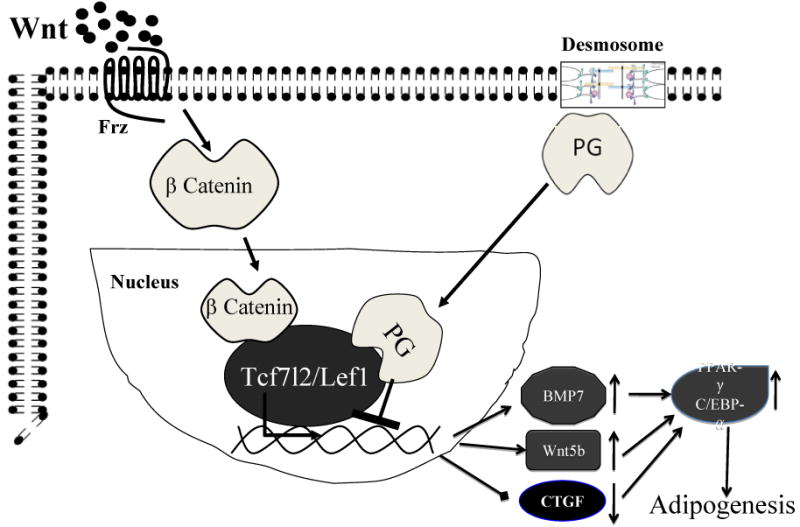
Pathogenesis of ARVC. Mutations in desmosomal proteins interfere with efficient and proper assembly of desmosomes, which allows the plakoglobin (PG) to translocate from desmosomes to the nucleus. In the nucleus, PG interferes with proper assembly of the Wnt canonical signaling protein complex and suppresses gene expression through the β-catenin/Lef7l2 transcriptional assembly.
The net effect is removal of the inhibitory effects of the canonical Wnt signaling and hence increased expression of bone morphogenic protein 7 (BMP7) and noncanonical Wnt5b, which are known promoters of adipogenesis. Likewise, suppression of the canonical Wnt signaling reduces expression of connective tissue growth factor (CTGF), which is a known inhibitor of adipogenesis. The transcriptional switch from myogenesis to adipogenesis promotes the differentiation of a subset of second heart field progenitor cells to adipocytes.
The cellular origin of adipocytes in ARVC has been an enigma. To identify the cellular origin of adipocytes we performed a series of genetic fate mapping (a.k.a. lineage tracing) experiments [11**]. We conditionally suppressed expression of DP in cardiac myocyte lineages while simultaneously labeling the cells with a reporter protein. These studies were based on our hypothesis that in desmosomal ARVC the cell type that gives rise to adipocytes must either express the mutant desmosomal protein or differentiate into adipocytes because of the expression of paracrine factors by the cells that express the mutant desmosomal protein. In the heart, the only cell type known to express desmosomal proteins is cardiac myocyte lineage. Adult cardiac myocytes are terminally differentiated and hence, not plausible candidates to dedifferentiate to adipocytes. However, the heart has an enormous capacity for regeneration, which is endowed by resident, and possibly circulating, progenitor cells. The results of lineage tracing experiments showed that adipocytes in ARVC originate from the second heart field progenitor cells that preferentially differentiate into adipocytes because of suppressed canonical Wnt signaling [11**] (Figure 3). These findings also explain the predominant involvement of the right ventricle in ARVC, which has also been an enigma. On this aspect, we note that the second heart field progenitors give rise to the right ventricle and its outflow tracts through mechanisms governed by canonical Wnt signaling [41]. Thus a subset of the second heart field cells differentiate into adipocytes in the presence ofsuppressed canonical Wnt signaling exerted by the nuclear PG [11**].
Treatment
Currently, there is no specific medical treatment for patients with ARVC. Asymptomatic patients are followed periodically and assessed for the risk of SCD, which unfortunately could be the first manifestation of the disease. The risk factors for SCD are not yet well defined. In general, a family history of SCD, history of aborted SCD, recurrent syncope, spontaneous sustained ventricular tachycardia particularly when associated with hypotension, and drug-refractory ventricular arrhythmias are considered major risk factors. In high-risk patients – individuals with 2 or more major risk factors implantation of an Automatic Internal Cardioverter/Defibrillator (AICD) is the best safeguard against SCD.
Pharmacological therapy in symptomatic patients is largely empiric and aimed at symptomatic relief. Anti-arrhythmic drugs, such as amiodarone, β-blockers and sotalol have been used to treat ventricular arrhythmias. However, none reliably prevents SCD in ARVC patients. Amiodarone may be more effective than sotalol and beta-blockers in preventing ventricular arrhythmias [42]. Catheter ablative therapy following electro-anatomical mapping could effectively abolish ventricular tachycardia in patients with ARVC. Catheter ablation is expected to be more effective in the early stages of the disease, where the pathological process is somewhat restricted to certain anatomical regions. Given the overall effectiveness of AICD in abrogating SCD, most symptomatic patients with ventricular arrhythmias should undergo AICD implantation. Likewise, AICD is probably the best choice in patients presenting with syncope, aborted SCD or found to have inducible ventricular tachycardia on electrophysiological studies. Overall, AICD implantation has been shown to be effective in improving the long-term prognosis in patients with ARVC [43, 44]. Patients with heart failure are treated with β-blockers, angiotensin-1 converting enzyme inhibitors and diuretics.
Conclusions
During the past few years there have been considerable advances in our understanding of the molecular genetics and pathogenesis of ARVC. Despite such advances, the causal gene remains unknown in approximately half of all cases. It is also unsettled whether ARVC is exclusive to mutant desmosomal proteins or could results from mutations in non-desmosomal proteins. Suppression of the canonical Wnt signaling in second heart field progenitor cells has emerged as the major responsible mechanism for the pathogenesis of ARVC. Whether or not the suppression of canonical Wnt signaling by nuclear PG is common to all forms of ARVC remains to be established. Recent discoveries have provided an excellent opportunity to further delineate the molecular genetics and pathogenesis of the ARVC phenotype, develop novel diagnostic markers and identify novel therapeutic targets for this uncommon and yet potentially deadly disease.
Acknowledgments
Supported in part by grants from the National Heart, Lung, and Blood Institute, Burroughs Wellcome Award In Translational Research, and a TexGen grant from Greater Houston Community Foundation.
Footnotes
There is no conflict of interest.
References
- 1.Frank R, Fontaine G, Vedel J, et al. Electrocardiology of 4 cases of right ventricular dysplasia inducing arrhythmia. Archives Des Maladies Du Coeur Et Des Vaisseaux. 1978;71:963–972. [PubMed] [Google Scholar]
- 2.Corrado D, Fontaine G, Marcus FI, et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: need for an international registry. Study Group on Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy of the Working Groups on Myocardial and Pericardial Disease and Arrhythmias of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the World Heart Federation. Circulation. 2000;101:E101–E106. doi: 10.1161/01.cir.101.11.e101. [DOI] [PubMed] [Google Scholar]
- 3.Norman M, Simpson M, Mogensen J, et al. Novel Mutation in Desmoplakin Causes Arrhythmogenic Left Ventricular Cardiomyopathy. Circulation. 2005;112:636–642. doi: 10.1161/CIRCULATIONAHA.104.532234. [DOI] [PubMed] [Google Scholar]
- 4.Hamid MS, Norman M, Quraishi A, et al. Prospective evaluation of relatives for familial arrhythmogenic right ventricular cardiomyopathy/dysplasia reveals a need to broaden diagnostic criteria. J Am Coll Cardiol. 2002;40:1445–1450. doi: 10.1016/s0735-1097(02)02307-0. [DOI] [PubMed] [Google Scholar]
- 5.Thiene G, Nava A, Corrado D, et al. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med. 1988;318:129–133. doi: 10.1056/NEJM198801213180301. [DOI] [PubMed] [Google Scholar]
- 6.Corrado D, Thiene G, Nava A, et al. Sudden death in young competitive athletes: clinicopathologic correlations in 22 cases. Am J Med. 1990;89:588–596. doi: 10.1016/0002-9343(90)90176-e. [DOI] [PubMed] [Google Scholar]
- 7.Tabib A, Loire R, Chalabreysse L, et al. Circumstances of death and gross and microscopic observations in a series of 200 cases of sudden death associated with arrhythmogenic right ventricular cardiomyopathy and/or dysplasia. Circulation. 2003;108:3000–3005. doi: 10.1161/01.CIR.0000108396.65446.21. [DOI] [PubMed] [Google Scholar]
- 8.Shen WK, Edwards WD, Hammill SC, et al. Sudden unexpected nontraumatic death in 54 young adults: a 30-year population-based study. Am J Cardiol. 1995;76:148–152. doi: 10.1016/s0002-9149(99)80047-2. [DOI] [PubMed] [Google Scholar]
- 9.Maron BJ, Shirani J, Poliac LC, et al. Sudden death in young competitive athletes. Clinical, demographic, and pathological profiles. JAMA: The Journal of the American Medical Association. 1996;276:199–204. [PubMed] [Google Scholar]
- 10.Sen-Chowdhry S, Syrris P, Ward D, et al. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation. 2007;115:1710–20. doi: 10.1161/CIRCULATIONAHA.106.660241. [DOI] [PubMed] [Google Scholar]
- 11**.Lombardi R, Dong J, Rodriguez G, et al. Genetic fate mapping identifies second heart field progenitor cells as a source of adipocytes in arrhythmogenic right ventricular cardiomyopathy. Circ Res. 2009;104:1076–84. doi: 10.1161/CIRCRESAHA.109.196899. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** The findings delineate the cellular origin of adiocytes in arrhythmogenic right ventricular cardiomyopathy (ARVC), which has been an enigma since the initial description of the phenotype more than 2 decades ago. The authors show that nuclear localization of plakoglobin, a desmosomal proteins, suppresses the canonical Wnt signaling and lead to differntiation of a subset of second heart field progenitor cells to adipocytes. The investigators further collaborate their findings in human heart samples from patients with ARVC.
- 12.Steriotis AK, Bauce B, Daliento L, et al. Electrocardiographic pattern in arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol. 2009;103:1302–8. doi: 10.1016/j.amjcard.2009.01.017. [DOI] [PubMed] [Google Scholar]
- 13.Piccini JP, Nasir K, Bomma C, et al. Electrocardiographic findings over time in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Am J Cardiol. 2005;96:122–6. doi: 10.1016/j.amjcard.2005.02.057. [DOI] [PubMed] [Google Scholar]
- 14.Auffermann W, Wichter T, Breithardt G, et al. Arrhythmogenic right ventricular disease: MR imaging vs angiography. AJR Am J Roentgenol. 1993;161:549–55. doi: 10.2214/ajr.161.3.8352102. [DOI] [PubMed] [Google Scholar]
- 15.Tandri H, Castillo E, Ferrari VA, et al. Magnetic resonance imaging of arrhythmogenic right ventricular dysplasia: sensitivity, specificity, and observer variability of fat detection versus functional analysis of the right ventricle. J Am Coll Cardiol. 2006;48:2277–84. doi: 10.1016/j.jacc.2006.07.051. [DOI] [PubMed] [Google Scholar]
- 16.Marcus FI, Zareba W, Calkins H, et al. Arrhythmogenic right ventricular cardiomyopathy/dysplasia clinical presentation and diagnostic evaluation: results from the North American Multidisciplinary Study. Heart Rhythm. 2009;6:984–92. doi: 10.1016/j.hrthm.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17*.Asimaki A, Tandri H, Huang H, et al. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2009;360:1075–84. doi: 10.1056/NEJMoa0808138. [DOI] [PubMed] [Google Scholar]; * The authors describe reduced plakoglobin at cardiac myocytes junctions, detected by immunoflourescence staining, as a diagnostic marker for arrythmogenic right ventricular cardiomyopathy.
- 18.Tandri H, Asimaki A, Abraham T, et al. Prolonged RV endocardial activation duration: a novel marker of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Heart Rhythm. 2009;6:769–75. doi: 10.1016/j.hrthm.2009.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burke AP, Farb A, Tashko G, Virmani R. Arrhythmogenic Right Ventricular Cardiomyopathy and Fatty Replacement of the Right Ventricular Myocardium : Are They Different Diseases? Circulation. 1998;97:1571–1580. doi: 10.1161/01.cir.97.16.1571. [DOI] [PubMed] [Google Scholar]
- 20.Finsterer J, Stollberger C. The Heart in Human Dystrophinopathies. Cardiology. 2003;99:1–19. doi: 10.1159/000068446. [DOI] [PubMed] [Google Scholar]
- 21.Tiso N, Stephan DA, Nava A, et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2) Human Molecular Genetics. 2001;10:189–194. doi: 10.1093/hmg/10.3.189. [DOI] [PubMed] [Google Scholar]
- 22.Priori SG, Napolitano C, Tiso N, et al. Mutations in the Cardiac Ryanodine Receptor Gene (hRyR2) Underlie Catecholaminergic Polymorphic Ventricular Tachycardia. Circulation. 2001;103:196–200. doi: 10.1161/01.cir.103.2.196. [DOI] [PubMed] [Google Scholar]
- 23.McKoy G, Protonotarios N, Crosby A, et al. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease) Lancet. 2000;355:2119–2124. doi: 10.1016/S0140-6736(00)02379-5. [DOI] [PubMed] [Google Scholar]
- 24.Gerull B, Heuser A, Wichter T, et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet. 2004;36:1162–1164. doi: 10.1038/ng1461. [DOI] [PubMed] [Google Scholar]
- 25.Heuser A, Plovie ER, Ellinor PT, et al. Mutant desmocollin-2 causes arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 2006;79:1081–1088. doi: 10.1086/509044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Norgett EE, Hatsell SJ, Carvajal-Huerta L, et al. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Human Molecular Genetics. 2000;9:2761–2766. doi: 10.1093/hmg/9.18.2761. [DOI] [PubMed] [Google Scholar]
- 27.Alcalai R, Metzger S, Rosenheck S, et al. A recessive mutation in desmoplakin causes arrhythmogenic right ventricular dysplasia, skin disorder, and woolly hair. J Am Coll Cardiol. 2003;42:319–327. doi: 10.1016/s0735-1097(03)00628-4. [DOI] [PubMed] [Google Scholar]
- 28.van Tintelen JP, Entius MM, Bhuiyan ZA, et al. Plakophilin-2 mutations are the major determinant of familial arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation. 2006;113:1650–1658. doi: 10.1161/CIRCULATIONAHA.105.609719. [DOI] [PubMed] [Google Scholar]
- 29.Pilichou K, Nava A, Basso C, et al. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation. 2006;113:1171–1179. doi: 10.1161/CIRCULATIONAHA.105.583674. [DOI] [PubMed] [Google Scholar]
- 30.Syrris P, Ward D, Evans A, et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in the desmosomal gene desmocollin-2. Am J Hum Genet. 2006;79:978–984. doi: 10.1086/509122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beffagna G, De Bortoli M, Nava A, et al. Missense mutations in desmocollin-2 N-terminus, associated with arrhythmogenic right ventricular cardiomyopathy, affect intracellular localization of desmocollin-2 in vitro. BMC Med Genet. 2007;8:65. doi: 10.1186/1471-2350-8-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beffagna G, Occhi G, Nava A, et al. Regulatory mutations in transforming growth factorbeta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc Res. 2005;65:366–373. doi: 10.1016/j.cardiores.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 33.Merner ND, Hodgkinson KA, Haywood AFM, et al. Arrhythmogenic Right Ventricular Cardiomyopathy Type 5 Is a Fully Penetrant, Lethal Arrhythmic Disorder Caused by a Missense Mutation in the TMEM43 Gene. The American Journal of Human Genetics. 2008;82:809–821. doi: 10.1016/j.ajhg.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garcia-Gras E, Lombardi R, Giocondo MJ, et al. Suppression of canonical Wnt/betacatenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic rightventricular cardiomyopathy. J Clin Invest. 2006;116:2012–21. doi: 10.1172/JCI27751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maeda O, Usami N, Kondo M, et al. Plakoglobin (gamma-catenin) has TCF/LEF family dependent transcriptional activity in beta-catenin-deficient cell line. Oncogene. 2004;23:964–972. doi: 10.1038/sj.onc.1207254. [DOI] [PubMed] [Google Scholar]
- 36.Klymkowsky MW, Williams BO, Barish GD, et al. Membrane-anchored plakoglobins have multiple mechanisms of action in Wnt signaling. Mol Biol Cell. 1999;10:3151–3169. doi: 10.1091/mbc.10.10.3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhurinsky J, Shtutman M, Ben Ze'ev A. Differential mechanisms of LEF/TCF familydependent transcriptional activation by beta-catenin and plakoglobin. Mol Cell Biol. 2000;20:4238–4252. doi: 10.1128/mcb.20.12.4238-4252.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ross SE, Hemati N, Longo KA, et al. Inhibition of adipogenesis by Wnt signaling. Science. 2000;289:950–953. doi: 10.1126/science.289.5481.950. [DOI] [PubMed] [Google Scholar]
- 39.Polesskaya A, Seale P, Rudnicki MA. Wnt Signaling Induces the Myogenic Specification of Resident CD45+ Adult Stem Cells during Muscle Regeneration. Cell. 2003;113:841–852. doi: 10.1016/s0092-8674(03)00437-9. [DOI] [PubMed] [Google Scholar]
- 40.Chen AE, Ginty DD, Fan CM. Protein kinase A signalling via CREB controls myogenesis induced by Wnt proteins. Nature. 2005;433:317–322. doi: 10.1038/nature03126. [DOI] [PubMed] [Google Scholar]
- 41.Ai D, Fu X, Wang J, et al. Canonical Wnt signaling functions in second heart field to promote right ventricular growth. Proc Natl Acad Sci USA. 2007;104:9319–9324. doi: 10.1073/pnas.0701212104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marcus GM, Glidden DV, Polonsky B, et al. Efficacy of antiarrhythmic drugs in arrhythmogenic right ventricular cardiomyopathy: a report from the North American ARVC Registry. J Am Coll Cardiol. 2009;54:609–15. doi: 10.1016/j.jacc.2009.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wichter T, Borggrefe M, Haverkamp W, et al. Efficacy of antiarrhythmic drugs in patients with arrhythmogenic right ventricular disease. Results in patients with inducible and noninducible ventricular tachycardia. Circulation. 1992;86:29–37. doi: 10.1161/01.cir.86.1.29. [DOI] [PubMed] [Google Scholar]
- 44.Corrado D, Leoni L, Link MS, et al. Implantable cardioverter-defibrillator therapy for prevention of sudden death in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation. 2003;108:3084–3091. doi: 10.1161/01.CIR.0000103130.33451.D2. [DOI] [PubMed] [Google Scholar]