

Summary
Chromatin structure affects the accessibility of DNA to transcription, repair and replication. Changes in chromatin structure occur during development, but less is known about changes during aging. We examined the state of chromatin structure and its effect on gene expression during aging in Drosophila at the whole genome and cellular level using whole genome tiling microarrays of activation and repressive chromatin marks, whole genome transcriptional microarrays and single cell immunohistochemistry. We found dramatic reorganization of chromosomal regions with age. Mapping of H3K9me3 and HP1 signals to fly chromosomes reveals in young flies the expected high enrichment in the pericentric regions, the 4th chromosome and islands of facultative heterochromatin dispersed throughout the genome. With age there is a striking reduction in this enrichment resulting in a nearly equivalent level of H3K9me3 and HP1 in the pericentric regions, the 4th chromosome, facultative heterochromatin and euchromatin. These extensive changes in repressive chromatin marks are associated with alterations in age-related gene expression. Large-scale changes in repressive marks with age are further substantiated by single cell immunohistochemistry that show changes in nuclear distribution of H3K9me3 and HP1 marks with age. Such epigenetic changes are expected to directly or indirectly impinge upon important cellular functions such as gene expression, DNA repair and DNA replication. The combination of genome-wide approaches such as whole genome chromatin immunoprecipitation and transcriptional studies in conjunction with single cell immunohistochemistry as shown here provide a first step toward defining how changes in chromatin may contribute to the process of aging in metazoans.
Keywords: epigenetics, heterochromatin
Introduction
In eukaryotes DNA associates with histones and protein complexes to form the compact structure of chromatin. The accessibility of DNA to transcriptional machinery is determined by the compaction of the nucleosomes that is in part affected by modifications of the amino terminal tails of the histone proteins, most notably H3 and H4 (Li et al. 2007). Heterochromatic regions representing transcriptionally silenced areas are characterized as having increased levels of di- or tri-methylation of lysine 9 of the H3 amino terminal tail (H3K9me2 or 3) (Elgin & Grewal 2003; Grewal & Jia 2007). Euchromatic regions that contain genes being actively transcribed are highly acetylated and/or tri-methylated at lysine 4 of the H3 amino terminal tail (H3K4me3) (Elgin & Grewal 2003; Grewal & Jia 2007).
The chromatin state of a specific region can be assessed by measuring the type and amount of histone modifications (e.g. H3K9me3) and accessory chromatin binding proteins (e.g. HP1) associated with specific DNA fragments in that genomic region. The presence of H3K9me2 or H3K9me3 and HP1 in chromatin immunoprecipitation (ChIP) experiments is associated with silenced heterochromatin regions, while transcriptionally active euchromatin regions are associated with H3K4me3 and H3K36me3 (Sims et al. 2003; Dion et al. 2005; Nègre et al. 2006; Kouzarides 2007; Peng & Karpen 2007; Schones & Zhao 2008; Hon et al. 2009). The association between these histone modifications or histone marks and their specific chromosomal location can be determined by ChIP followed by whole genome tiling arrays (ChIP-chip) (Barski et al. 2007; Park 2008; Schones & Zhao 2008; Hon et al. 2009; Park 2009).
Little is understood about potential changes in chromatin that occur with age in organisms such as Drosophila. Assessing the state of histone and other chromatin-related marks with age associated with transcriptionally active (euchromatin) and transcriptionally repressed or silenced (heterochromatin) regions at the local and global level would be a valuable first step toward understanding the relationship between aging and changes in chromatin structure. In this report we make use of ChIP-chip to assess the relative active or repressed state of chromatin with age by concentrating on three activation marks--RNA polymerase II, H3K4me3, and H3K36me3--and two repressive marks--H3K9me3 and HP1.
Results
Activation marks associated with genes and exonic regions decrease with age
A ChIP-chip approach was used to examine the changes that take place in the chromatin of the female adult Drosophila melanogaster head as it ages: at 10d (young) or 40d (old). The genomic binding profiles of activation marks including RNA polymerase II (RNA pol II) and H3K4me3, associated with transcriptional initiation, and H3K36me3, associated with active transcriptional elongation, were examined using the 4H8 antibody which recognizes active and inactive RNA pol II and antibodies specific for the H3K4me3 and H3K36me3 marks on a high-resolution genomic tiling array covering all unique regions of the Drosophila genome.
A hidden Markov model (HMM) (Rabiner 1989) was used to examine the association between RNA pol II, H3K4me3 and H3K36me3 and focal regulatory regions such as promoters and enhancers (Fig. S1, Supplemental Methods). HMM analysis indicates that all three activation marks are almost exclusively found associated with the transcriptional start site (TSS) and 90% of the high state probesets are within 750 nucleotides of a gene (Table S1). The percent occupancy of all three activation marks at TSS and gene regions does not significantly change with age.
RNA pol II is present at the promoter of most genes with an enrichment profile of a sharp peak at the TSS, and signal across exonic regions (Fig. 1A). Only a slight reduction in overall average signal was seen at 40d as compared to background.
Fig. 1. Average signal of activating marks across length of gene decreases with age.
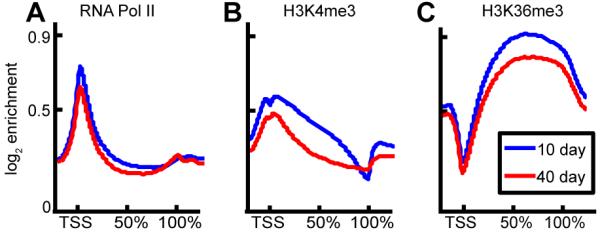
Enrichment profile is shown for (A) RNA Polymerase II, (B) H3K4me3, and (C) H3K36me3 for 10 day old (blue) and 40 day old (red) samples. Mean log2 ChIP signal is shown on a composite gene representing the exonic regions of all ~14,000 genes in the Drosophila genome scaled from transcriptional start site (TSS, 0%) to transcriptional stop (100%) with a +/− 2kbp flank.
The binding profile of H3K4me3 revealed the characteristic double peak around the TSS with a localized signal depression at the TSS (Fig. 1B) (Barski et al. 2007), and a significant signal in gene exons, with signal highest at the promoter and declining toward the 3′ end of the gene. When averaged across all genes and compared to background the 40d sample showed a lower overall signal than the 10d sample.
Enrichment of H3K36me3 levels over the spliced transcript (Barski et al. 2007; Kolasinska-Zwierz et al. 2009) rises across the gene in a 5′ to 3′ direction, peaking at 50-60% of the length of the spliced gene (Fig. 1C), and exhibiting a localized loss of signal directly at the TSS. Similar to H3K4me3, the overall signal at 40d was lower than at 10d when compared to background.
A clustering analysis was performed on the combined data from the 10d and 40d flies for each mark that divided the genes into two groups based on similarities in binding profile. The RNA pol II and H3K4me3 signal at the TSS and across all gene exons was considered separately for purposes of the clustering analysis, resulting in five different sets of clusters: RNA pol II at TSS, RNA pol II across gene, H3K4me3 at TSS, H3K4me3 across gene, and H3K36me3 across gene (Fig. S2). For each mark, genes in cluster 1 exhibited a high level of signal and genes in cluster 2 exhibited a flat, low-level binding profile (Fig. S2). Using the clusters, we scored each gene for the presence (cluster 1) or absence (cluster 2) of each of the five marks. The 40d samples consistently showed a lower level of binding across each of these five parameters, with more genes appearing in cluster 1 (high-binding) from the 10d sample, and more genes in cluster 2 (non-binding) from the 40d sample in each case (Fig. S3). ANOVA analysis supports the finding of a decrease in binding of activation marks across genes with age (Supplemental Methods). Hence, while localization of these marks does not change significantly on average between young and old age as revealed by the HMM approach, the ANOVA analysis suggests that the enrichment of these marks at those locations decreases with age.
Repressive marks change with age
Repressive chromatin modifications such as H3K9me3 and HP1 are associated with constitutive and facultative heterochromatin and tend to be enriched over broad regions rather than the discrete foci associated with activation marks. Consistent with this we found no general enrichment of H3K9me3 or HP1 above background at the TSS of either young or old flies (data not shown). Mapping of the H3K9me3 and HP1 ChIP-chip signals to the fly chromosomes, averaged over 50kb, revealed that in young flies, as expected, the enrichment of H3K9me3 and HP1 was higher, in comparison to euchromatin in the pericentric regions and the 4th chromosome (Fig. 2, S5, S6) (Yasuhara & Wakimoto 2008). However, in older flies the differential enrichment of H3K9me3 and HP1 between euchromatin and heterchromatin regions (pericentric regions, 4th chromosome and islands of facultative heterochromatin dispersed throughout the genome) is strikingly reduced leaving all chromosomal regions at almost an equivalent level of H3K9me3 and HP1 enrichment (Fig. 2, S5, S6). A direct comparison of the binding of the two marks in these regions shows that with age the H3K9me3 signal is lost to a greater extent than HP1 (Fig. 2C). Differences in binding throughout non-pericentric chromatin are also seen, as signals from H3K9me3 and HP1 do not completely overlap. H3K9me3 marks tend to cluster into “hotspots” that are fairly randomly distributed throughout the genome, while HP1 signal appears more evenly distributed (Fig. 2C).
Fig. 2. Enrichment of repressive marks are lost with age, especially in pericentric regions and 4th chromosome.

(A) Comparison of H3K9me3 marks averaged over 50 kb regions for each chromosome at 10 days (top) and 40 days (bottom). The data are expressed as a heat map with brighter colors representing greater binding of H3K9me3 in that region: the greatest signal changes occur at the pericentric heterochromatin of chr 2L, 2R, and 3L (marked with blue bounding box), and chr 4. (B) Comparison of HP1 signal at 10 days (top) and 40 days (bottom). (C) Two color heat map comparing relative signal of H3K9me3 (red) and HP1 (green). Areas of high H3K9me3 appear red, high HP1 green, high in both marks yellow, and low in both marks black. All chromosomes are drawn to relative scale except chr 4, which is enlarged.
Quantitative analysis of H3K9me3 and HP1 with age
ChIP-chip allows us to compare the relative amount of H3K9me3 and HP1 in the euchromatin region with the pericentric region and 4th chromosome, but is unable to provide information on absolute values. Thus the results showing an equilibration of the enrichment of H3K9me3 and HP1 in the pericentric and 4th chromosome with euchromatin cannot distinguish between the change being due to a decrease in pericentric and 4th chromosome repressive markers or an increase in repressive marks throughout euchromatin. Western blot analyses were performed to determine whether there is a change in the level of H3K9me3 and HP1 with age relative to total histones. As seen in Fig. 3C, with age the amount of H3K9me3 relative to H3 greatly increases, while the level of HP1 to H3 appears to stay nearly the same with age or slightly increases. We did not observe any significant difference in total histone H3 with age when normalized to total protein or DNA.
Figure 3. Nuclear distribution of H3K9me3 and HP1 changes with age.

(A) H3K9me3 immunofluorescence signal (green) in four representative fat body nuclei of young (10 days) (left) and old (40-70 days) (right) flies. DAPI stain (red) is presented for reference of nuclear boundary. Displayed scale bar is 5 microns. H3K9me3 signal changes from a single bright focus in each nucleus at 10 days, to a more diffuse staining pattern throughout the nucleus at 40-70 days (arrows). (B) HP1 immunofluorescence signal (red) at 10 days (left) and 40-70 days (right) in fat body nuclei. HP1 signal changes from diffuse in young flies to more concentrated in older flies (arrows). (C) Western blots showing relative amount of H3K9me3 and HP1 in 10 day and 40 day males and females. Total histone H3 is presented as a loading control.
Nuclear distribution of H3K9me3 and HP1 changes with age
Single cell studies have several advantages that complement the ChIP-chip whole genome approach. While ChIP-chip requires pooling and homogenization of many different cells to obtain sufficient chromatin material, single cell immunohistochemistry can be performed on tissue section allowing for resolution at the individual cellular level.
Using the same H3K9me3 and HP1 antibodies as the ChIP-chip studies, we examined the nuclear staining pattern of H3K9me3 and HP1 in fat body cells of sections of young and old flies. This confirmed that there is an age-related change in the nuclear organization of H3K9me3 and HP1 (Fig. 3). Nuclei of fat body cells from young animals (10d) show a characteristic intensely concentrated staining of H3K9me3 in one or two foci within the nucleus, near the chromocenter or nucleoli (Fig. 3A). In older animals (40-70d) the nuclei of fat body cells show a more diffuse H3K9me3 staining pattern that begins to encompass most of the nucleus (Fig. 3A). The compact heterochromatic foci of H3K9me3 at a young age become less coherent with age. HP1 staining also shows changes with age. Staining of HP1 in nuclei of fat body cells from young animals (10d) show a diffuse relatively uniform staining of HP1 throughout the nucleus (Fig. 3B) that becomes more concentrated and punctate in older animals (40-70 days) (Fig. 3B).
Association of gene expression with chromatin marks
We also examined the relationship between chromatin marks and gene expression. As previously described in both flies and humans we found a weak correlation between ChIP-chip RNA pol II binding to a gene and the level of gene expression (Kim et al. 2005; Lee et al. 2006; Muse et al. 2007). Examination of either H3K4me3 or H3K36me3 alone did not further improve the ability to predict the level of gene expression. However, the combined use of a small number of histone modifications can improve gene expression prediction (Karlic et al.). Scoring each gene based on the presence or absence of each of the 5 different activation marks, as in the clustering analysis described above, improved the correlation between activation markers and gene expression at both 10d (Fig. 4A) and 40d (Fig. S7). Genes with a higher activation mark score tend to be expressed at a higher level.
Figure 4. Correlation of active and repressive marks with gene expression reveals loss of repressive marks with age is associated with increased gene expression.
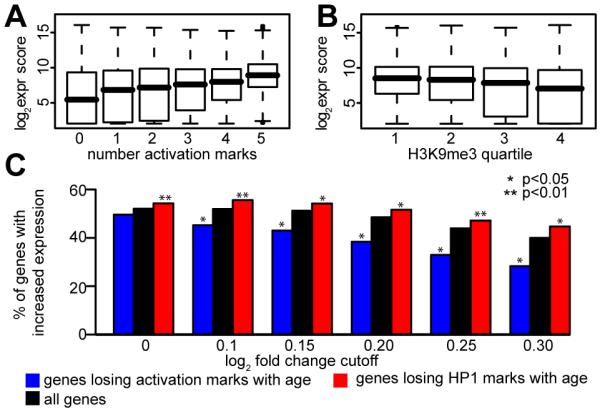
(A) Correlation of active marks with gene expression at 10 days. Genes were assigned an activation mark score based on clustering analysis (see text for details). Box plot shows distribution of genes for each group plotted against GCRMA expression score. Thick black line is median, top and bottom of box are 25th and 75th percentile, dashed lines show two standard deviation coverage (~95%). (B) H3K9me3 signal is anticorrelated to gcRMA expression score at 10 days. Genes were divided into quartiles based on H3K9me3 signal and distributions plotted as in (A). (C) Correlation of changes between 10 days and 40 days with expression score. Genes were filtered based on a log2 fold change cutoff between 10 days and 40 days (x axis). Only genes changing by at least the indicated amount are present in each group. The percentage of genes in each group whose expression is increased at 40 days relative to 10 days is shown on the y axis. Three series are plotted: all genes (black), genes losing at least two activation marks with age (blue), and genes whose HP1 signal moves down at least two quartiles with age (red). The method used to test for statistical significance between percentages is described in the supplemental materials and methods.
To examine the relationship between repressive marks and gene expression, the signal from H3K9me3 or HP1 was divided into quartiles and the expression scores of the genes within each quartile was examined. An inverse correlation with expression was found, such that genes with higher levels of H3K9me3 marks tended to be expressed at a lower level, at both 10d (Fig. 4B) and 40d (Fig. S7). Interestingly, a similar correlation between HP1 signal and gene expression was not seen (Fig. S7).
Loss of repressive marks with age is associated with increased gene expression
We examined how gene expression changed between our 10d and 40d samples, and how well this correlated with the changes we observed with chromatin marks. Examination of changes in chromatin mark binding between young and old samples and linkage with changes in gene expression showed that most genes tended to keep the same activation and repressive mark scores as they aged. Of the many genes that had changes in chromatin marks between 10d and 40d, the most common change was a loss of marks with age (3.5% of genes lose at least two activation marks, 0.5% gain at least two).
We next examined whether the trend of decreasing marks with age was correlated with any changes in gene expression. In order to investigate this effect, we examined the fold-change of the genes that lost activation marks or one of the repressive marks with age in comparison to the entire population of genes that changed between 10d and 40d. Among all the genes whose expression changed between 10d and 40d, we computed the fraction Fall of genes whose expression is larger at 40d than at 10d for different fold-change cutoffs (Fig. 4C). For example, at a log2-fold-change cutoff of 0 (i.e. all genes selected), 52% of genes go up at 40d, giving Fall(0) = 52%, but of the genes with a log2-fold-change of at least 0.3 in either direction, only 40% go up at 40 days. Investigation of how losing at least two activation marks affected gene expression fold-change was done by computing the same fraction, Factivation, for the subset of genes that lost activation marks (Fig. 4C) and testing the significant difference between Fall and Factivation (see Supplementary Methods). For all log2-fold-changes above 0 there was a significant difference between the two fractions, with Factivation being consistently lower than Fall. This indicates that genes that lose activation marks have a greater tendency to lose expression than the entire population of genes that change.
Loss of repressive marks was defined as a change in enrichment for that mark by at least two quartiles (e.g. from the 4th quartile at 10d to either the 1st or 2nd quartile at 40d, or from the 3rd quartile at 10d to the 1st quartile at 40d). A similar analysis to that described above was performed on the subset of genes that lost HP1 with age (Fig. 4C). This fraction Frepressive was significantly greater than Fall at all fold changes, indicating that genes that lose repressive marks with age tend to have increased expression compared to the entire population of genes. The same trend was observed for H3K9me3, when defining the loss as a change in enrichment for that mark by at least three quartiles, although no cutoff reached statistical significance due to the very small number of genes in this category (Fig. S8).
Discussion
The silencing of specific chromosomal regions is dynamically changing during development (Ebert et al. 2004; Ebert et al. 2006; Talbert & Henikoff 2006), but less is known about changes in chromatin state during aging. Since changes in chromatin state can alter gene transcription, resulting in expression of genes otherwise silenced (Elgin & Grewal 2003; Berger 2007; Grewal & Jia 2007; Sedivy et al. 2008; Dang et al. 2009) or repression of genes otherwise expressed, it has the potential to significantly alter cellular physiology. During aging in yeast, loss of silencing at the mating locus leads to re-expression of the silenced mating type gene, causing sterility, and loss of silencing near telomeres results in expression of previously silenced genes in the subtelomeric region (Kim et al. 1996; Smeal et al. 1996; Berger 2007; Dang et al. 2009). These and other examples have suggested that changes in chromatin with age are related to, and may be responsible for, some elements of the aging process.
We used Drosophila to examine the changes in chromatin state globally and locally with age. An HMM analysis confirmed the expectation that the activation marks, including RNA pol II, H3K4me3 (associated with transcript initiation) and H3k36me3 (associated with transcript extension), are almost exclusively associated with the transcriptional start site (TSS) or gene regions. Consistent with the nearly ubiquitous nature of RNA pol II binding to TSS in other organisms (Barski et al. 2009), only a small but statistically significant decrease was seen in the enrichment of RNA pol II at the TSS and over genes in older flies. A larger statistically significant decrease in the average enrichment of histone modifications associated with active transcription, H3K4me3 and H3K36me3 at TSS and over genes, was seen in older flies (Fig. 1).
Comparisons between ChIP-chip for total RNA pol II (active and inactive) and whole genome RNA transcriptional microarrays shows the same weak correlation between RNA pol II binding and gene expression that has been reported by others (Kim et al. 2005; Lee et al. 2006; Muse et al. 2007). The addition of ChIP-chip information from two other histone activation marks (H3K4me3 and H3K36me3) improves the correlation between activation marks and gene expression, although the improved correlation remains weak and is insufficient to confidently make definitive predictions. The poor correlation seen between activation marks and gene expression is probably not due to the inherent “noise” in the experimental procedures of using whole genome microarrays. Similar difficulties of correlating activation marks with gene expression continue to be seen with deep-sequencing methodologies that should significantly improve the signal-to-noise ratio (Barski et al. 2009).
Unlike activation marks, repressive marks such as H3K9me3 and HP1 are not found closely associated with the TSS of individual genes. In general, repressive marks were seen enriched in broader regions than activation marks and in areas devoid of known transcriptional units. It is thought that HP1 is recruited to a chromosomal region in response to addition of the H3K9me3 mark, resulting in an increase in chromatin packing and gene silencing. This is consistent with our finding of a stronger association between enrichment of HP1 and decreased gene expression, than for H3K9me3 (Fig. 4C, S8).
With age we found that the relative enrichment of both H3K9me3 and HP1 is greatly diminished in the pericentric heterochomatin regions, the heterochromatic 4th chromosome and in islands of facultative heterochromatin in comparison to euchromatin (Fig. 2, S5, S6). For H3K9me3, more than for HP1, the large difference in enrichment in these heterochromatin regions in young flies is reduced to being virtually equivalent to the euchromatin regions in older flies (Fig. 2, S5, S6). However, since ChIP-chip is not able to measure absolute values, and because of the need of inter-array normalization, these data alone are not able to definitively state whether H3K9me3 or HP1 is lost from these “heterochromatin” regions with age. The observed change with age could be due to a decrease in H3K9me3 and HP1 in the heterochromatin-like regions or an increase in H3K9me3 and HP1 ubiquitously throughout euchromatin. Either scenario would lead to the apparent equilibration of signal between the euchromatin and heterochromatin regions seen in older flies.
Western blots show a significant increase in the level of H3K9me3 and a small increase in the level of HP1 in older flies as compared with total levels of histone H3 (Fig. 3C). This absolute increase in H3K9me3 and HP1 with age suggests that either repressive marks increase throughout euchromatin or are sequestered to the ~30% of the genome of Drosophila made up of repetitive heterochromatic DNA which is not examined by ChIP-chip methodology. Single cell immunohistochemistry clearly shows evidence of H3K9me3’s altered distribution to more extensive chromosomal regions with age, although it does not distinguish between the change in distribution being targeted preferentially to euchromatin or repetitive chromosomal regions. The extent of the spread of the H3K9me3 signal throughout most of the nuclear region in older cells suggests the change in H3K9me3 is not limited to repetitive chromosomal regions and likely includes an increase of H3K9me3 in euchromatin.
The finding of extensive changes in repressive H3K9me3 and HP1 marks with age in the absence of a corresponding large-scale change in gene expression suggests that if there is an increase in repressive marks throughout euchromatin it is ineffective in modifying global gene expression. Alternatively, if the level of H3K9me3 and HP1 does not change significantly in euchromatin with age, then many of the changes seen in the level and distribution of repressive marks may reflect a redistribution or sequestration of repressive marks to the repetitive regions of the fly genome. It is possible that there may be significant changes with age in RNAs from repetitive DNA regions or transposons, which we were not able to detect using transcriptional microarrays.
Taken together these studies demonstrate that there are dramatic changes in chromatin structure with age in Drosophila. Although the specific functional consequences of these age-related changes have not been directly determined, it is likely that they alter gene expression and have important effects on cell physiology. Such changes are expected to directly or indirectly impinge upon important cell functions such as gene expression, DNA repair and DNA replication. The combination of genome-wide approaches such as whole genome ChIP and transcriptional studies in conjunction with single cell immunohistochemistry as shown here provides a first step toward defining how changes in chromatin may contribute to the process of aging in metazoans.
Experimental Procedures
The specificity of the H3K9me3 and HP1 antibodies was validated by showing it primarily localized to the centromere of 3rd instar polytene chromosomes (data not shown) and examination of the ChIP in the transition zone between euchromatin and pericentric heterochromatin. (Fig. S4) (Yasuhara & Wakimoto 2008).
Chromatin IP
Mixed sex Canton-S flies were grown in cages for 10 days or 40 days on standard food (6% yeast, 12% sucrose, 5% cornmeal), in a humidified (50%), incubator with 12 hour on/off light cycle at 25°C, changing food every 2-3 days. Flies were kept at −80°C, then sexed, and heads removed by vortexing. 200 mg of heads were used to prepare chromatin (Nègre et al. 2006),with sonication to an average size of 500 bp. 250 μl chromatin extract was immunoprecipitaed with one of the following antibodies: 4H8 (Abcam ab5408, 2 μl/IP), H3K4me3 (Abcam ab8580, 4 μl/IP), H3K9me3 (Millipore 07-442, 4 μl/IP), H3K36me3 (Abcam ab9050, 4 μl/IP), or HP1 (Developmental Studies Hybridoma Bank C1A9, 2 μl/IP). After elution, DNA was purified with the MinElute PCR cleanup kit (Qiagen) and amplified with the WGA2 kit (Sigma).
ChIP-chip
Chromatin IP samples were hybridized to Affymetrix Drosophila Tiling 2.0R arrays. Background probes were used to calculate median background signal which was subtracted from the signal of each genomic probe based on GC content. All non-unique probe sequences were removed from further analysis. The intensities were quantile normalized together, grouped by antibody. The Hodges-Lehman estimate of triplicate IP samples - triplicate input samples in a 500 bp window centered on the probe was calculated as signal. For each factor, the mean signal from the euchromatic region of the genome was subtracted from each probe.
RNA extraction, whole genome transcriptional arrays and preprocessing of the data
Total head mRNA was extracted from 75-100 heads of three independent biological replicates of 10 and 40 day old flies using Qiagen RNeasy total RNA isolation kit and hybridized to Affymetrix Drosophila 2 arrays according to the Affymetrix Protocol. See supplemental methods.
Probes were mapped to CG numbers using the drosophila2.db annotation package from Bioconductor. The data were quantile normalized and summarized using GCRMA (Wu et al. 2004) to obtain expression scores in the log2 scale.
Bioinformatic analysis
To plot the average trend of the marks over the exonic regions (Fig. 1), a construct including 2kbp upstream of the transcription start site, UTRS, the exons, and 2kbp downstream was made for each RefSeq gene location (genome.ucsc.edu). Then the Hodges-Lehman estimate of the ChIP signal (IP-input) was linearly interpolated over this region to give a fixed-length signal vector for each gene. k-means clustering using 2 clusters for each factor was performed on the signal after linear interpolation using the squared Euclidean distance. Transcripts with fewer than 10 probes in the window were removed before clustering. This left 16,760 unique transcription site windows (+/−1500 base pairs) and 20,486 unique whole-gene constructs. See supplemental methods.
Immunofluorescence
10, 40, 50, 60 and 70 day old male and female flies were fixed in 4% paraformaldehyde in PBS, embedded and cryosectioned. 10 micron sections containing fat body cells were incubated with H3K9me3 (1:500) or HP1 (1:1000) antibody, followed by the appropriate Alexa 488 or 568 -conjugated secondary antibody and DAPI staining, and imaging with a Zeiss Axioimager Z.1 microscope. Images of representative nuclei were selected and processed identically in Adobe Photoshop.
Western blots
Chromatin extracts were prepared as above from 10 and 40 day heads, except samples were not crosslinked with formaldehyde. Samples were normalized to total amount of chromatin, run on SDS-PAGE gels, blotted and probed with H3K9me3 (1:500), HP1 (1:1000), and total histone H3 (Epitomics 1326-1, 1:1000) antibodies.
Supplementary Material
Acknowledgements
The authors would like to thank Yiannis Savva and Robert A. Reenan for polytene chromosome staining to confirm the anti-H3K9me3 and anti-HP1 specificity, Adam Kroll for technical assistance and contributions from Stuart Geman and William Thompson. This work was supported by NIA grants AG16667, AG24353 and AG25277 to SLH and NIA AG028753 to NN and an Ellison Medical Foundation New Scholar award to ASB. SLH is an Ellison Medical Research Foundation Senior Investigator and recipient of a Glenn Award for Research in Biological Mechanisms of Aging.
Abbreviations
- ChIP-chip
chromatin immunoprecipitation whole genome tiling array
References
- Barski A, Cuddapah S, Cui K, Roh T-Y, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Barski A, Jothi R, Cuddapah S, Cui K, Roh TY, Schones DE, Zhao K. Chromatin poises miRNA- and protein-coding genes for expression. Genome Res. 2009;19:1742–1751. doi: 10.1101/gr.090951.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- Dang W, Steffen KK, Perry R, Dorsey JA, Johnson FB, Shilatifard A, Kaeberlein M, Kennedy BK, Berger SL. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature. 2009;459:802–807. doi: 10.1038/nature08085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dion MF, Altschuler SJ, Wu LF, Rando OJ. Genomic characterization reveals a simple histone H4 acetylation code. Proc Natl Acad Sci USA. 2005;102:5501–5506. doi: 10.1073/pnas.0500136102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert A, Lein S, Schotta G, Reuter G. Histone modification and the control of heterochromatic gene silencing in Drosophila. Chromosome Res. 2006;14:377–392. doi: 10.1007/s10577-006-1066-1. [DOI] [PubMed] [Google Scholar]
- Ebert A, Schotta G, Lein S, Kubicek S, Krauss V, Jenuwein T, Reuter G. Su(var) genes regulate the balance between euchromatin and heterochromatin in Drosophila. Genes Dev. 2004;18:2973–2983. doi: 10.1101/gad.323004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgin SCR, Grewal SIS. Heterochromatin: silence is golden. Curr Biol. 2003;13:R895–898. doi: 10.1016/j.cub.2003.11.006. [DOI] [PubMed] [Google Scholar]
- Grewal SIS, Jia S. Heterochromatin revisited. Nature Reviews Genetics. 2007;8:35–46. doi: 10.1038/nrg2008. [DOI] [PubMed] [Google Scholar]
- Hon G, Wang W, Ren B. Discovery and annotation of functional chromatin signatures in the human genome. PLoS Comput Biol. 2009;5:e1000566. doi: 10.1371/journal.pcbi.1000566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlic R, Chung HR, Lasserre J, Vlahovicek K, Vingron M. Histone modification levels are predictive for gene expression. Proc Natl Acad Sci U S A. 107:2926–2931. doi: 10.1073/pnas.0909344107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Villeponteau B, Jazwinski SM. Effect of replicative age on transcriptional silencing near telomeres in Saccharomyces cerevisiae. Biochem Biophys Res Commun. 1996;219:370–376. doi: 10.1006/bbrc.1996.0240. [DOI] [PubMed] [Google Scholar]
- Kim TH, Barrera LO, Zheng M, Qu C, Singer MA, Richmond TA, Wu Y, Green RD, Ren B. A high-resolution map of active promoters in the human genome. Nature. 2005;436:876–880. doi: 10.1038/nature03877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolasinska-Zwierz P, Down T, Latorre I, Liu T, Liu XS, Ahringer J. Differential chromatin marking of introns and expressed exons by H3K36me3. Nat Genet. 2009;41:376–381. doi: 10.1038/ng.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Lee TI, Jenner RG, Boyer LA, Guenther MG, Levine SS, Kumar RM, Chevalier B, Johnstone SE, Cole MF, Isono K, Koseki H, Fuchikami T, Abe K, Murray HL, Zucker JP, Yuan B, Bell GW, Herbolsheimer E, Hannett NM, Sun K, Odom DT, Otte AP, Volkert TL, Bartel DP, Melton DA, Gifford DK, Jaenisch R, Young RA. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006;125:301–313. doi: 10.1016/j.cell.2006.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- Muse GW, Gilchrist DA, Nechaev S, Shah R, Parker JS, Grissom SF, Zeitlinger J, Adelman K. RNA polymerase is poised for activation across the genome. Nat Genet. 2007;39:1507–1511. doi: 10.1038/ng.2007.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nègre N, Hennetin J, Sun LV, Lavrov S, Bellis M, White KP, Cavalli G. Chromosomal distribution of PcG proteins during Drosophila development. PLoS Biol. 2006;4:e170. doi: 10.1371/journal.pbio.0040170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park PJ. Epigenetics meets next-generation sequencing. Epigenetics. 2008;3:318–321. doi: 10.4161/epi.3.6.7249. [DOI] [PubMed] [Google Scholar]
- Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nature Reviews Genetics. 2009;10:669–680. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng JC, Karpen GH. H3K9 methylation and RNA interference regulate nucleolar organization and repeated DNA stability. Nat Cell Biol. 2007;9:25–35. doi: 10.1038/ncb1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabiner LR. A tutorial on hidden Markov Models and selected applications in speech recognition. Proceedings of the IEEE. 1989;77:257–286. [Google Scholar]
- Schones DE, Zhao K. Genome-wide approaches to studying chromatin modifications. Nature Reviews Genetics. 2008;9:179–191. doi: 10.1038/nrg2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedivy JM, Banumathy G, Adams PD. Aging by epigenetics--a consequence of chromatin damage? Exp Cell Res. 2008;314:1909–1917. doi: 10.1016/j.yexcr.2008.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims RJ, Nishioka K, Reinberg D. Histone lysine methylation: a signature for chromatin function. Trends Genet. 2003;19:629–639. doi: 10.1016/j.tig.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Smeal T, Claus J, Kennedy B, Cole F, Guarente L. Loss of transcriptional silencing causes sterility in old mother cells of S. cerevisiae. Cell. 1996;84:633–642. doi: 10.1016/s0092-8674(00)81038-7. [DOI] [PubMed] [Google Scholar]
- Talbert PB, Henikoff S. Spreading of silent chromatin: inaction at a distance. Nature Reviews Genetics. 2006;7:793–803. doi: 10.1038/nrg1920. [DOI] [PubMed] [Google Scholar]
- Wu Z, Irizarry RA, Gentleman R, Martinez Murillo F, Spencer F. A Model-Based Background Adjustment for Oligonucleotide Expression Arrays. Journal of the American Statistical Association. 2004;99 [Google Scholar]
- Yasuhara JC, Wakimoto BT. Molecular landscape of modified histones in Drosophila heterochromatic genes and euchromatin-heterochromatin transition zones. PLoS Genet. 2008;4:e16. doi: 10.1371/journal.pgen.0040016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.