

Abstract
Human galactokinase (GALK) is the first enzyme in the Leloir pathway, converting α-D-galactose into galactose-1-phosphate (Gal-1-P). Recently, there is increasing interest in targeting GALK as a novel therapy to ameliorate the disease manifestations in patients with Classic Galactosemia as it would, in combination with (ga-)lactose restriction reduce accumulation of Gal-1-P, a cytotoxic agent. Previously, we identified 34 small molecule compounds that inhibited GALK in vitro using experimental high-throughput screening. In order to isolate useful lead compounds, we characterized these hits with regards to their kinase selectivity profiles, potency and capability to reduce Gal-1-P accumulation in patient cell lines, and their modes of action. We found that the majority of these compounds had IC50s ranging from 0.7μM to 33.3μM. When tested against other members of the GHMP kinase family, three compounds (1, 4, and 24) selectively inhibited GALK with high potency. Through alignment of GALK and mevalonate kinase (MVK) crystal structures, we identified that eight amino acid residues and an L1 loop were different within the ATP-binding pockets of these two closely related kinases. By site-directed mutagenesis experiments, we identified one amino acid residue required for the inhibitory function of two of the three selective compounds. Based on these results, we generated binding models of these two compounds using a high-precision docking program. Compounds 4 and 24 inhibited GALK in a mixed model, while compound 1 exhibited parabolic competitive inhibition. Most importantly, using cells from galactosemic patients we found that selected compounds lowered Gal-1-P concentrations.
1. Introduction
In all living cells, the metabolism of α-D-galactose requires its conversion to galactose-1-phosphate (Gal-1-P) by the enzyme galactokinase (GALK). In the presence of galactose-1-phosphate uridyltransferase (GALT), Gal-1-P will react with UDP-Glucose to form UDP-Galactose and Glucose-1-phosphate [1]. Classic Galactosemia is an autosomal recessive metabolic disorder caused by the deficiency of galactose-1-phosphate uridyltransferase (GALT) [2], consequently leading to accumulation of Gal-1-P and deficiency of UDP-Galactose and UDP-Glucose in patient cells [3, 4]. If untreated, Classic Galactosemia can result in severe disease in the newborn period, including liver failure, coagulopathy, coma, and death [5–7]. Classic Galactosemia is included in newborn screening panels in the United States, since a galactose-restricted diet prevents the neonatal lethality of this disorder [8]. However, many well-treated newborns continue to develop complications such as premature ovarian insufficiency (POI), ataxia, speech dyspraxia, and mental retardation [7]. The causes of these organ-specific complications remain unknown, but there is a strong association with the intracellular accumulation of Gal-1-P. But what is the source of Gal-1-P in these patients with Classic Galactosemia if they limit their galactose intake? Recent studies have shown that the patients on a galactose-restricted diet are never really “galactose-free”. A significant amount of galactose is found in non-dairy foodstuffs such as vegetables and fruits [9, 10]. More importantly, galactose is produced endogenously from the natural turnover of glycolipids and glycoproteins [11]. Using isotopic labeling, Berry et al. demonstrated that a 50kg adult male could produce up to 2 grams of galactose per day [11, 12]. Once galactose is formed intracellularly, it is converted to Gal-1-P by GALK and in GALT-deficient patients' cells, Gal-1-P is concentrated more than one order of magnitude above normal, even with strict adherence to a galactose-restricted diet. Accumulation of Gal-1-P is regarded as a major, if not sole, factor for the chronic complications seen in patients with Classic Galactosemia, as suggested by both clinical observation and experimental results from the yeast models. First, patients with inherited deficiency of GALK, who do not accumulate Gal-1-P, do not experience the brain and ovary complications seen in GALT-deficient patients [13–15]. Second, while gal7 (i.e., GALT-deficient) mutant yeast stops growing upon galactose challenge, a ga17 ga11 double mutant strain (i.e., GALT- and GALK-deficient) is no longer sensitive to galactose [16, 17]. Based on these observations, in conjunction with dietary therapy, inhibiting GALK activity with a safe small-molecule inhibitor might prevent the sequelae of chronic Gal-1-P exposure in patients with Classic Galactosemia. Previously, we identified 34 GALK inhibitors from experimental high-throughput screening of 50,000 small molecule compounds with diverse structural scaffolds [18]. In this study, we define the selectivity, sensitivity, toxicity, in vitro kinetics and in vivo effects of selected inhibitors for GALK.
Although GALK phosphorylates galactose, a six-carbon monosaccharide, this human enzyme does not belong to the sugar kinase family. It is, in fact, an archetype of the GHMP kinase family (GHMP: Galactokinase, Homoserine kinase, Mevalonate kinase and Phosphomevalonate kinase) [19, 20]. Proteins belonging to this family have a different structure compared to other kinase families. All members of the GHMP kinase family have three conserved motifs (I, II and III). Motif II is the most conserved motif with a typical sequence of Pro-X-X-X-Gly-Leu-X-Ser-Ser-Ala and is involved in nucleotide binding [19]. The characterization of selective GALK inhibitors can therefore offer novel insights into the structural biology of the GHMP kinases.
2. Materials and Methods
2.1 Enzymes over-expression and purification
Cloning, over-expression and purification of human GALK were performed as described [18]. The plasmid containing human mevalonate kinase (MVK) cDNA was purchased from Invitrogen and the MVK cDNA was later sub-cloned into bacterial expression vector pET-21d. The expression plasmid clone of Methanococcus jannaschii homoserine kinase (HSK) was kindly shared by Dr. Hong Zhang, UT Southwestern Medical Center. The 4-diphosphocytidyl-2C-methyl-D-erythritol (CDP-ME) kinase gene from Escherichia coli DH5α was isolated by PCR in our laboratory and was subsequently sub-cloned into the vector pET-21d. The expression and purification protocols for MVK, HSK and CDP-ME kinase were the same as described for human GALK except there was no galactose in the purification buffers. Glucokinase (cat. #G8887) from Bacillus stearothermophilus and hexokinase (cat. #H4502) from Saccharomyces cerevisiae were purchased from Sigma-Aldrich (St. Louis, MO).
2.2 Site-directed Mutagenesis of human GALK cDNA
Primers for site-directed mutagenesis of human GALK cDNA are listed in Table 1. The loop mutation primers that replace the larger L1 loop of GALK into the smaller L1 loop of MVK are included. Using these primers, we introduced the mutations into the wild-type GALK cDNA sequence in pET-21d with QuickChange II Site-directed Mutagenesis Kit (cat. #200523, Stratagene (La Jolla, CA)). The introduced sequence changes were confirmed by direct DNA sequencing and the mutated proteins were expressed and purified as mentioned above.
Table 1.
Oligonucleotide primers used in site-directed mutagenesis experiments
| Residues of GALK | Residues of MVK | Site-directed mutagenesis primers* |
|---|---|---|
| Thr77 | Leu53 | T77L P1 5'-ggtgtctctcctcctcacctctgagggtgccgatg- 3' |
| T77L P2 5'-caccctcagaggtgaggaggagagacaccagcccatcc-3' | ||
|
| ||
| Ser79 | Asn57 | S79N P1 5'- ctcctcctcaccaatgagggtgccgatgagccccag -3' |
| S79N P2 5'- Cat cggcaccctcattggtgaggaggagagaca cc -3' | ||
|
| ||
| Leu145 | Tyr149 | L145Y P1 5'-ccagctcagcatcctacgaagtggccacgtacaccttcc-3' |
| L145Y P2 5'-cgtggccacttcgtaggatgctgagctggacaggcc-3' | ||
| L145A P1 5'-ccagctcagcatccgcggaagtggccacgtacaccttcc-3' | ||
| L145A P2 5'-cgtggccacttccgcggatgctgagctggacaggcc-3' | ||
|
| ||
| Trp106 | Thr104 | W106T P1 5'-ggactcctcggacggccaactatgtcaagggagtg-3' |
| W106T P2 5'-acatagttggccgtccgaggagtcccaggctccag-3' | ||
| W106A P1 5'-ggactcctcgggcggccaactatgtcaagggagtg-3' | ||
| W106A P2 5'-acatagttggccgcccgaggagtcccaggctccag-3' | ||
|
| ||
| Tyr109 | Leu107 | Y109L P1 5'- gtgggccaacttggtcaagggagtgattcagtacta -3' |
| Y109L P2 5'- actcccttgaccaagttggcccaccgaggagtcc -3 | ||
| Y109A P1 5'- gtgggccaacgctgtcaagggagtgattcagtacta -3 | ||
| Y109A P2 5'- actcccttgacagcgttggcccaccgaggagtcc -3 | ||
|
| ||
| Ser140 | Gly144 | S140G P1 5'-ggggtggcctgggcagctcagcatccttggaagtggc -3' |
| S140G P2 5'- aggatgctgagctgcccaggccaccccccaggggc -3' | ||
|
| ||
| Leu135 | Pro139 | L135P P1 5'- ctcagtgcccccggggggtggcctgtccagctcagc -3' |
| L135P P2 5'- ggccaccccccgggggcactgagctgaccaccac -3' | ||
|
| ||
| Arg37 | Lys7 | R37K P1 5'- cagcgccgggcaaagtcaacctcatcggggaacacacgg -3' |
| R37K P2 5'- atgaggttgactttgcccggcgctgacacggccagctcggg -3' | ||
| R37A P1 5'- cagcgccgggcgccgtcaacctcatcggggaacacacgg -3' | ||
| R37A P2 5'- atgaggttgacggcgcccggcgctgacacggccagctcggg -3' | ||
|
| ||
| Loop L1 | Loop L1 | Loop P1 5'- ggctggtgtctctcctcacccccaacattggtcggctgcagtttccactgcccacagcccag -3' |
| Loop P2 5'-gcagtggaaactgcagccgaccaatgttgggggtgaggagagacaccagcccatccttgcg-3' | ||
2.3 Enzyme activity assays and inhibition kinetic studies
The IC50 of all compounds were tested with the same two-step ATP-depletion assay for GALK developed with the Kinase-Glo™ reagent (Promega, Madison, WI) as described previously [18]. An alternative assay, the pyruvate kinase (PK)/lactate dehydrogenase (LDH)-coupled assay, which measures the steady-state velocity of the reaction, was also employed to determine the IC50 of compounds and the modes of inhibition. IC50 values were determined from normalized data from both assays using the values obtained for the corresponding controls (i.e., no inhibitor) as 100%. These data were fitted with a standard dose-response inhibition model using GraphPad Prism 5.01 software (GraphPad Software, Inc. La Jolla, CA), and a log[inhibitor] versus normalized response model, y = 100/[1 + 10(x–logIC50)], was established for each inhibitor. The models demonstrated adequate fit, with R2 values of all curves greater than 0.90.
For kinetic analyses of the GALK inhibitors, either galactose or ATP was held in excess while the other was changed under different inhibitor concentrations. The velocity of the reaction was measured by monitoring the change of absorbance at 340nm, and was plotted against the substrate concentration, then the curve was fit to the equation V= VMapp S/ (KMapp+S) by Sigma Plot 10.0 (Systat Softerware, Inc. San Jose, CA), where V was the measured reaction velocity, S was the substrate concentration, VMapp was the apparent VM and KMapp was the apparent KM. Then an inhibition pattern was assigned based on plotting 1/ VMapp against the inhibitor concentration and KMapp / VMapp or KMapp against the inhibitor concentration.
2.4 Cell culture, compounds toxicity assessment and measurement of Gal-1-P accumulation in cultured cells
Primary fibroblasts and SV40-transformed fibroblasts derived from GALT-deficient patients were maintained in galactose-free DMEM medium supplemented with 10% fetal bovine serum (FBS). For primary fibroblasts, only the ones less than 32 passages were used for the experiments, but for transformed fibroblasts there was no such limitation.
Compounds were incubated with primary fibroblasts at concentration of 10μM, 50μM and 100μM for three days, and then the cellular toxicity of the compounds was evaluated by estimating the cell survival ratio under light microscope.
Before galactose challenge, inhibitors were added to the medium at designated concentrations and incubated at 37°C for 2 to 4 hours. Then galactose was added to reach 0.05% in the medium. After 4 hours of challenge, cells were collected and washed with PBS twice. Then the cells were disrupted in 300μl of ice cold hypotonic buffer containing 25mM Tris-HCl (pH 7.4), 25mM NaCl, 0.5mM EDTA and protease inhibitor cocktail (Roche, cat. # 11 697 498 001). The lysates were passed five times through a 30 gauge needle and centrifuged for 20 minutes at 16,000g and 4°C. A small portion of supernatant was saved for protein concentration measurement. Gal-1-P level was measured using the methods previously described [21]. The Gal-1-P concentration was normalized to protein concentration.
2.5 Molecular docking experiments and crystal structure alignment
All experiments of docking compounds to the human GALK crystal structure were performed using GLIDE docking software from Schrodinger (GLIDE version 5.0, Schrödinger, New York) [22–24]. C subunit of human GALK crystal structure 1wuu was prepared for the docking through Schrödinger Maestro protein preparation wizard. First, all the water and metal molecules were removed and bond orders were assigned, necessary hydrogen was added. After the hydrogen bond assignment was optimized, the whole structure was minimized. Then the docking grid was generated on the prepared structure. Two grids were prepared: one centered at the AMPPNP and the other at the galactose moiety in the crystal structure. The compound structures were prepared with Maestro Ligpreparation. Possible ionization statistics were generated at pH from 5.0 to 9.0, then the structures were desalted, and different tautomers and stereoisomers were generated. Docking was preformed with the prepared Grid file and ligand file in extra precision modes (XP) [24].
Crystal structure alignment was also performed within Schrodinger Maestro software. Crystal structures for human GALK (1wuu), human mevalonate kinase (1kvk) and homoserine kinase (HSK) from Methanococcus jannaschii (1fwk) were downloaded. These structures were aligned according to αC helix where Motif II, one of the three conserved motifs originally used to define the GHMP kinases superfamily, located.
3. Results
3.1 IC50 and toxicity of human GALK inhibitors
Only 29 out of the 34 compounds identified from an experimental high-throughput screen were commercially available, and their IC50s were determined by both a two-step ATP-depletion assay and the PK/LDH-coupled assay. At the end, there was no difference between the results of these two assays, and the results from the two-step ATP-depletion assay were shown in Table 2. The IC50s of the compounds ranged from 0.7μM to 33μM. Three compounds (2, 15 and 25) had IC50s much higher than 33μM and were not considered for further characterization. We assessed the toxicity of compounds by incubating 10 – 100 μM of each compound with human diploid fibroblasts for 3 to 6 days and observed their effects on cell viability. The majority of selected compounds had mild to severe toxicity, but two compounds (10, 32) were not toxic at all up to 100μM. Two compounds (18, 23) had comparatively low toxicity at their IC50 range (Table 2).
Table 2.
In vitro IC50 of selected inhibitors against GALK
| Compound | IC50 (μM) | LD50 (μM) | Compound | IC50 (μM) | LD50 (μM) | Compound | IC50 (μM) | LD50(μM) | Compound | IC50 (μM) | LD50 (μM) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.7 | <10 | 10 | 6.5 | >100 | 19 | N/A* | 28 | 20 | 10 | |
| 2 | >40 | N/T** | 11 | 33.3 | 50 | 20 | 25 | <10 | 29 | N/A* | |
| 3 | 10.5 | 10 | 12 | N/A* | 21 | 18.5 | 10 | 30 | 21.1 | 50 | |
| 4 | 1.5 | <10 | 13 | 33.3 | 100 | 22 | 30 | 10 | 31 | 21 | 20 |
| 5 | 12 | 10 | 14 | 27.1 | 50 | 23 | 11.5 | 100 | 32 | 20.3 | >100 |
| 6 | 16.5 | 50 | 15 | >40 | N/T** | 24 | 6.3 | <10 | 33 | N/A* | |
| 7 | N/A* | 16 | 30 | 50 | 25 | >40 | N/T** | 34 | N/A* | ||
| 8 | 24 | 50 | 17 | 12.1 | 50 | 26 | 19.3 | 10 | |||
| 9 | 5.2 | 10 | 18 | 12.5 | 60 | 27 | 32 | 50 |
Not commercially available for study.
Not tested
3.2 Selectivity Profiles of human GALK inhibitors
To further prioritize the compounds, five kinases, glucokinase from Bacillus stearothermophilus, hexokinase from Saccharomyces cerevisiae, mevalonate kinase (MVK) from humans, homoserine kinase (HSK) from Methanococcus jannaschii and 4-Diphosphocytidyl-2C-methyl-D-erythritol (CDP-ME) kinase from E. coli DH5α were selected to serve as a small kinase panel to test the selectivity of the compounds. All five kinases catalyze a similar reaction to GALK by transferring the γ-phosphoryl group of ATP to a six-carbon sugar or their respective small molecule substrates. Despite GALK phosphorylates galactose, it does not belong to the same sugar kinase family as hexokinase and glucokinase. In fact, GALK and the other three kinases (MVK, HSK, CDP-ME kinase) belong to the GHMP kinase superfamily.
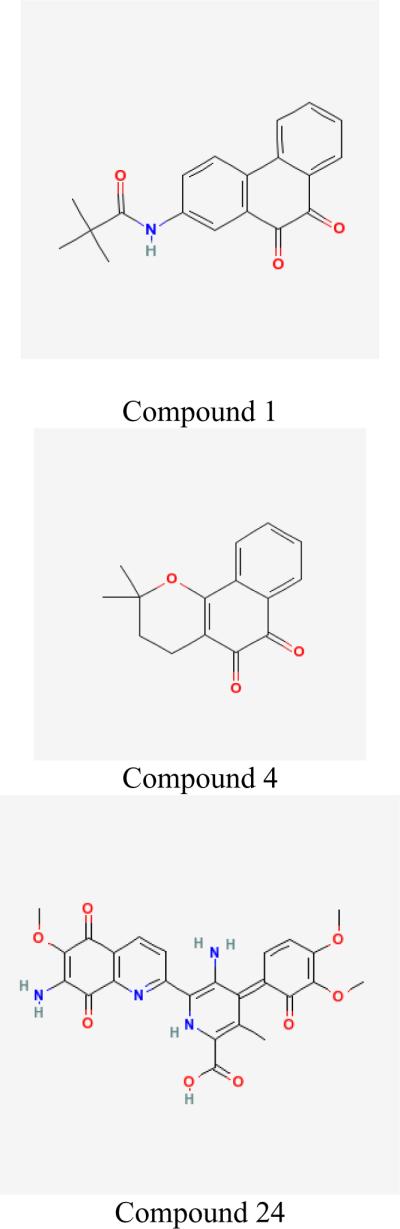
Results in Table 3 effectively described the selectivity profiles of the GALK inhibitors. None of the selected inhibitors interfered with hexokinase or glucokinase, except for compound 30. Compound 30 inhibited glucokinase weakly (IC50 > 60μM), but not hexokinase. When we tested the GALK inhibitors against other three GHMP kinases, we found a significant number of them also inhibited human MVK with IC50s similar to that of GALK (Table 3). Four compounds weakly inhibited HSK (IC50s>60μM) and eight inhibited bacterial CDP-ME kinase. However, six compounds (1, 4, 6, 23, 24, and 32) had no detectable inhibition against any of the tested GHMP kinases at a concentration up to 60μM, thus indicating that the unique selectivity of these six inhibitors against human GALK. Based on both potency and selectivity, we chose compound 1, 4, and 24 for further investigation (Fig. 1). Although compound 32 is selective and more importantly, less toxic, it could not be re-synthesized in large quantity, which made further investigation impossible.
Table 3.
Comparing IC50s of inhibitors to Galactokinase (GALK), Mevalonate kinase (MVK), Homoserine kinase (HSK), CDP-ME kinase, Glucokinase and Hexokinase
| GALK (μM) | MVK (μM) | HSK (μM) | CDP-ME kinase(μM) | Glucokinase/Hexokinase (μM) | GALK (μM) | MVK (μM) | HSK (μM) | CDP-ME kinase(μM) | Glucokinase/Hexokinase (μM) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.7 | >60 | >60 | >60 | >60 | 18 | 12.5 | 23 | >60 | >60 | >60 |
| 3 | 10.5 | 24.5 | >60 | >60 | >60 | 20 | 25 | 13 | >60 | 5.5 | >60 |
| 4 | 1.5 | >60 | >60 | >60 | >60 | 21 | 18.5 | 15 | >60 | 5.5 | >60 |
| 5 | 12 | 20 | >60 | 18 | >60 | 22 | 30 | 21.3 | >60 | >60 | >60 |
| 6 | 16.5 | >60 | >60 | >60 | >60 | 23 | 11.5 | >60 | >60 | >60 | >60 |
| 8 | 24 | 25.2 | >60 | 5 | >60 | 24 | 6.3 | >60 | >60 | >60 | >60 |
| 9 | 5.2 | 8.2 | >60 | 11 | >60 | 26 | 19.3 | 11.5 | >60 | >60 | >60 |
| 10 | 6.5 | 10.3 | 15.8 | >60 | >60 | 27 | 32 | 30 | >60 | >60 | >60 |
| 11 | 33.3 | 40 | >60 | >60 | >60 | 28 | 20 | 21 | >60 | >60 | >60 |
| 13 | 33.3 | 20 | >60 | 17 | >60 | 30 | 21.1 | 14.5 | >60 | >60 | Weak inhibition for Glucokinase |
| 14 | 27.1 | 16 | >60 | >60 | >60 | 31 | 21 | 18.5 | >60 | >60 | >60 |
| 16 | 30 | 40 | >60 | Not tested | >60 | 32 | 20.3 | >60 | >60 | >60 | >60 |
| 17 | 12.1 | 15.2 | >60 | 28 | >60 |
Note for the Table: Compounds were test against Mevalonate kinase (MVK), Homoserine kinase (HSK), CDP-ME kinase, Glucokinase and Hexokinase, and IC50S are shown here. IC50S for GALK also listed
Fig. 1. Structure of three compounds specifically inhibit human GALK.
3.3 Site-directed mutagenesis of human GALK
One interesting observation is that except for the six kinase inhibitors mentioned above, the other compounds inhibited both GALK and MVK with similar potency. On the other hand, the other two GHMP kinases (i.e., HSK and CDP-ME kinase) cross-reacted with fewer GALK inhibitors. Based on this observation, we hypothesized that GALK shared more structural similarity with MVK than with the two other GHMP kinases. To test this hypothesis, we aligned the protein sequences of human MVK and Methanococcus jannaschii HSK to that of the human GALK separately with ClustaIW2 [25]. Human GALK and MVK shared 70 identical residues and 69 conserved residues; for HSK, only 56 residues were identical to those of GALK and 67 residues were conserved. We then aligned the crystal structures of human MVK and Methanococcus jannaschii HSK separately to the crystal structure of human GALK (Figs. 2a, b). As shown in Fig. 2a, the ATP-binding site of GALK aligned well with that of MVK, except the L1 loop of GALK (red arrow) was larger than that of MVK (blue arrow) and covered a larger area. Note that the ATP (MVK) and the AMPPNP (GALK) co-crystallized with these enzymes were proximal to each other. However, as shown in Fig. 2b, when the L1 loop (blue arrow) of GALK was compared to the L1 loop of HSK (blue arrow), the L1 loop of HSK was shifted down and the structures are different. The L1 loop of HSK was closer to the αC helix as well as the adjacent β3 sheet as compared with GALK. These changes led to a narrower binding pocket for the adenine of ADP in HSK, which might explain why the ADP adopted a different binding pose for HSK. In fact, the adenine flipped almost 180 degrees away in the HSK crystal structure.
Fig. 2. Crystal structure alignment of GHMP kinases and amino acid residues which are different in ATP binding pockets of GALK and MVK.


a, human GALK (1wuu, green) aligned with human MVK (1kvk, orange), red arrow: L1 loop of GALK; blue arrow: L1 loop of MVK; AMPPNP in red color was the molecule from GALK structure; ATP in gray color was the molecule from MVK structure. b. human GALK (1wuu, green) aligned with HK (1fwk, maroon), red arrow: L1 loop of GALK; blue arrow: L1 loop of MVK; AMPPNP in red color was the molecule from GALK structure; ADP in light purple color was the molecule from HK structure. c. Residues which are different in ATP binding pocket of GALK. d. Residues which are different in ATP binding pocket of GALK.
To explain why the six compounds selectively inhibited human GALK but not human MVK, we compared the crystal structures of these two kinases in more detail, focusing on their ATP-binding pockets. Except for the L1 loop mentioned above, we found that eight amino acid residues were different within ATP binding pockets (Figs. 2c, d and Table 1). Based on this, we designed primers that changed the residues of GALK to corresponding residues of MVK (Table 1). Some of the residues were also changed to alanine.
We expressed these mutated proteins and tested their enzymatic properties. As shown in Table 4, the KM of ATP for eight of ten active GALK mutants increased and six out of eight increased more than ten-fold. This decrease in apparent affinity correlated with the fact that these residues are in the ATP-binding pocket. The KM for the other two mutants, S140G and R37K, decreased. Four of ten mutant GALK enzymes, T77L, S79N, Y109L and GALK Loop to MVK Loop, had an increased KM for galactose, but they are all less than five-fold. All kcat of the mutants decreased except Y109L, which increased more than two-fold. Among all active GALK mutants, the one that has its L1 loop changed had the worst effects, both the KM for galactose and ATP increased the most and kcat decreased the most.
Table 4.
Effect of amino acid changes in human GALK on their enzymatic properties and the IC50 of inhibitors 1, 4 and 24
| Mutations | kcat (s−1) | KM of ATP (μM) | KM of Galactose (μM) | Effects on IC50 of compound 1 | Effects on IC50 of compound 4 | Effects on IC50 of compound 24 |
|---|---|---|---|---|---|---|
| T77L | 4.1 | 218.4 | 1305.2 | None | None | None |
| S79N | 4.8 | 303.4 | 1227.3 | None | None | None |
| L145Y | 11.6 | 259.7 | 222.7 | None | None | None |
| LI 45 A | 6.4 | 379.9 | 356.8 | None | None | None |
| W106A | No protein expression | - | - | - | - | - |
| W106T | No protein expression | - | - | - | - | - |
| Y109L | 43.2 | 70.2 | 963.2 | None | None | None |
| Y109A | 8.7 | 579.3 | 268.7 | None | None | None |
| GALK Loop to MVK Loop | 0.1 | 695.4 | 1857.3 | None | None | None |
| SMOG | 2.1 | 8.2 | 141.9 | None | Increased 10-fold | Increased 20-fold |
| L135P | 13.3 | 51.1 | 544.9 | None | None | None |
| R37K | 0.4 | 6.4 | 623.8 | None | None | None |
| R37A | No activity | - | - | - | - | - |
| WT | 17.5 | 20.9 | 319 | - | - | - |
Surprisingly, none of the mutations affected the IC50 of compound 1. One mutation, S140G, increased the IC50 of compounds 4 and 24. Although the L1 loop is the most noticeable structural difference of these two closely related enzymes, and the substitution of this loop significantly impaired GALK activity, it did not affect IC50 of the three inhibitory molecules with human GALK. Ser140 of GALK resides at the signature motif of the GHMP kinase family, Motif II, but this amino acid is not conserved among the GHMP kinases and GALK is the only member that has a serine at this site. This could explain the selectivity of compound 4 and 24 for GALK (see Table 3). As for compound 1, we cannot postulate which residue or residues confer its selectivity for GALK relative to the other GHMP kinases based on the site-directed mutagenesis data.
3.4 Kinetic studies of the compounds
We analyzed the kinetics of inhibition by compounds 1, 4 and 24. Human GALK catalyzes the reaction in an ordered mechanism which first binds ATP and then galactose [26]. If the galactose concentration were held at saturated levels (1.5mM) and the ATP concentration were varied, compound 1 showed an unique inhibitory pattern known as parabolic inhibition (Figs. 3a and b). As shown in Fig. 3, the KMapp / VMapp increased nonlinearly as the concentration of the inhibitor increased: at higher compound concentrations the KMapp / VMapp increased dramatically compared with these kinetic parameters at lower concentrations. Since the 1/ VMapp did not change, compound 1 was regarded as a parabolic competitive inhibitor of ATP and described as Scheme 1 and Equation 1.
Fig. 3. Enzymatic study of compounds.



Enzyme inhibition by compound 1, 4 and 24 were tested under saturated galactose and varied ATP concentration (a, b, e, f, g, h) or saturated ATP and varied galactose concentration(c, d). Triplicate data were analyzed with Sigma Plot 10.0 software. a. Plots of Km / Vmax vs compound 1 concentration when galactose was saturated and ATP varied. b. Plot of 1/ Vmax vs. compound 1 concentration under the same condition as a. c. Plot of 1/ Vmax vs. compound 1concentration when ATP was saturated and galactose varied. d. Plot of Km vs. compound 1concentration under the same condition as c. e. Plots of 1/ Vmax vs compound 4 concentration under the same condition as a. f. Plot of Km / Vmax vs. compound 4 concentration under the same condition as a. g. Plot of 1/ Vmax vs compound 24 concentration under the same condition as a. h. Plot of Km / Vmax vs. compound 24 concentration under the same condition as a.
Scheme 1.

Parabolic competitive inhibition
Four equations can be derived from the above scheme, which represent parabolic competitive inhibition.
| Equation 1 |
| Equation 2 |
| Equation 3 |
| Equation 4 |
Equations 2, 3 & 4 define the results shown in Fig. 1a & b in which KMapp / VMapp increased parabolicly when compound 1 increased and 1/ VMapp was kept constant.
If Equation 1 were rearranged according to B, where ATP was at a saturated level with varying galactose concentration, then we will have the followings
| Equation 5 |
and
| Equation 6 |
| Equation 7 |
When we performed the experiments, ATP level was saturated; As a result, A was much larger than KA, so KA +A+I KA /Ki1+I2 KA /Ki1Ki2 ≈ A+I KA /Ki1+I2 KA /Ki1Ki2. KMapp could be regarded as constant and 1/ VMapp increased parabolically as inhibitor concentration increased just as shown in Figs. 3c and 3d. These results confirmed that the assumption of Scheme 1 was correct and compound 1 was a parabolic competitive inhibitor for GALK.
The kinetic results for compounds 4 and 24 indicated that they are typical mixed inhibitors for GALK as shown in Figs. 3e – h. The unusual behavior of compound 1 may explain the reason why changes of amino acids at the presumptive ATP-binding site of GALK did not reveal any specific residues that interacted with this compound, as it probably has two binding sites for GALK. The sperm-specific isozyme of lactate dehydrogenase (LDH-C4) also exhibited “parabolic mixed-type” inhibition by gossypol [27]. The kinetic data and site-directed mutagenesis data of compounds 4 and 24 are consistent and suggest that these two inhibitors interact with the same amino acid residue in GALK and inhibit the enzyme by similar mechanisms.
3.5 Docking of compounds to GALK
Based on the information from site-directed mutagenesis and kinetic studies, we used a molecular docking program to generate possible binding models for compounds 4 and 24. Docking programs have been applied widely in drug development for virtual screening of enzyme inhibitors and predicting interactions of compounds and target proteins. Among the software programs cited in literature, GLIDE (Schrödinger, LLC, New York) is regarded as one of the best [28].
The protein structure we used was human GALK protein co-crystallized with galactose and the ATP analogue AMPPNP (PDB: 1wuu). First, we evaluated the software by self dockings: Galactose and AMPPNP were docked back to the protein structure which was already prepared as mentioned in the methods section. The docking poses of galactose and AMPPNP were identical to the poses determined in the crystal structure (data not shown). The root mean square deviations (RMSD) between docked poses and crystal structure were less then 1.5 Å for both galactose and AMPPNP. These results supported our desire to use the GLIDE docking program for further virtual modeling.
Next, compounds 4 and 24 were prepared and docked to the virtual protein structure. At first, we did not set any bond constraints for the docking exercises. Surprisingly, both compounds 4 and 24 did not form interactions with Ser140 as suggested from the site-directed mutagenesis data. Instead, they formed hydrogen-bonds with the hydroxyl group of Ser141 which is conserved among the GHMP kinase family (Figs. 4c and d). Interestingly, the structures of GALK and MVK associated with this site are nearly identical and the two serines of GALK and MVK at position 141 overlapped. It is highly unlikely that the change from serine to glycine at amino acid-140 of GALK would cause any significant change of Ser141. Also, as shown in Figs. 4c and d, the two compounds were close to other residues in the mutagenesis studies and if these docking poses are correct, it is likely that some of these mutations in these residues will affect the binding of these two compounds. Yet, the results from our site-directed mutagenesis experiments did not support this hypothesis. Further, since these two compounds appeared to dock to the middle of ATP-binding site in these docking poses, they should act as ATP competitive inhibitors, instead of mixed inhibitors as shown in our kinetic studies. Then we performed the docking experiments again with constraints that the compounds need to form interactions with Ser140. The new binding models are shown in Figs. 4a and b. For compound 4, as shown in Fig. 4a, two dione oxygen atoms of these compounds faced residue Ser140, and form hydrogen bonds with the hydroxyl group and the backbone hydrogen atom of Ser140. More importantly, the docking pose of compound 4 did not interact with other residues included in the mutagenesis studies such as Tyr109, Leu145, Thr77, Ser79 and Leu135. These GALK mutations did not affect the binding affinity of compound 4. Also, the docking site which compound 4 occupied was close to the ATP-binding site, which explained its' mixed competition to ATP determined by the kinetic studies (Figs. 3e and f).
Fig.4. Docking of compound 4 and 24 to GALK (1wuu).


Docking of compound 4 (a, c) and 24 (b,d ) to GALK, with constrains (a, b) and without constrains (c,d). Molecules in green are compounds. Ser140 is in dark blue, Ser141 is in purple, other residues of the site-directed mutagenesis studies are in yellow. Other residues surrounding the compounds are in gray. Yellow dot lines are hydrogen-bond formed between the compounds and protein.
Since the binding affinity of compound 24 was affected by the same amino acid residue, Ser140, as compound 4, it is reasonable that the docking pose of compound 24 also formed hydrogen-bond with the hydroxyl hydrogen of Ser140, as shown in Fig. 4b. In addition, this docking model indicated that compound 24 formed extensive hydrogen-bond interactions with other residues: Arg228, Glu174, Tyr 236, Ser142 and Arg105. Again, compound 24 had no interaction with other residues included in the mutagenesis studies. Similar to compound 4, compound 24 occupied the ATP phosphate binding domain and the area close to the catalytic center, which might also explain its mixed inhibition as shown in kinetic studies (Fig. 4d and Figs. 3g, h).
3.6 Lowering Gal-1-P in primary and transformed fibroblasts
Earlier attempts to lower intracellular Gal-1-P concentrations in cultured diploid primary GALT-deficient fibroblasts challenged with galactose were not successful due to the inherent toxicity of compounds (data not published). This motivated us to try an SV40-transformed GALT-deficient fibroblast cell line, GM00638A, which was more robust in culture and tolerant to compound 1 and 24. These cells remain viable in the presence of 10μM of compound 1 and 30μM of compound 24. More importantly, these GALT-deficient cells accumulated Gal-1-P upon galactose challenge (Figs. 5a, b). When treated with compound 1, there was a dose-dependent effect of this GALK inhibitor on Gal-1-P accumulation. At an external concentration of 6μM, Compound 1 lowered intracellular Gal-1-P to a level that approached that of the non-treated control (Fig. 5a). Similarly, treatment of compound 24 at 20μM decreased the accumulation of Gal-1-p by 70% as compared to the positive control (Fig. 5b). This is the proof-of-concept data that small molecule inhibitors of GALK can lower Gal-1-P in galactose-challenged GALT-deficient cells. Unfortunately, long-term exposure of these first-generation GALK inhibitors was too toxic for diploid cells. The kinetic and structural interactions of this first group of selective GALK inhibitors will, however, enable future development of non-toxic and more specific GALK inhibitors.
Fig. 5. Gal-1-P level upon compound 1 and 24 treatment in GM00638A.

Gal-1-p levels were measured after the cells were treated with compound for 4 hours with increasing concentration and then challenged with 0.05% galactose. Cells in glucose served as negative control.
4. Discussion
Elevated concentrations of Gal-1-P in GALT-deficient cells is considered the major culprit of chronic, organ-specific dysfunction in patients with Classic Galactosemia. As Gal-1-P is the product of galactokinase (GALK), at least four groups of investigators proposed to inhibit the GALK enzyme with specific, non-toxic, low molecular weight compounds as a rational and novel approach to treat Classic Galactosemia [18, 29–31]. We are aware of the suggested possibility that some of the GALK inhibitors might also inhibit N-acetylgalactosamine kinase (GALK2) due to the structural similarity [32], and potentially impair the biosynthesis of N-acetylgalactosamine-1-phosphate - an important intermediate in the biosynthesis of UDP-N-acetylgalactosamine. However, as UDP-galactose 4-epimerase (GALE) catalyses the interconversion between UDP-N-acetylgalactosamine and UDP-N-acetylglucosamine [33], any inhibition of GALK2 is unlikely to result in deficiency of UDP-N-acetylgalactosamine. To date, our group has screened different chemical compound libraries composed of about 50,000 small molecules with diverse structural scaffolds for their inhibitory properties against activity of purified GALK. Thus far, we have identified nearly 150 small molecules (or hits) that inhibited human GALK activity in vitro at the level of 86.5% or more [18]. We selected 34 of these compounds for further characterization in this study. For the first time, we demonstrated the reduced accumulation of Gal-1-P in GALT-deficient cells treated with two small molecule compounds (compounds 1 and 24) selectively inhibiting GALK (Fig. 5). This is a proof-of-concept that stronger, non-toxic, and more specific small molecule GALK inhibitors might provide adjunct therapy for patients with Classic Galactosemia. Nevertheless, we must recognize that like most drug discovery projects, the identification of these first-generation lead compounds only marks the beginning of a long journey of drug development. To optimize these lead compounds and make them applicable as therapeutics in the future, one must thoroughly characterize them with respect to their potency (Table 2.), modes of inhibition (Fig. 3), selectivity/ toxicity profiles (Table 3.), and interactions with the GALK target at the structural level (Fig. 4).
Yet, one must not assume that the characterization of these inhibitors is only useful for the discovery of more potent and safe therapeutics. In fact, as we showed in this study, the characterization process also offers novel insights into the fundamental knowledge on the structure-functions of GALK and other members of the GHMP kinases. For instance, the lack of cross-inhibition for nearly all GALK inhibitors identified for the highly conserved glucokinase and hexokinase confirmed the fact that despite GALK phosphorylates galactose, it is remotely related to the members of the sugar kinase family (Table 3). As we examined the selectivity profiles of the GALK inhibitors against other closely-related GHMP kinases, we identified six compounds that uniquely inhibited GALK (Table 3). Although these results were not entirely unexpected, we were intrigued by them, especially after we compared the crystal structures of human GALK and human MVK (Fig. 2). From Fig. 2, it is clear that the active sites of these two closely-related GHMP kinases are highly similar, which is not surprising as human GALK and human MVK shared 70 identical residues and 69 conserved residues. So what are the structural determinants of the two GHMP kinases giving rise to the observed selectivity of the GALK inhibitors? We decided to address this fundamental question with site-directed mutagenesis experiments and computer modeling.
From our initial structural analysis, the difference in size of the L1 loops in human GALK and human MVK became obvious and presented a logical explanation for the difference in the selectivity of inhibitors towards the two closely-related GHMP kinases (Figs. 2a, b). However, further investigation revealed that this was not the case. Although this loop affects the GALK activity, maybe through affecting ATP binding, it did not appear to interact with the compounds we tested here (Table 4). Through site-directed mutagenesis, we altered the non-conserved amino acids at the ATP-binding site of GALK and re-tested the inhibitors with the resulting variant GALK proteins. We found that one amino acid residue, Ser140, conferred the selectivity for two compounds of interest, compound 4 and 24, respectively. Using this knowledge, we then generated binding models of these two compounds with the high precision docking program called GLIDE (Schrödinger, LLC New York, 2009). The virtual models correlated with the actual results from site-directed mutagenesis and kinetic studies, and gave us the first impression of how these inhibitors might interact with GALK. One interesting observation is that the residue, Ser140, and the computer models indicated that the selectivity of these two compounds for GALK was conferred by the area near the phosphate-binding region, while in protein kinases the two hydrophobic regions close to adenine binding region and the sugar-binding pocket often determine the selectivity of kinase inhibitors [34].
The most potent inhibitor identified from high-throughput screening, compound 1, inhibited GALK as a parabolic competitive inhibitor as shown in kinetic studies (Figs. 3a – d). This suggested there are two binding sites for compound 1 in the GALK protein. We could not identify specific residue interacts with this compound and therefore, we could not confirm a binding model. Interestingly, the structures of compound 1 and 4 are very similar. Compound 1 is a derivative of dioxophenanthren and compound 4 is very similar to dioxophenanthren except it has a nitro substitution. Yet they behave so differently with regard to GALK inhibition.
In conclusion, we identified three compounds which selectively inhibited GALK with high potency, and illustrated the inhibition modes of these compounds to GALK. Moreover, through site-directed mutagenesis we identified one amino acid residue required for the inhibitory function of two of the three selective compounds. Based on these results, we generated binding models of these two compounds using a high-precision docking program. Lastly, we showed for the first time two of the GALK inhibitor prevented Gal-1-P accumulation in galactose-challenged GALT-deficient cells.
Acknowledgement
Research grant support to Kent Lai includes NIH grant 5R01 HD054744-04, 5R01 HD054744-04S1 and 7R03 MH085689-02.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Leloir LF. The enzymatic transformation of uridine diphosphate glucose into a galactose derivative. Arch Biochem. 1951;33(2):186–90. doi: 10.1016/0003-9861(51)90096-3. [DOI] [PubMed] [Google Scholar]
- 2.Isselbacher KJ, et al. Congenital galactosemia, a single enzymatic block in galactose metabolism. Science. 1956;123(3198):635–6. doi: 10.1126/science.123.3198.635. [DOI] [PubMed] [Google Scholar]
- 3.Gitzelmann R, Curtius HC, Schneller I. Galactitol and galactose-1-phosphate in the lens of a galactosemic infant. Exp Eye Res. 1967;6(1):1–3. doi: 10.1016/s0014-4835(67)80047-2. [DOI] [PubMed] [Google Scholar]
- 4.Lai K, et al. GALT deficiency causes UDP-hexose deficit in human galactosemic cells. Glycobiology. 2003;13(4):285–94. doi: 10.1093/glycob/cwg033. [DOI] [PubMed] [Google Scholar]
- 5.Sitzmann FC, Kaloud H. [Congenital disorders of galactose metabolism] Med Klin. 1975;70(12):491–8. [PubMed] [Google Scholar]
- 6.Rennert OM. Disorders of galactose metabolism. Ann Clin Lab Sci. 1977;7(6):443–8. [PubMed] [Google Scholar]
- 7.Dogan K, et al. [Inborn disorders of galactose metabolism: report of a case of “classical galactosemia” (author's transl)] Lijec Vjesn. 1977;99(10):589–94. [PubMed] [Google Scholar]
- 8.Kaye CI, et al. Newborn screening fact sheets. Pediatrics. 2006;118(3):e934–63. doi: 10.1542/peds.2006-1783. [DOI] [PubMed] [Google Scholar]
- 9.Berry GT, et al. The effect of dietary fruits and vegetables on urinary galactitol excretion in galactose-1-phosphate uridyltransferase deficiency. J Inherit Metab Dis. 1993;16(1):91–100. doi: 10.1007/BF00711320. [DOI] [PubMed] [Google Scholar]
- 10.Acosta PB, Gross KC. Hidden sources of galactose in the environment. Eur J Pediatr. 1995;154(7 Suppl 2):S87–92. doi: 10.1007/BF02143811. [DOI] [PubMed] [Google Scholar]
- 11.Berry GT, et al. Endogenous synthesis of galactose in normal men and patients with hereditary galactosaemia. Lancet. 1995;346(8982):1073–4. doi: 10.1016/s0140-6736(95)91745-4. [DOI] [PubMed] [Google Scholar]
- 12.Berry GT, et al. The rate of de novo galactose synthesis in patients with galactose-1-phosphate uridyltransferase deficiency. Mol Genet Metab. 2004;81(1):22–30. doi: 10.1016/j.ymgme.2003.08.026. [DOI] [PubMed] [Google Scholar]
- 13.Gitzelmann R. Letter: Additional findings in galactokinase deficiency. J Pediatr. 1975;87(6 Pt 1):1007–8. doi: 10.1016/s0022-3476(75)80937-1. [DOI] [PubMed] [Google Scholar]
- 14.Stambolian D, et al. Cataracts in patients heterozygous for galactokinase deficiency. Invest Ophthalmol Vis Sci. 1986;27(3):429–33. [PubMed] [Google Scholar]
- 15.Gitzelmann R, Wells HJ, Segal S. Galactose metabolism in a patient with hereditary galactokinase deficiency. Eur J Clin Invest. 1974;4(2):79–84. doi: 10.1111/j.1365-2362.1974.tb00376.x. [DOI] [PubMed] [Google Scholar]
- 16.Douglas HC, Hawthorne DC. Enzymatic Expression And Genetic Linkage Of Genes Controlling Galactose Utilization In Saccharomyces. Genetics. 1964;49:837–44. doi: 10.1093/genetics/49.5.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Douglas HC, Hawthorne DC. Regulation of genes controlling synthesis of the galactose pathway enzymes in yeast. Genetics. 1966;54(3):911–6. doi: 10.1093/genetics/54.3.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wierenga KJ, et al. High-throughput screening for human galactokinase inhibitors. J Biomol Screen. 2008;13(5):415–23. doi: 10.1177/1087057108318331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bork P, Sander C, Valencia A. Convergent evolution of similar enzymatic function on different protein folds: the hexokinase, ribokinase, and galactokinase families of sugar kinases. Protein Sci. 1993;2(1):31–40. doi: 10.1002/pro.5560020104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bork P, Sander C, Valencia A. An ATPase domain common to prokaryotic cell cycle proteins, sugar kinases, actin, and hsp70 heat shock proteins. Proc Natl Acad Sci U S A. 1992;89(16):7290–4. doi: 10.1073/pnas.89.16.7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lai K, Willis AC, Elsas LJ. The biochemical role of glutamine 188 in human galactose-1-phosphate uridyltransferase. J Biol Chem. 1999;274(10):6559–66. doi: 10.1074/jbc.274.10.6559. [DOI] [PubMed] [Google Scholar]
- 22.Friesner RA, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47(7):1739–49. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 23.Halgren TA, et al. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem. 2004;47(7):1750–9. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 24.Friesner RA, et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem. 2006;49(21):6177–96. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 25.Larkin MA, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–8. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 26.Ballard FJ. Kinetic studies with liver galactokinase. Biochem J. 1966;101(1):70–5. doi: 10.1042/bj1010070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stephens DT, et al. Kinetic characterization of the inhibition of purified cynomolgus monkey lactate dehydrogenase isozymes by gossypol. J Androl. 1986;7(6):367–77. doi: 10.1002/j.1939-4640.1986.tb00946.x. [DOI] [PubMed] [Google Scholar]
- 28.Michino M, et al. Community-wide assessment of GPCR structure modelling and ligand docking: GPCR Dock 2008. Nat Rev Drug Discov. 2009;8(6):455–63. doi: 10.1038/nrd2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bosch AM. Classical galactosaemia revisited. J Inherit Metab Dis. 2006;29(4):516–25. doi: 10.1007/s10545-006-0382-0. [DOI] [PubMed] [Google Scholar]
- 30.Timson DJ. GHMP Kinases - Structures, Mechanisms and Potential for Therapeutically Relevant Inhibition. Current Enzyme Inhibition. 2007;3(1):77–94. [Google Scholar]
- 31.Fridovich-Keil J. Toward Improved Intervention for Classic Galactosemia. 2007 http://www.galactosemia.org/PGC_awards.asp.
- 32.Agnew A, Timson D. Mechanistic studies on human N-acetylgalactosamine kinase. J Enzyme Inhib Med Chem. 2009 doi: 10.3109/14756360903179492. [DOI] [PubMed] [Google Scholar]
- 33.Thoden JB, et al. Molecular basis for severe epimerase deficiency galactosemia. X-ray structure of the human V94m-substituted UDP-galactose 4-epimerase. J Biol Chem. 2001;276(23):20617–23. doi: 10.1074/jbc.M101304200. [DOI] [PubMed] [Google Scholar]
- 34.Cherry M, Williams DH. Recent kinase and kinase inhibitor X-ray structures: mechanisms of inhibition and selectivity insights. Curr Med Chem. 2004;11(6):663–73. doi: 10.2174/0929867043455792. [DOI] [PubMed] [Google Scholar]